
Efficient Consecutive Synthesis of Ethyl-2-(4-Aminophenoxy) Acetate, a Precursor for Dual GK and PPARγ Activators, X-ray Structure, Hirshfeld Analysis, and DFT Studies



 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Equipments
2.2. Synthesis of Ethyl-2-(4-Aminophenoxy) Acetate (4)
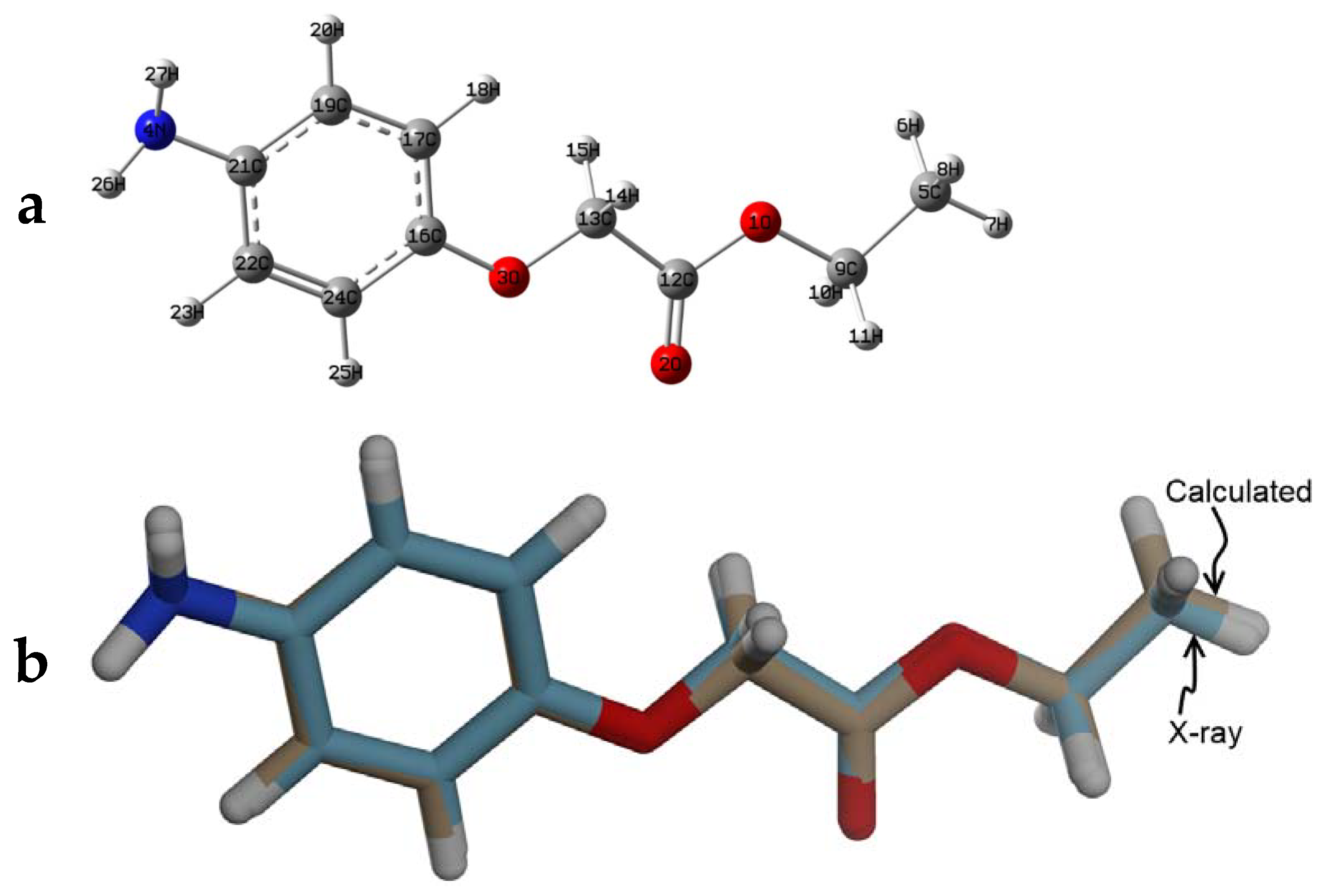
2.3. X-ray Structure Determination
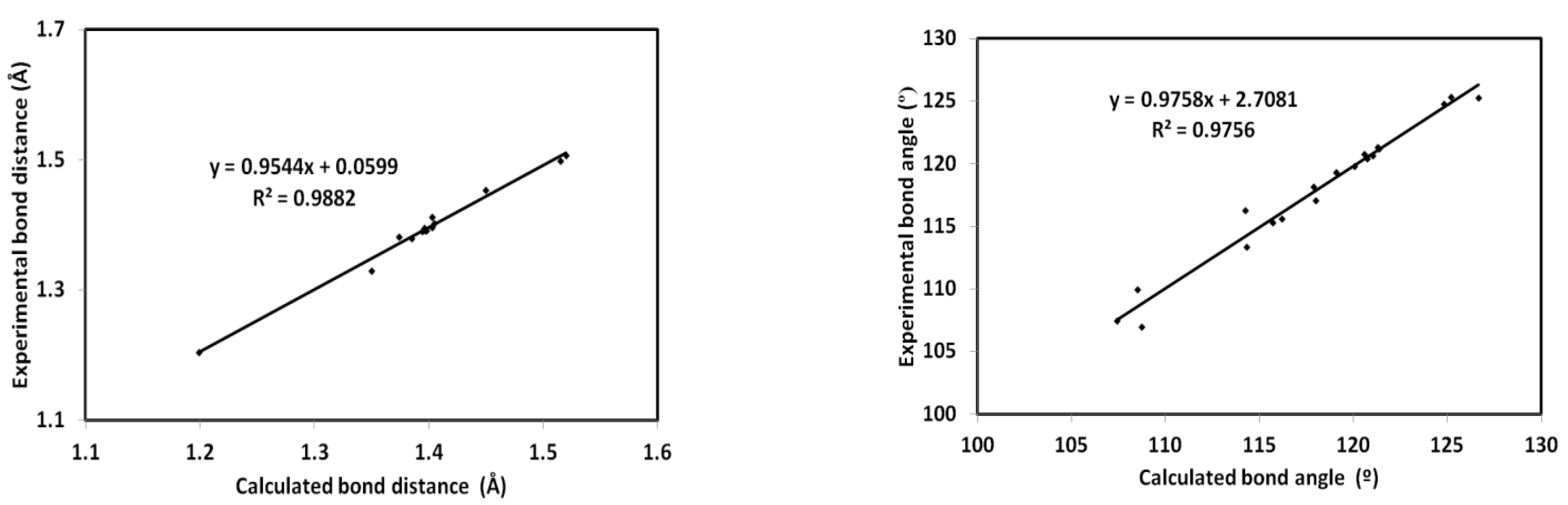
2.4. Computational Methods
3. Results and Discussion
3.1. Chemistry
3.2. X-ray Structure
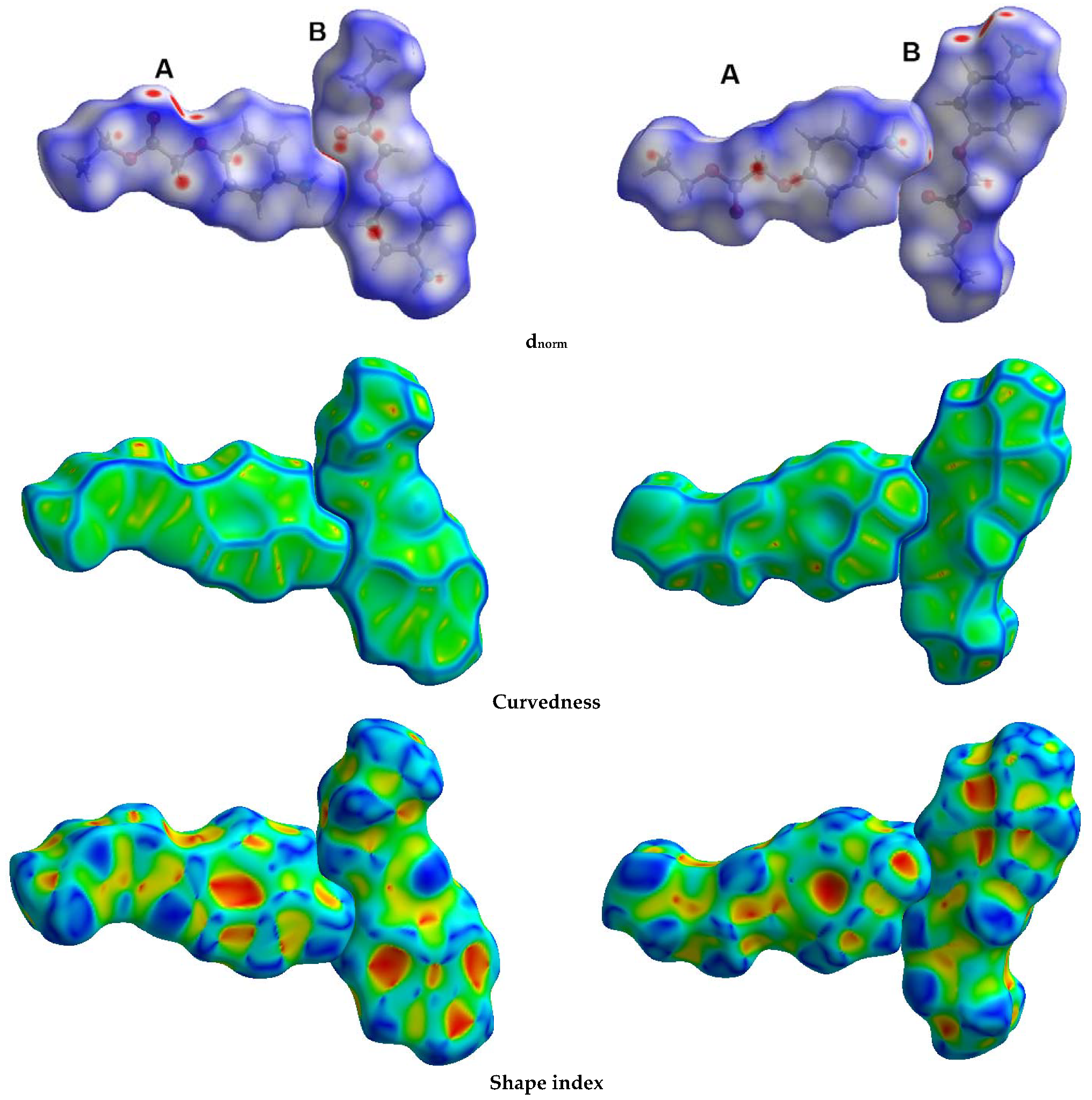
3.3. Hirshfeld Surface Analysis
3.4. DFT Studies
3.5. NBO Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Appel, S.J.; Wadas, T.M. Characterization of Type 2 Diabetes into Five Sub-Types. Curr. Res. Diabetes Obes. J. 2018, 8, 27–29. [Google Scholar] [CrossRef]
- American Diabetes Association. Diagnosis and Classification of Diabetes Mellitus. Diabetes Care 2009, 32, S62–S67. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Chen, L. Berberinein type 2 diabetes therapy: A new perspective for an old antidiarrheal drug? Acta Pharm. Sin. B 2012, 2, 378–386. [Google Scholar] [CrossRef]
- Matschinsky, F.M.; Glaser, B.; Magnuson, M.A. Pancreatic beta-cell glucokinase: Closing the gap between theoretical concepts and experimental realities. Diabetes 1998, 47, 307–315. [Google Scholar] [CrossRef]
- Moller, D.E. New drug targets for type2 diabetes and the metabolic syndrome. Nature 2001, 414, 821–827. [Google Scholar] [CrossRef]
