
Electronic, Magnetic and Optical Properties of Double Perovskite Compounds: A First Principle Approach


Abstract
:1. Introduction
2. Methodology
3. Results and Discussions
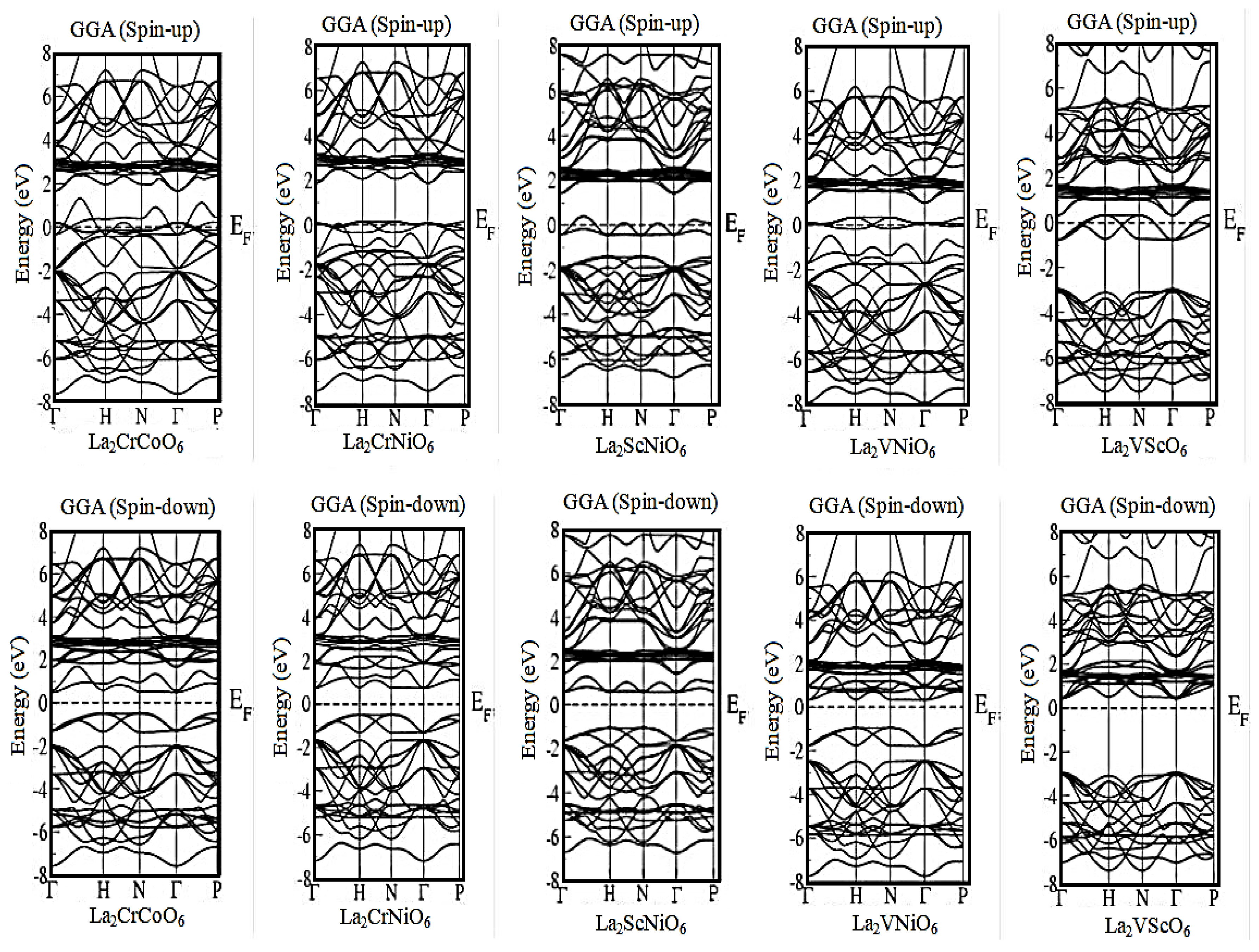
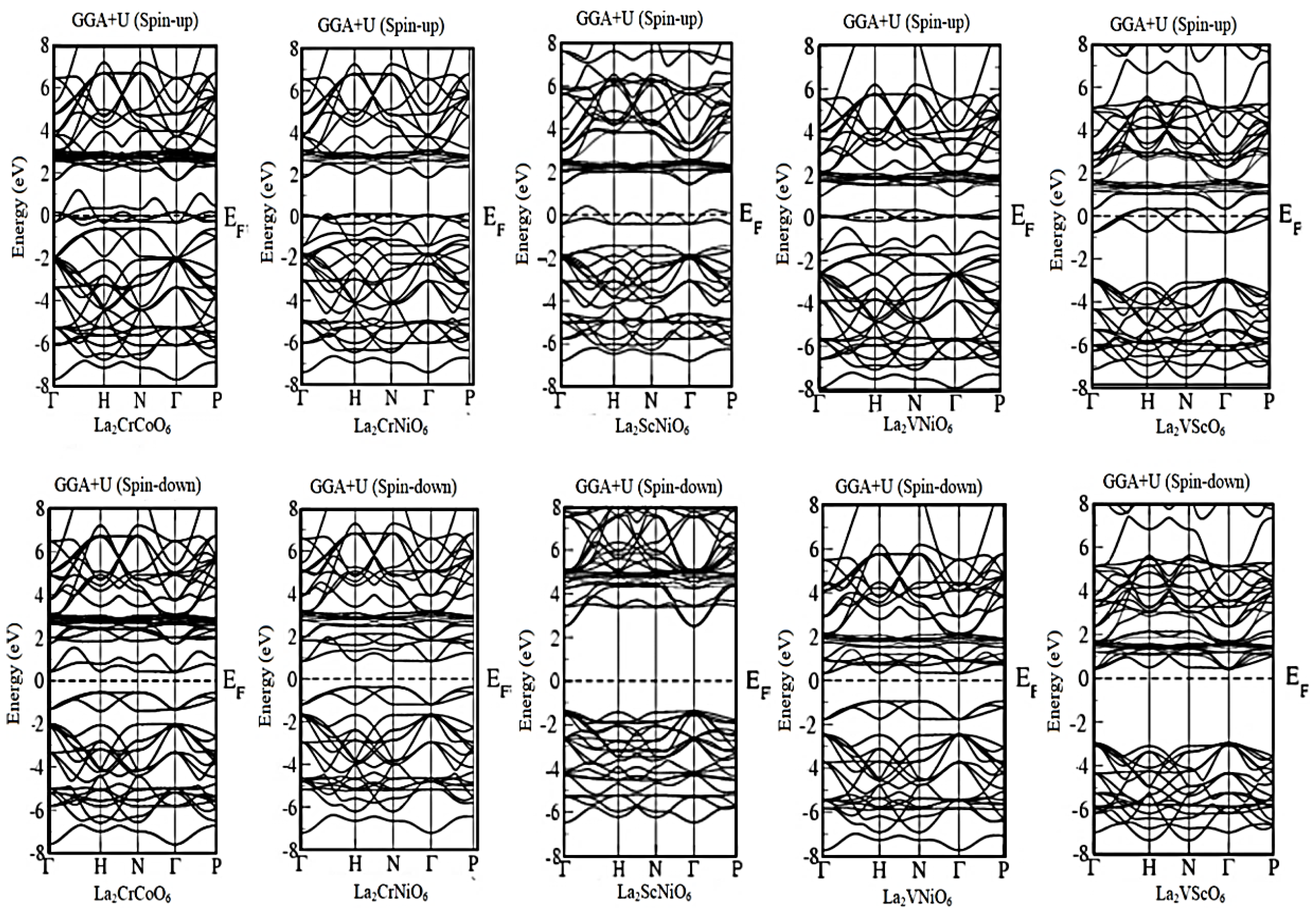
3.1. Band Structure Calculations
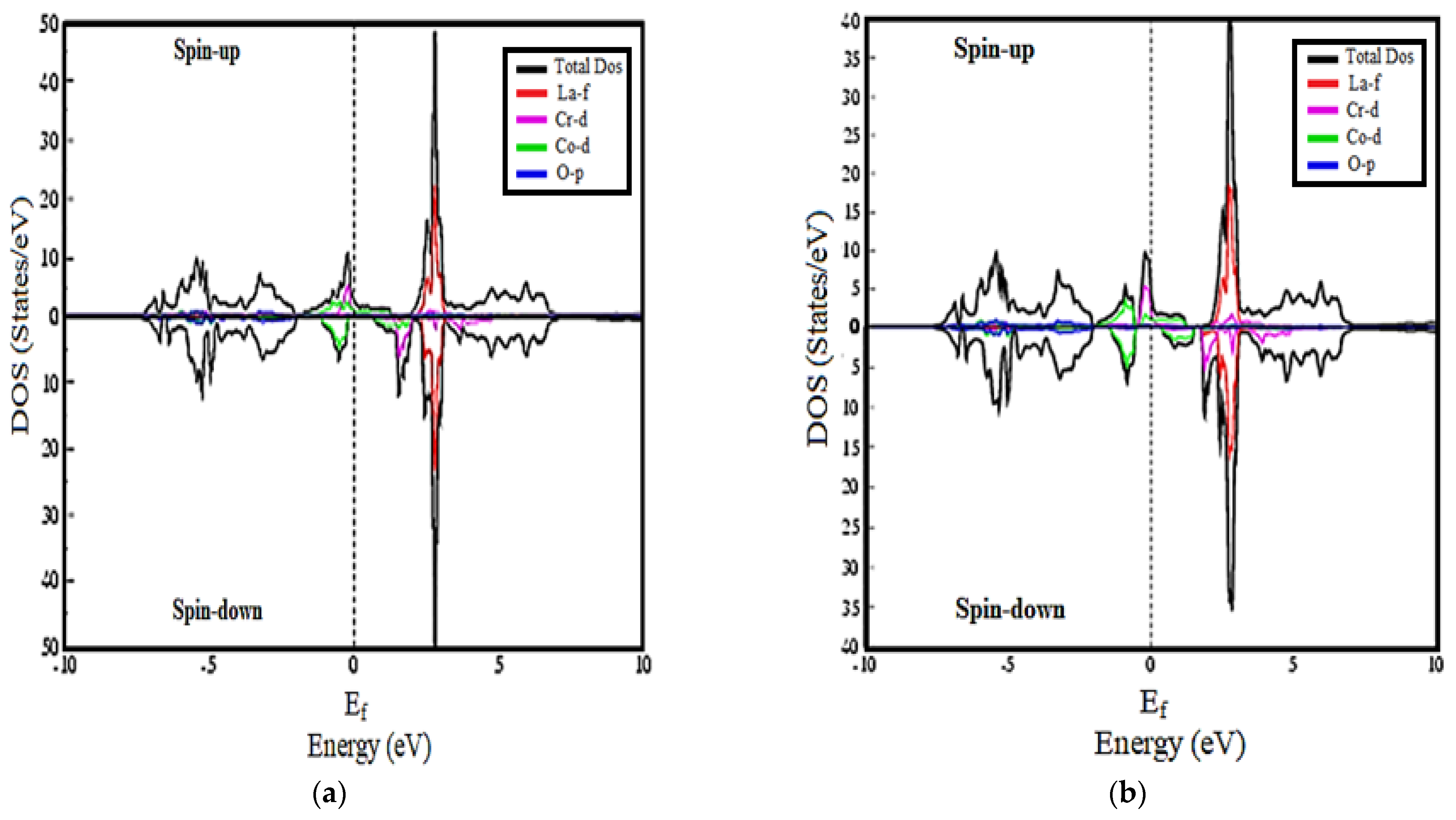
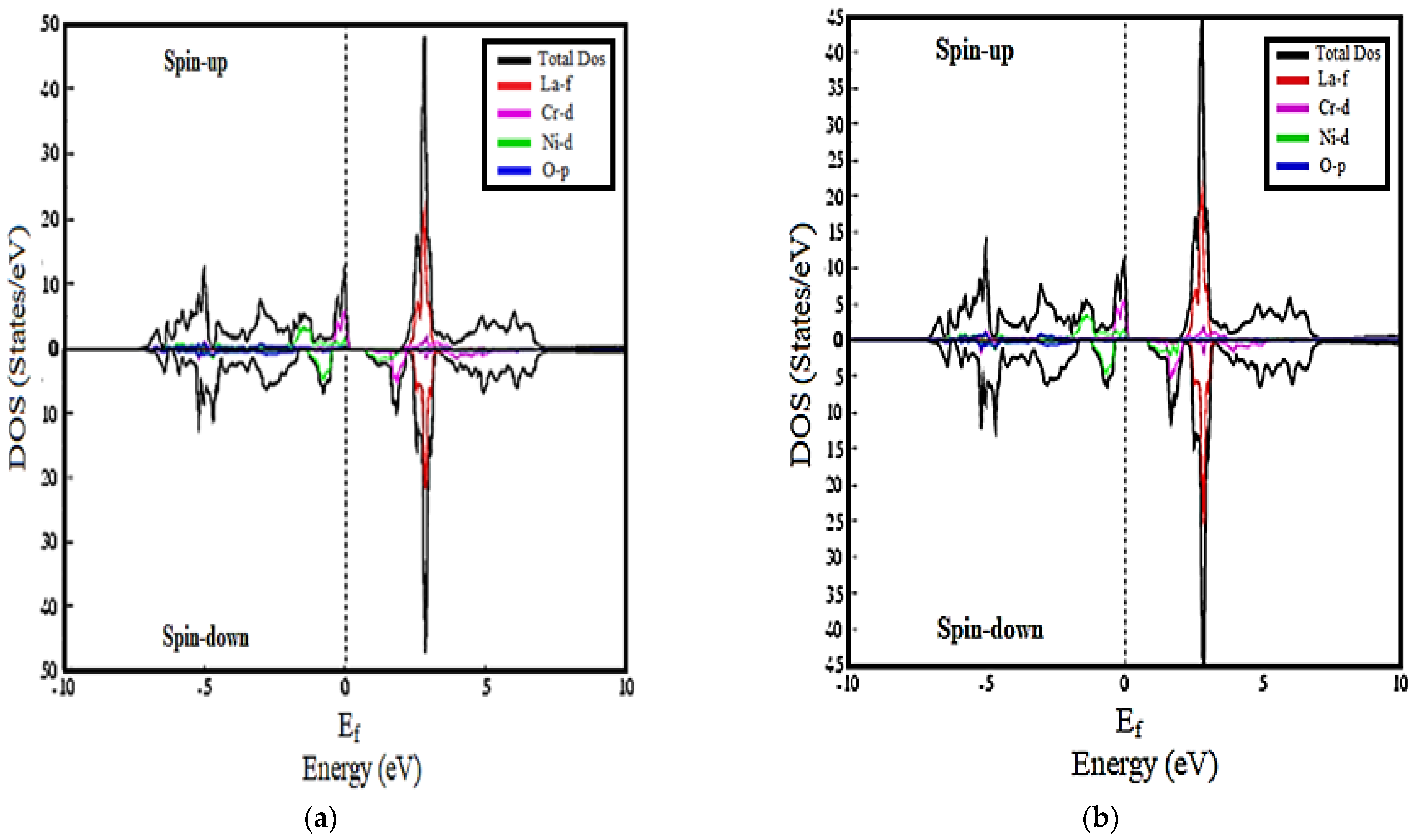
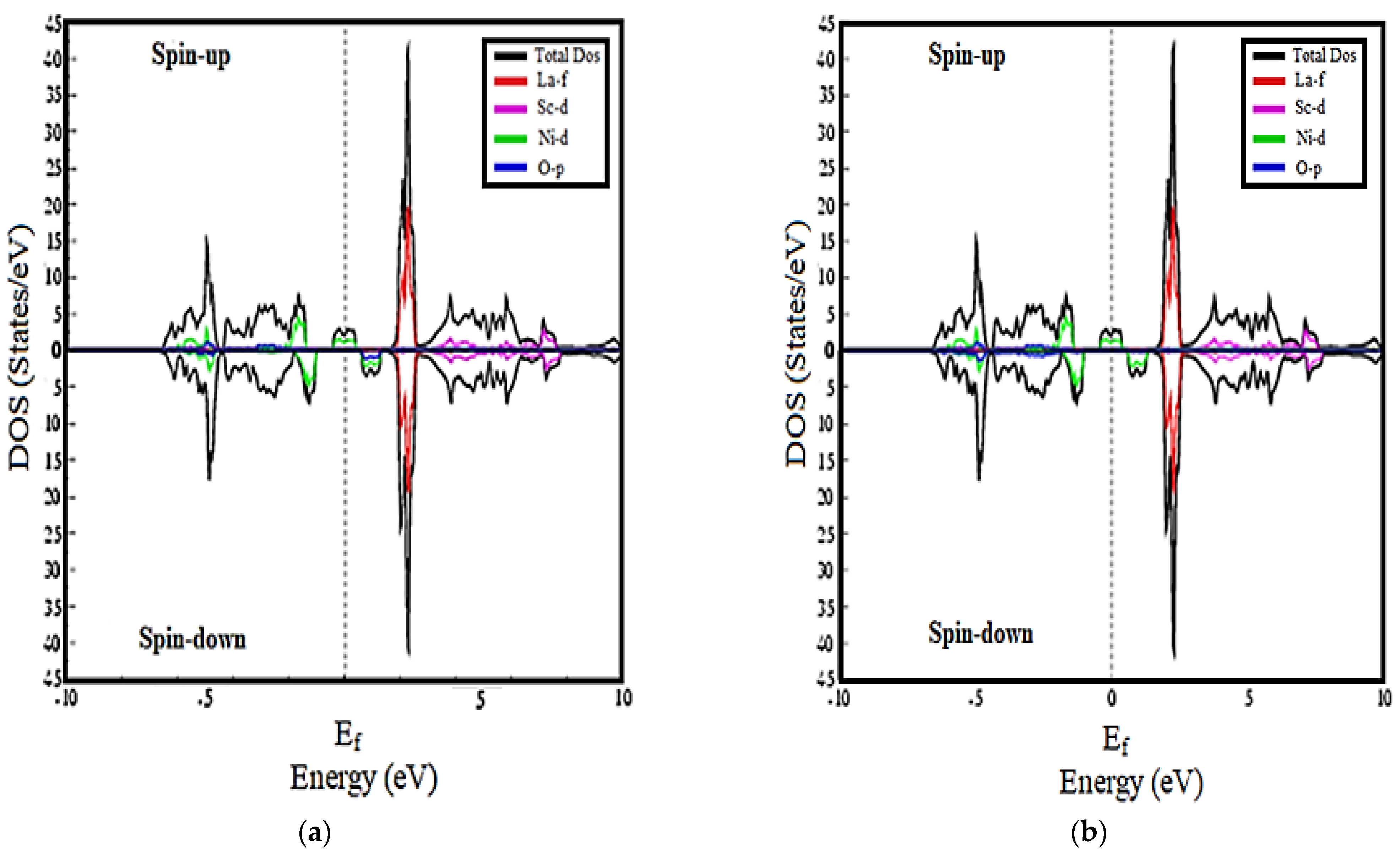
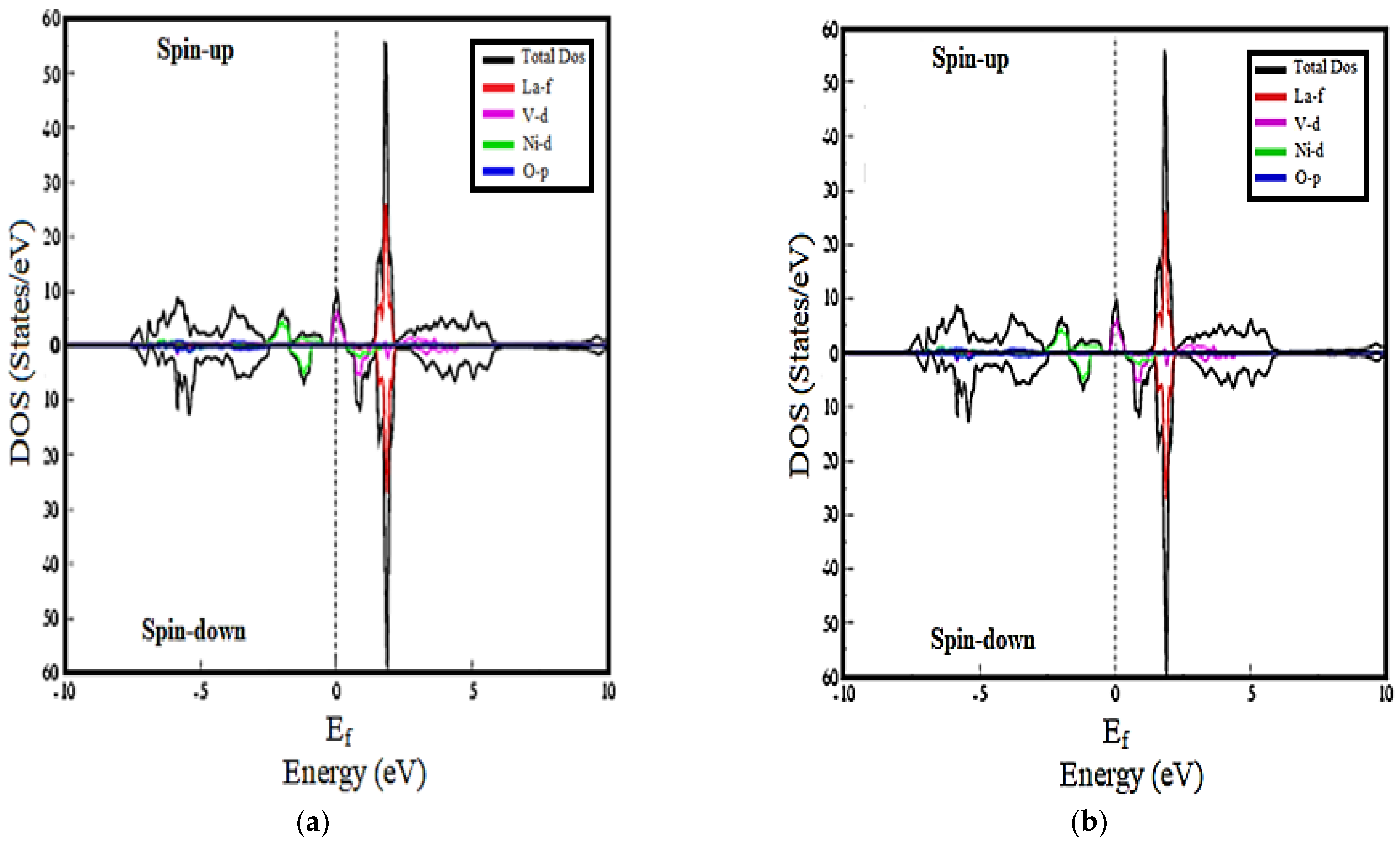
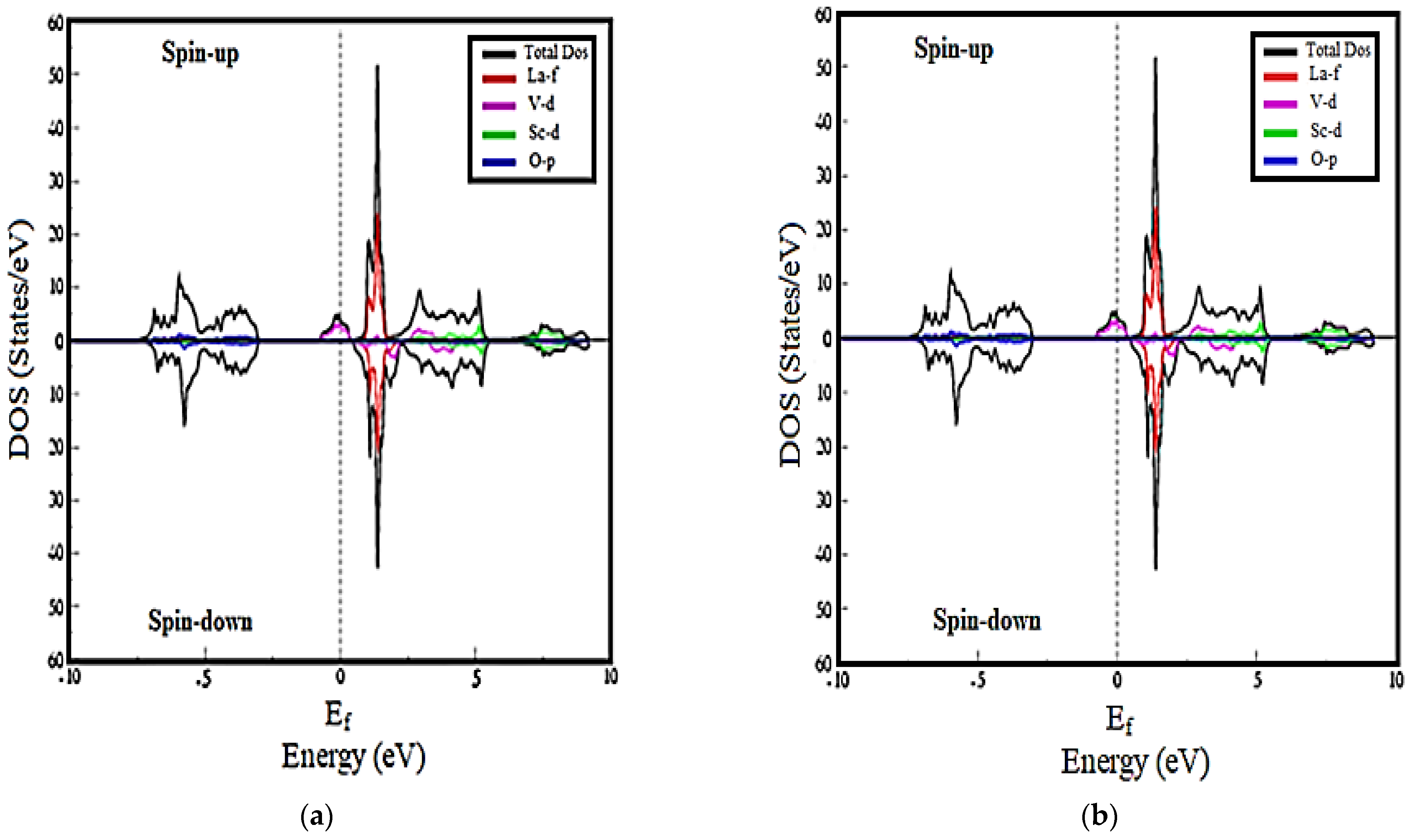
3.2. Density of States Calculations
3.3. Magnetic Properties Calculations
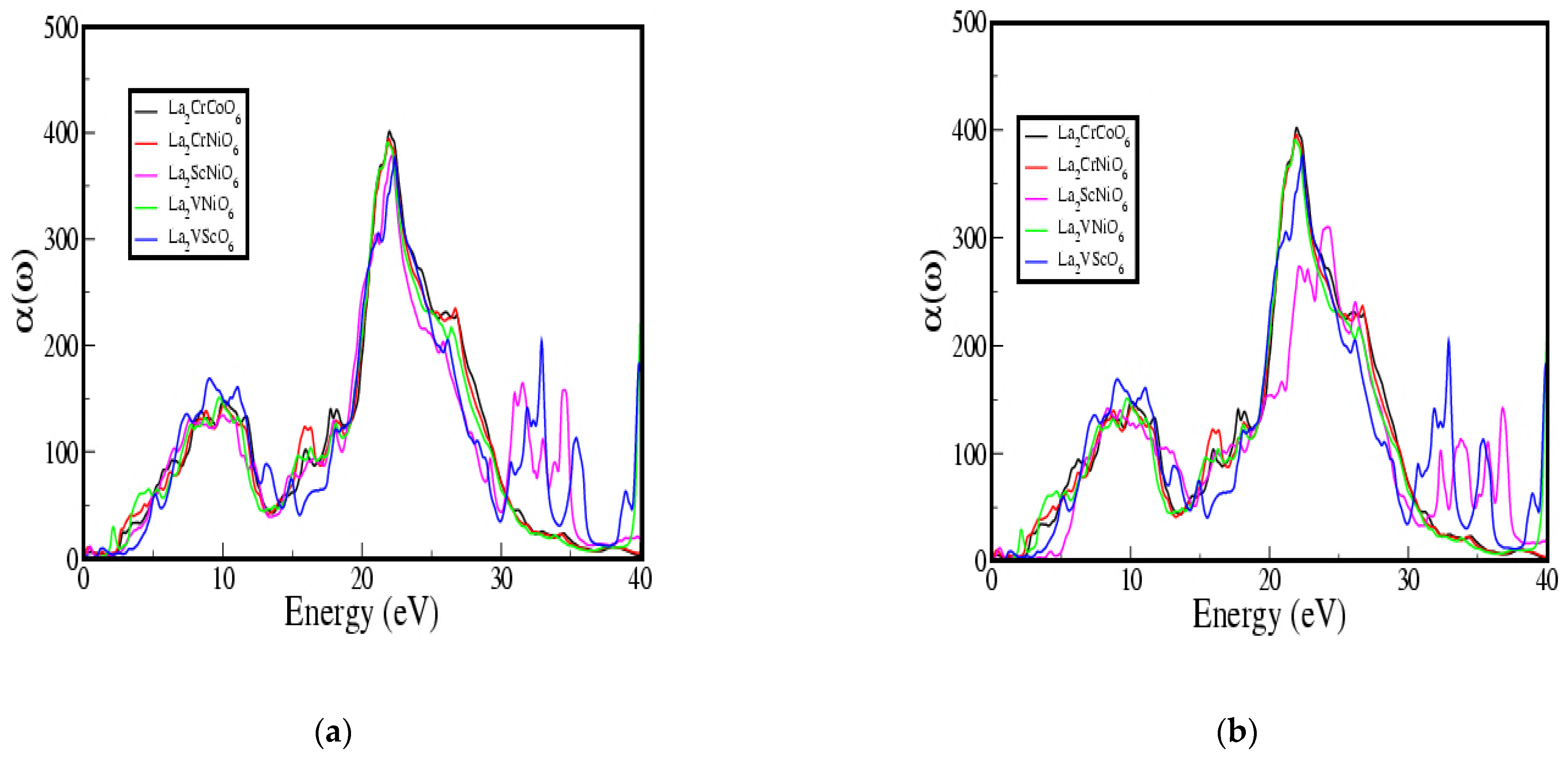
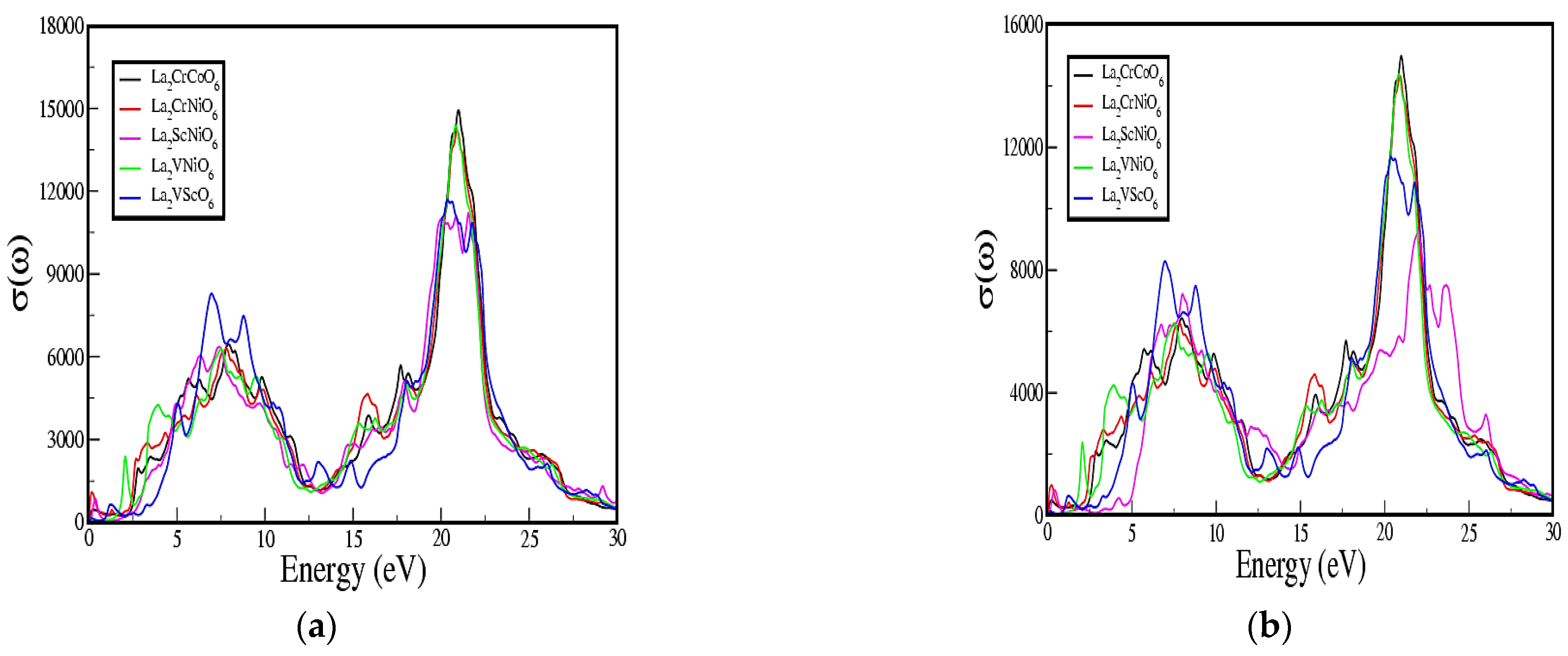
3.4. Optical Properties Calculations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sarma, D.D. A new class of magnetic materials: Sr2FeMoO6 and related compounds. Curr. Opin. Solid State Mater. Sci. 2001, 5, 261–268. [Google Scholar] [CrossRef]
- Vasala, S.; Karppinen, M. A2B′ B″O6 perovskites: A review. Prog. Solid State Chem. 2015, 43, 1–36. [Google Scholar] [CrossRef]
