
Synthesis, Crystal and Electronic Structure of the New Ternary Compound Ca3InAs3

Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis
2.2. Single-Crystal X-ray Diffraction (SC-XRD)
2.3. Powder X-ray Diffraction
2.4. Electronic Structure Calculations
3. Results and Discussion
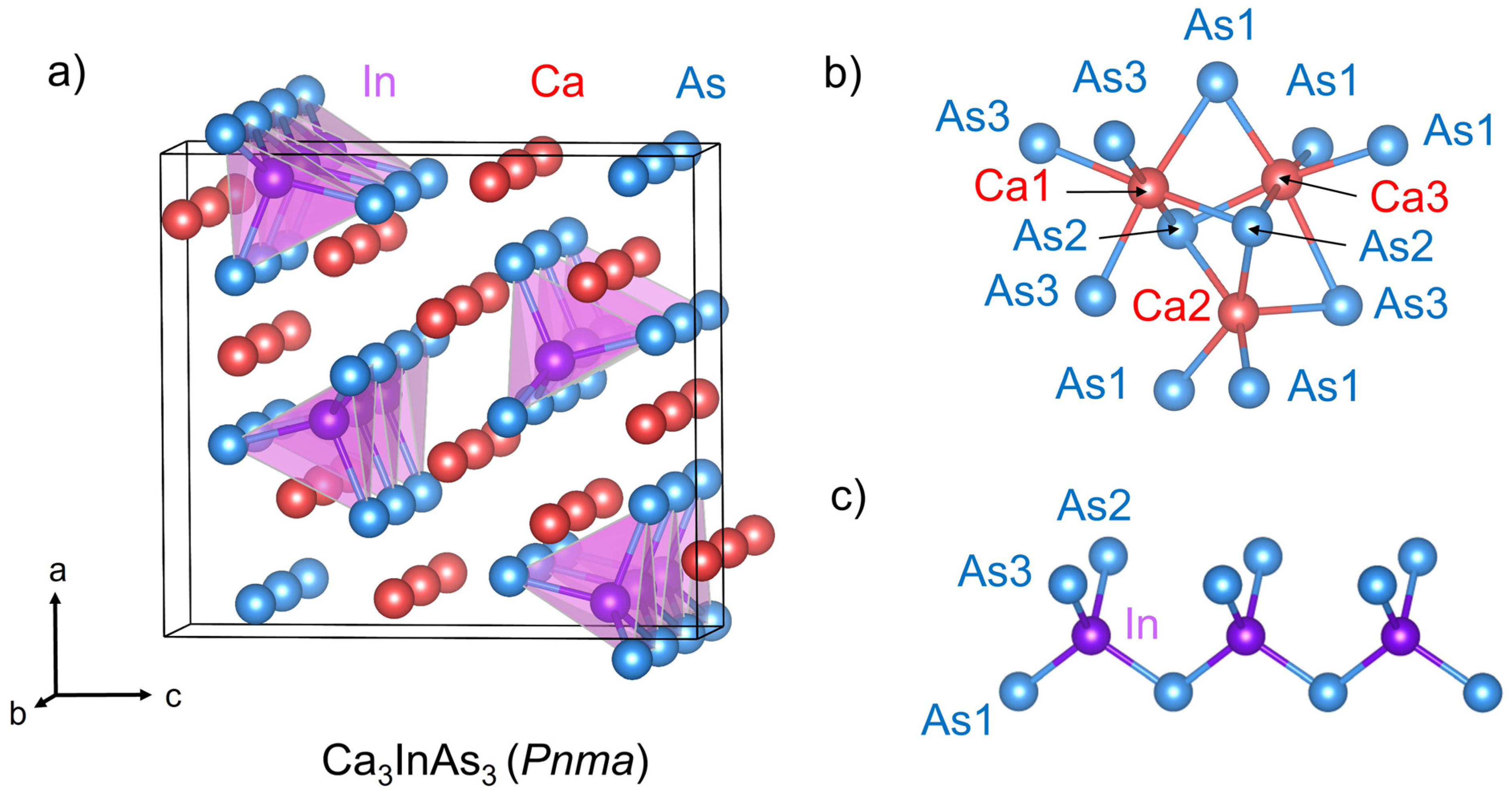
3.1. Crystal Structure
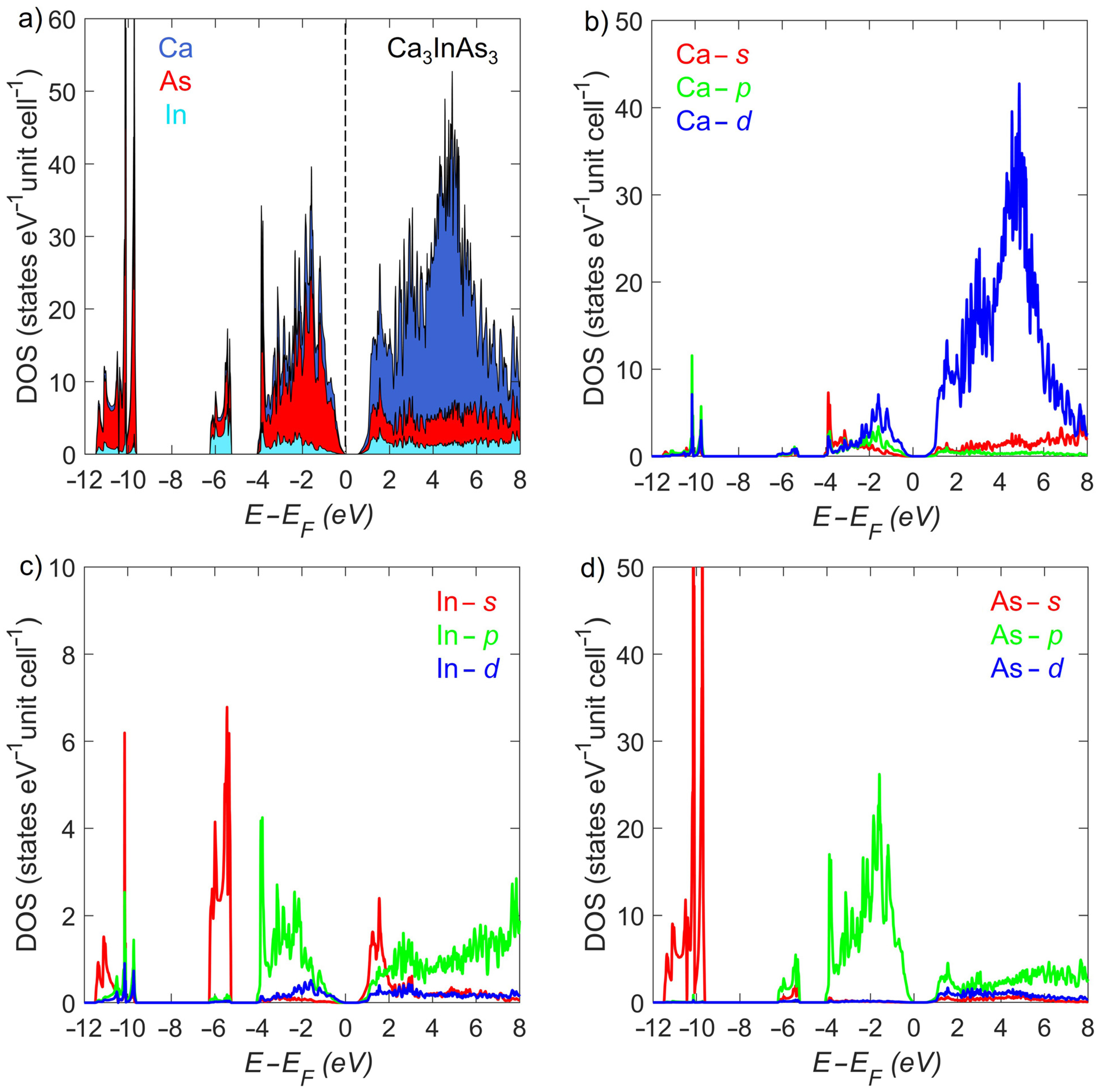
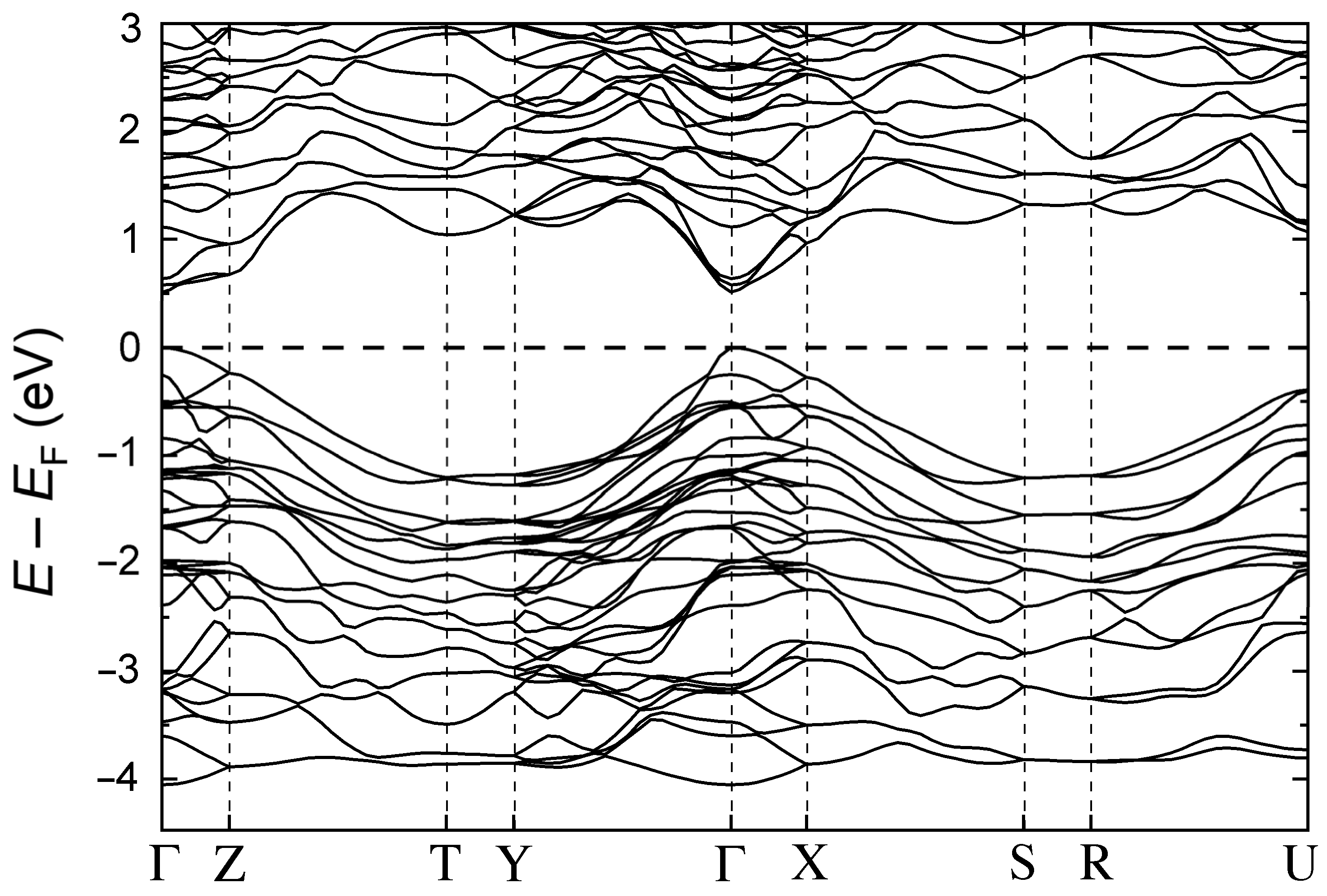
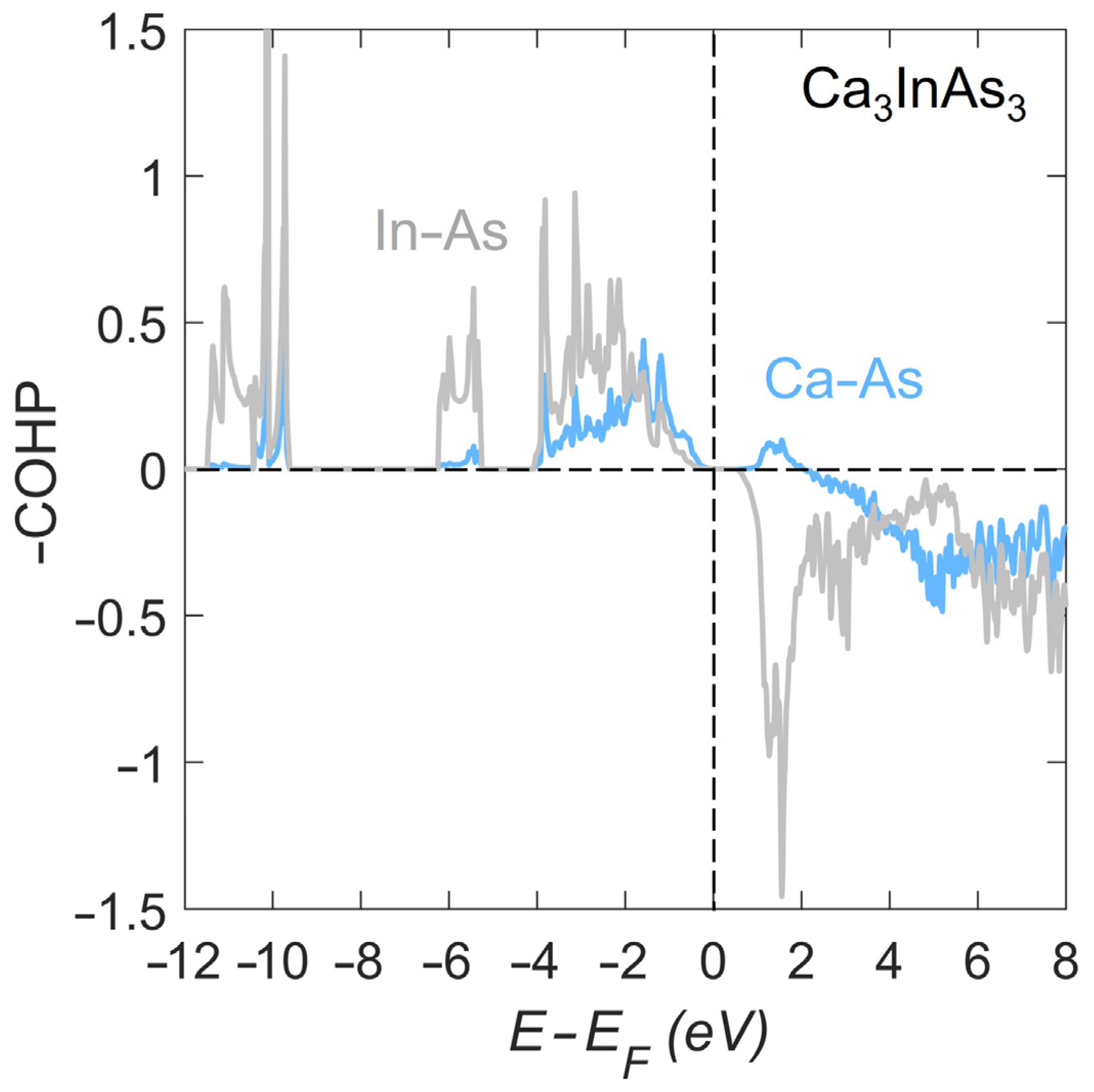
3.2. Electronic Structure
4. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stoyko, S.; Voss, L.; He, H.; Bobev, S. Synthesis, crystal and electronic structures of the pnictides AE3TrPn3 (AE = Sr, Ba; Tr = Al, Ga; Pn = P, As). Crystals 2015, 5, 433–446. [Google Scholar] [CrossRef] [Green Version]