- Janani, C.; Kumari, B.R. PPAR gamma gene—A review. Diabetes Metab. Syndr. Clin. Res. Rev. 2015, 9, 46–50. [Google Scholar] [CrossRef]
- Chen, D.; Guo, D.; Yan, Z.; Zhao, Y. Allenamide as a bioisostere of acrylamide in the design and synthesis of targeted covalent inhibitors. Med. Chem. Commun. 2018, 9, 244–253. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Samanta, I.; Mondal, A.; Liu, W.R. Covalent inhibition in drug discovery. ChemMedChem 2019, 14, 889–906. [Google Scholar] [CrossRef]
- Serrano-Sterling, C.; Becerra, D.; Portilla, J.; Rojas, H.; Macías, M.; Castillo, J.C. Synthesis, biological evaluation and X-ray crystallographic analysis of novel (E)-2-cyano-3-(het) arylacrylamides as potential anticancer agents. J. Mol. Struct. 2021, 244, 130944. [Google Scholar] [CrossRef]
- Rikagu Oxford Diffraction. CrysAlisPro; Agilent Technologies Inc.: Oxfordshire, UK, 2018. [Google Scholar]
- Sheldrick, G.M. SHELXT-Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt graphical user interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. Crystal Explorer17; University of Western Australia: Crawley, WA, Australia, 2017; Available online: http://hirshfeldsurface.net (accessed on 20 May 2017).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. GAUSSIAN 09; Revision A02; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- GaussView; Dennington, R., II; Keith, T.; Millam, J. (Eds.) Semichem Inc.: Shawnee Mission, KS, USA, 2007; Version 4.1. [Google Scholar]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