- Mahesh, R.; Kannan, K.R.; Rao, C.N.R. Electrochemical synthesis of ferromagnetic LaMnO3 and metallic NdNiO3. J. Solid State Chem. 1995, 114, 294–296. [Google Scholar] [CrossRef]
- Sleight, A.W.; Gillson, J.L.; Bierstedt, P.E. High-temperature superconductivity in the BaPb1-xBixO3 system. Solid State Commun. 1993, 88, 841–842. [Google Scholar] [CrossRef]
- MacChesney, J.B.; Sherwood, R.C.; Potter, J.F. Electric and magnetic properties of the strontium ferrates. J. Chem. Phys. 1965, 43, 1907–1913. [Google Scholar] [CrossRef]
- Aylett, B.J. Transition Metal Oxides: Structure, Properties and Synthesis of Ceramic Oxides, 2nd ed.; Rao, C.N.R., Raveau, B., Eds.; Wiley-VCH: New York, NY, USA; Weinheim, Germany, 1998; p. 80. ISBN 0-471-18971-5. [Google Scholar]
- Hikami, S.; Matsuda, Y. High Tc superconductors of the perovskite structure oxides. Jpn. J. Appl. Phys. 1987, 26, 1027. [Google Scholar] [CrossRef]
- Bhalla, A.S.; Guo, R.; Roy, R. The perovskite structure—A review of its role in ceramic science and technology. Mater. Res. Innov. 2000, 4, 3–26. [Google Scholar] [CrossRef]