- Cordier, G.; Savelsberg, G.; Schäfer, H. Zintl phasen mit komplexen anionen: Zur kenntnis von Ca3AlAs3 und Ba3AlSb3. Z. Naturforsch. B 1982, 37, 975–980. [Google Scholar] [CrossRef]
- Cordier, G.; Schäfer, H.; Stelter, M. Sr3GaSb3 und Sr3InP3, zwei neue Zintl phasen mit komplexen anionen. Z. Naturforsch. B 1982, 42, 1268–1272. [Google Scholar] [CrossRef]
- Cordier, G.; Schäfer, H.; Steher, M. Neue Zintl phasen: Ba3GaSb3, Ca3GaAs3 und Ca3InP3. Z. Naturforsch. B 1985, 40, 1100–1104. [Google Scholar] [CrossRef] [Green Version]
- Cordier, G.; Schäfer, H.; Stelter, M. Ca3AlSb3 und Ca5Al2Bi6, zwei neue Zintl phasen mit kettenförmigen anionen. Z. Naturforsch. B 1984, 39, 727–732. [Google Scholar] [CrossRef]
- Rajput, K.; Baranest, S.; Bobev, S. Observation of an unexpected n-type semiconducting behavior in the new ternary Zintl phase Eu3InAs3. Chem. Mater. 2020, 32, 9444. [Google Scholar] [CrossRef]