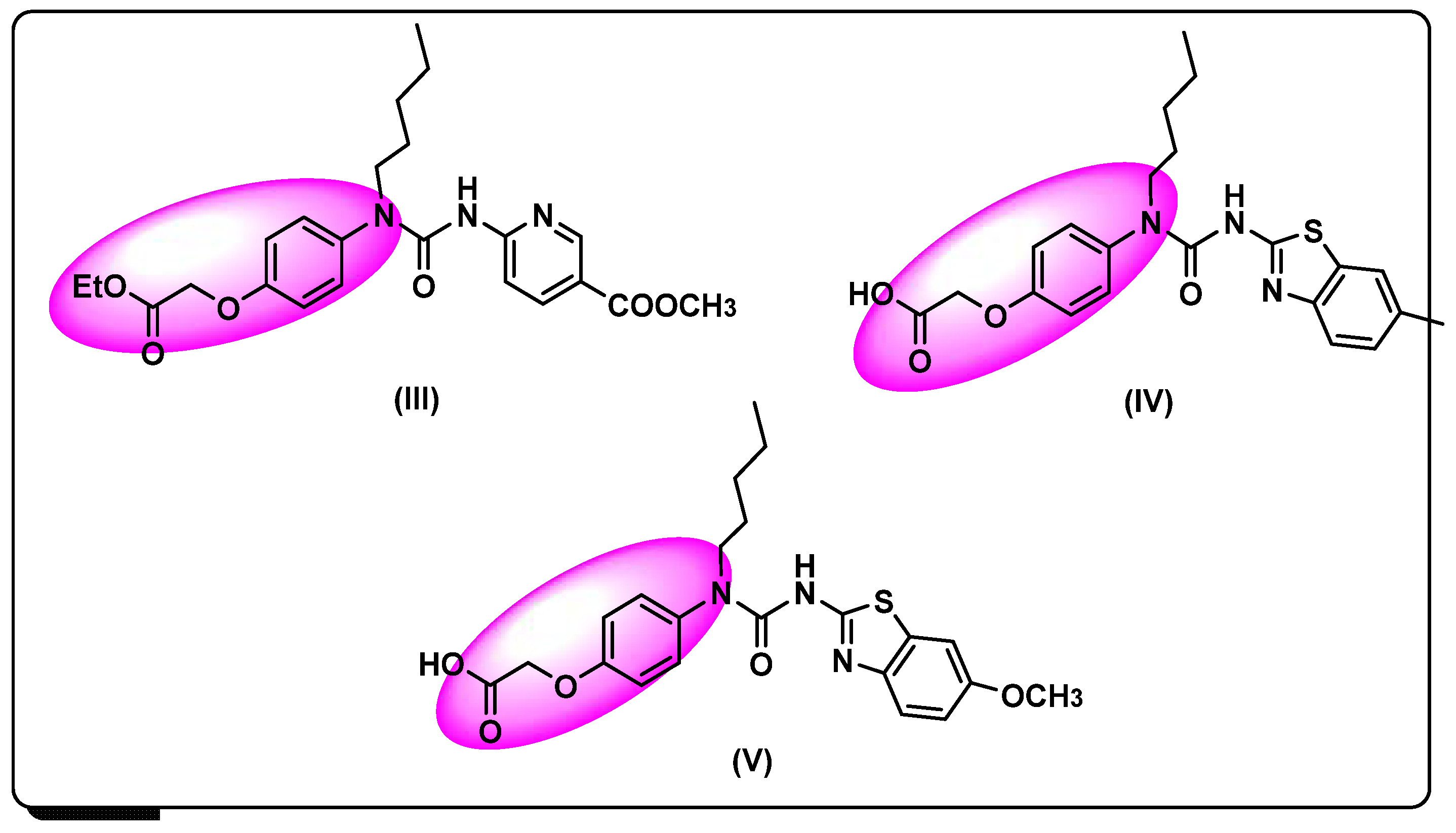
- Zhang, L.; Tian, K.; Li, Y.; Lei, L.; Qin, A.; Zhang, L.; Song, H.; Huo, L.; Zhang, L.; Jin, X.; et al. Novel phenyl-urea derivatives as dual-target ligands that can activate both GK and PPARγ. Acta Pharm. Sin. B 2012, 2, 588–597. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Spackman, M.A.; Mitchell, A.S. Novel tools for visualizing and exploring intermolecular interactions in molecular crystals. Acta Crystallogr. Sect. B Struct. Sci. 2004, 60, 627–668. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces. Chem. Commun. 2007, 37, 3814–3816. [Google Scholar] [CrossRef] [PubMed]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Foresman, J.B.; Frisch, E. Exploring Chemistry with Electronic Structure Methods, 2nd ed.; Gaussian: Pittsburgh, PA, USA, 1996. [Google Scholar]
- Chang, R. Chemistry, 7th ed.; McGraw-Hill: New York, NY, USA, 2001. [Google Scholar]
- Kosar, B.; Albayrak, C. Spectroscopic investigations and quantum chemical computational study of (E)-4-methoxy-2-[(p-tolylimino) methyl] phenol. Spectrochim. Acta 2011, 78, 160–167. [Google Scholar] [CrossRef]
- Koopmans, T.A. Ordering of wave functions and eigenenergies to the individual electrons of an atom. Physica 1933, 1, 104–113. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Parr, R.G.; Szentpaly, L.V.