- Saha-Dasgupta, T. Double perovskites with 3d and 4d/5d transition metals: Compounds with promises. Mater. Res. Express 2020, 7, 014003. [Google Scholar] [CrossRef]
- Galasso, F.; Pyle, J. Ordering in compounds of the A (B′0.33 Ta0. 67) O3 type. Inorganic Chem. 1963, 2, 482–484. [Google Scholar] [CrossRef]
- Lemmens, P.; Millet, P. Spin—Orbit—Topology, a triptych. Quantum Magn. 2004, 645, 433–477. [Google Scholar]
- Wu, H. Electronic structure study of double perovskites A 2 FeReO 6 (A = B a, S r, C a) and Sr 2 M MoO6 (M = C r, M n, F e, C o) by LSDA and LSDA+ U. Phys. Rev. B 2001, 64, 125126. [Google Scholar] [CrossRef]
- Jin, X.; Wang, Q.; Jadoon, A.M. Opto-Electronic Properties of Methyl-Ammonium Lead Halide: A First Principle Approach. In Journal of Physics: Conference Series; IOP Publishing: Bristol, UK, 2020; Volume 1622, p. 012105. [Google Scholar]
- Ghaithan, H.M.; Alahmed, Z.A.; Qaid, S.M.; Aldwayyan, A.S. Density Functional Theory Analysis of Structural, Electronic, and Optical Properties of Mixed-Halide Orthorhombic Inorganic Perovskites. ACS Omega 2021, 6, 30752–30761. [Google Scholar] [CrossRef] [PubMed]
- Kabbour, H.; Gauthier, G.H.; Tessier, F.; Huvé, M.; Pussacq, T.; Roussel, P.; Mentre, O. Topochemical reduction of YMnO3 into a composite structure. Inorg. Chem. 2017, 56, 8547–8553. [Google Scholar] [CrossRef] [PubMed]
- Jeng, H.T.; Guo, G.Y. First-principles investigations of orbital magnetic moments and electronic structures of the double perovskites Sr2 FeMoO6, Sr2 FeReO6, and Sr2 CrWO6. Phys. Rev. B 2003, 67, 094438. [Google Scholar] [CrossRef]
- Holman, K.L.; Huang, Q.; Klimczuk, T.; Trzebiatowski, K.; Bos, J.W.G.; Morosan, E.; Cava, R.J. Synthesis and properties of the double perovskites La2NiVO6, La2CoVO6, and La2CoTiO6. J. Solid State Chem. 2007, 180, 75–83. [Google Scholar] [CrossRef]
- Alias, F.I.H.; Maulud, M.F.; Mohamed, Z. Structural and optical properties of tellurium-based double perovskite Sr2ZnTeO6. In AIP Conference Proceedings; AIP Publishing LLC: New York, NY, USA, 2021; Volume 2368, p. 030001. [Google Scholar]
- Neelu, N.; Pandey, N.; Chakrabarti, S. Synthesis and optical study of ultra stable inorganic double perovskite Cs2CuBiCl6 for optoelectronic applications. In Proceedings of the Organic, Hybrid, and Perovskite Photovoltaics XXIII, San Diego, CA, USA, 22 August 2022; Volume 12209, pp. 39–45. [Google Scholar]
- Malyshkin, D.; Novikov, A.; Ivanov, I.; Sereda, V.; Tsvetkov, D.; Zuev, A. The origin of triple conductivity and water uptake in layered double perovskites: A case study on lanthanum-substituted GdBaCo2O6− δ. J. Alloys Compd. 2020, 845, 156309. [Google Scholar] [CrossRef]
- Bhowmik, T.K.; Sheikh, M.S.; Sakhya, A.P.; Dutta, A.; Sinha, T.P. Synthesis, structural and electrical conductivity of half-metallic perovskite oxide La2CrNiO6. In AIP Conference Proceedings; American Institute of Physics: College Park, MD, USA, 2021; Volume 2369, p. 020080. [Google Scholar]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822. [Google Scholar] [CrossRef]
- Schwarz, K. DFT calculations of solids with LAPW and WIEN2k. J. Solid State Chem. 2003, 176, 319–328. [Google Scholar] [CrossRef]
- Schwarz, K.; Blaha, P.; Trickey, S.B. Electronic structure of solids with WIEN2k. Mol. Phys. 2010, 108, 3147–3166. [Google Scholar] [CrossRef]
- Slater, J.C. Introduction to Chemical Physics; Read Books Ltd.: Redditch, UK, 2011. [Google Scholar]
- Anisimov, V.I.; Zaanen, J.; Andersen, O.K. Band theory and Mott insulators: Hubbard U instead of Stoner I. Phys. Rev. B 1991, 44, 943. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.P.; Chen, S.H.; Fuh, H.R.; Wang, Y.K. First-principle calculations of half-metallic double perovskite La2BBO6 (B, B = 3d transition metal). Commun. Comput. Phys. 2013, 14, 174–185. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DPC | GGA (Current) | GGA+U (Current) | GGA (Other) | GGA+U (Other) | ||||
|---|---|---|---|---|---|---|---|---|
| Spin-Up | Spin-Down | Spin-Up | Spin-Down | Spin-Up | Spin-Down | Spin-Up | Spin-Down | |