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Singh, R.N.; Kumar, A.; Tiwari, R.K.; Rawat, P.; Gupta, V.P. A combined experimental and quantum chemical (DFT and AIM) study on molecular structure, spectroscopic properties, NBO and multiple interaction analysis in a novel ethyl 4-[2-(carbamoyl) hydrazinylidene]-3, 5-dimethyl-1H-pyrrole-2-carboxylate and its dimer. J. Mol. Strut. 2013, 1035, 427–440. [Google Scholar] [CrossRef]
- Joe, I.H.; Kostova, I.; Ravikumar, C.; Amalanathan, M.; CîntǎPînzaru, S. Theoretical and vibrational spectral investigation of sodium salt of acenocoumarol. J. Raman Spectrosc. 2009, 40, 1033–1038. [Google Scholar] [CrossRef]
- Sebastian, S.; Sundaraganesan, N. The spectroscopic (FT-IR, FT-IR gas phase, FT-Raman and UV) and NBO analysis of 4-Hydroxypiperidine by density functional method. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2010, 75, 941–952. [Google Scholar] [CrossRef] [PubMed]
- Marten, B.; Kim, K.; Cortis, C.; Friesner, R.A.; Murphy, R.B.; Ringnalda, M.N.; Sitkoff, D.; Honig, B. New Model for Calculation of Solvation Free Energies: Correction of Self-Consistent Reaction Field Continuum Dielectric Theory for Short-Range Hydrogen-Bonding Effects. J. Phys. Chem. 1996, 100, 11775–11788. [Google Scholar]
- Tannor, D.J.; Marten, B.; Murphy, R.; Friesner, R.A.; Sitkoff, D.; Nicholls, A.; Ringnalda, M.; Goddard, W.A.; Honig, B. Accurate first principles calculation of molecular charge distributions and solvation energies from ab initio quantum mechanics and continuum dielectric theory. J. Am. Chem. Soc. 1994, 116, 11875–11882. [Google Scholar]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atoms | Distance | Atoms | Distance |