| La2CrCoO6 | Metallic | 0.945 eV | Metallic | 1.667 eV | Metallic | 0.762 eV | Metallic | 2.471 eV |
| La2CrNiO6 | Metallic | 1.149 eV | Metallic | 1.211 eV | Metallic | 1.007 eV | Metallic | 3.293 eV |
| La2ScNiO6 | Metallic | 1.632 eV | Metallic | 3.855 eV | Metallic | 1.497 eV | Metallic | 3.293 eV |
| La2VNiO6 | Metallic | 1.225 eV | Metallic | 2.008 eV | Metallic | 1.116 eV | Metallic | 3.65 eV |
| La2VScO6 | Metallic | 3.537 eV | Metallic | 3.566 eV | Metallic | 3.238 eV | Metallic | 3.238 eV |
| DPC | mINTERS | mS1 | mS2 | mS3 | mS4 | mS5 | mCell (µB) |
|---|---|---|---|---|---|---|---|
| La2CrCoO6 | 0.271 | 0.012 | 2.325 | 0.184 | 0.013 | 0.012 | 2.883 µB |
| La2CrNiO6 | 0.282 | 0.014 | 1.998 | 1.389 | 0.048 | 0.050 | 4.000 µB |
| La2ScNiO6 | 0.011 | −0.002 | 0.045 | 0.776 | 0.028 | 0.028 | 0.998 µB |
| La2VNiO6 | 0.153 | 0.013 | 0.892 | 1.481 | 0.069 | 0.069 | 2.971 µB |
| La2VScO6 | 0.427 | 0.073 | 1.423 | 0.036 | −0.004 | −0.004 | 2.008 µB |
| DPC | mINTERS | mS1 | mS2 | mS3 | mS4 | mS5 | mCell (µB) |
|---|---|---|---|---|---|---|---|
| La2CrCoO6 | 0.323 | 0.021 | 2.388 | 0.154 | 0.018 | 0.015 | 3.007 µB |
| La2CrNiO6 | 0.263 | 0.012 | 1.974 | 1.402 | 0.056 | 0.057 | 4.006 µB |
| La2ScNiO6 | −0.016 | 0.002 | 0.017 | 1.319 | −0.057 | −0.052 | 1.000 µB |
| La2VNiO6 | 0.153 | 0.013 | 0.895 | 1.483 | 0.068 | 0.069 | 2.972 µB |
| La2VScO6 | 0.429 | 0.073 | 1.427 | 0.036 | −0.005 | −0.005 | 2.010 µB |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rehman, M.U.; Wang, Q.; Yu, Y. Electronic, Magnetic and Optical Properties of Double Perovskite Compounds: A First Principle Approach. Crystals 2022, 12, 1597. https://doi.org/10.3390/cryst12111597
Rehman MU, Wang Q, Yu Y. Electronic, Magnetic and Optical Properties of Double Perovskite Compounds: A First Principle Approach. Crystals. 2022; 12(11):1597. https://doi.org/10.3390/cryst12111597
Chicago/Turabian StyleRehman, Mehtab Ur, Qun Wang, and Yunfei Yu. 2022. "Electronic, Magnetic and Optical Properties of Double Perovskite Compounds: A First Principle Approach" Crystals 12, no. 11: 1597. https://doi.org/10.3390/cryst12111597