|---|---|---|---|
| O1–C3 | 1.3301(15) | O1B–C3B | 1.3381(14) |
| O1–C2 | 1.4534(16) | O1B–C2B | 1.4566(15) |
| O2–C3 | 1.2037(16) | O2B–C3B | 1.2052(16) |
| O3–C5 | 1.3822(15) | O3B–C5B | 1.3801(15) |
| O3–C4 | 1.4123(15) | O3B–C4B | 1.4143(14) |
| N1–C8 | 1.3961(17) | N1B–C8B | 1.4050(17) |
| Atoms | Angle | Atoms | Angle |
| C3–O1–C2 | 115.59(10) | O3–C4–C3 | 106.98(10) |
| C5–O3–C4 | 117.06(9) | O3–C5–C6 | 125.35(11) |
| O1–C2–C1 | 107.47(10) | O3–C5–C10 | 115.33(10) |
| O2–C3–O1 | 124.80(12) | C6–C5–C10 | 119.32(11) |
| O2–C3–C4 | 125.26(12) | C5–C6–C7 | 119.82(11) |
| O1–C3–C4 | 109.94(10) | C8–C7–C6 | 121.22(11) |
| D–H···A | d(D–H) | d(H···A) | d(D···A) | <(DHA) |
|---|---|---|---|---|
| N1–H1A···O2B | 0.930(19) | 2.234(19) | 3.1130(14) | 157.2(16) |
| N1–H1A···O3B | 0.930(19) | 2.549(19) | 3.2622(15) | 133.8(14) |
| N1B–H1BA···O2#1 | 0.89(2) | 2.32(2) | 3.1706(16) | 157.9(17) |
| N1B–H1BA···O3#1 | 0.89(2) | 2.55(2) | 3.2487(14) | 135.7(16) |
| Contact | Distance | Contact | Distance |
|---|---|---|---|
| O3B…H1A | 2.494 | O1…H17 | 2.555 |
| O2B…H1A | 2.163 | N1B…H2B | 2.572 |
| O2…H9 | 2.42 | N1…H4B | 2.573 |
| O2B…H4B | 2.459 | C10B…H4A | 2.627 |
| O3…H1BA | 2.467 | C5…H1C | 2.7 |
| O2…H1BA | 2.219 | H1D…H9B | 2.279 |
| O2…H9B | 2.418 | H1C…H10B | 2.456 |
| (NBO)i a | (NBO)j b | E(2) | (NBO)i a | (NBO)j b | E(2) |
|---|---|---|---|---|---|
| BD(2)C19–C21 | BD*(2)C16–C17 | 20.60 | LP(1)O1 | BD*(1)O2–C12 | 6.69 |
| BD(2)C19–C21 | BD*(2)C22–C24 | 18.37 | LP(2)O2 | BD*(1)O1–C12 | 32.42 |
| BD(2)C16–C17 | BD*(2)C19–C21 | 18.22 | LP(2)O2 | BD*(1)C12–C13 | 21.13 |
| BD(2)C16–C17 | BD*(2)C22–C24 | 19.13 | LP(1)O3 | BD*(1)C16–C17 | 6.87 |
| BD(2)C22–C24 | BD*(2)C16–C17 | 18.73 | LP(2)O1 | BD*(2)O2–C12 | 45.95 |
| BD(2)C22–C24 | BD*(2)C19–C21 | 19.80 | LP(2)O3 | BD*(2)C16–C17 | 27.10 |
| LP(1)N4 | BD*(2)C19–C21 | 24.28 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Altowyan, M.S.; Soliman, S.M.; Ismail, M.M.F.; Haukka, M.; Barakat, A.; Ayoup, M.S. Efficient Consecutive Synthesis of Ethyl-2-(4-Aminophenoxy) Acetate, a Precursor for Dual GK and PPARγ Activators, X-ray Structure, Hirshfeld Analysis, and DFT Studies. Crystals 2022, 12, 227. https://doi.org/10.3390/cryst12020227
Altowyan MS, Soliman SM, Ismail MMF, Haukka M, Barakat A, Ayoup MS. Efficient Consecutive Synthesis of Ethyl-2-(4-Aminophenoxy) Acetate, a Precursor for Dual GK and PPARγ Activators, X-ray Structure, Hirshfeld Analysis, and DFT Studies. Crystals. 2022; 12(2):227. https://doi.org/10.3390/cryst12020227
Chicago/Turabian StyleAltowyan, Mezna Saleh, Saied M. Soliman, Magda M. F. Ismail, Matti Haukka, Assem Barakat, and Mohammed Salah Ayoup. 2022. "Efficient Consecutive Synthesis of Ethyl-2-(4-Aminophenoxy) Acetate, a Precursor for Dual GK and PPARγ Activators, X-ray Structure, Hirshfeld Analysis, and DFT Studies" Crystals 12, no. 2: 227. https://doi.org/10.3390/cryst12020227