- Shang, R.; Nguyen, A.T.; He, A.; Kauzlarich, S.M. Crystal structure characterization and electronic structure of a rare-earth-containing Zintl phase in the Yb–Al–Sb family: Yb3AlSb3. Acta Crystallogr. C 2021, 77, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Radzieowski, M.; Stegemann, F.; Klenner, S.; Zhang, Y.; Fokwa, B.P.; Janka, O. On the divalent character of the Eu atoms in the ternary Zintl phases Eu5In2Pn6 and Eu3MAs3 (Pn = As–Bi; M = Al, Ga). Mater. Chem. Front. 2020, 4, 1231–1248. [Google Scholar] [CrossRef]
- Ogunbunmi, M.O.; Baranets, S.; Childs, A.B.; Bobev, S. The Zintl phases AIn2As2 (A = Ca, Sr, Ba): New topological insulators and thermoelectric material candidates. Dalton Trans. 2021, 50, 9173–9184. [Google Scholar] [CrossRef] [PubMed]
- Bruker Analytical X-ray Systems; Inc. SMART & SAINT: Madison, WI, USA, 2003.
- Sheldrick, G.M. SHELXT–Integrated space group and crystal structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Gelato, L.; Parthé, E. STRUCTURE TIDY—A computer program to standardize crystal structure data. J. Appl. Crystallogr. 1987, 20, 139–143. [Google Scholar] [CrossRef]
- Tank, R.; Jepsen, O.; Burkhardt, A.; Andersen, O. TB-LMTO-ASA Program; Max-Planck-Institut für Festkörperforschung: Stuttgart, Germany, 1994. [Google Scholar]
- von Barth, U.; Hedin, L. A local exchange-correlation potential for the spin polarized case. J. Phys. C Solid State Phys. 1972, 5, 1629. [Google Scholar] [CrossRef]
- Steinberg, S.; Dronskowski, R. The crystal orbital Hamilton population (COHP) method as a tool to visualize and analyze chemical bonding in intermetallic compounds. Crystals 2018, 8, 225. [Google Scholar] [CrossRef] [Green Version]
- Cordier, G.; Schäfer, H.; Stelter, M. Sr3In2P4 and Ca3In2As4 Zintl phases with strings of InP4 and InAs4 tetrahedra, resp., sharing edges and corners. Z. Naturforsch. B 1986, 41, 1416–1419. [Google Scholar] [CrossRef]
- Villars, P.; Calvert, L.D. (Eds.) Pearson’s Handbook of Crystallographic Data for Intermetallic Compounds, 2nd ed.; American Society for Metals: Materials Park, OH, USA, 1991. [Google Scholar]
- Somer, M.; Carrillo-Cabrera, W.; Peters, K.; von Schnering, H.-G.; Cordier, G. Crystal structure of tribarium triarsenidoindate, Ba3InAs3. Z. Kristallogr. New Cryst. Struct. 1996, 211, 632. [Google Scholar] [CrossRef]
- Childs, A.; Baranets, S.; Bobev, S. Five new ternary indium–arsenides discovered. Synthesis and structural characterization of the Zintl phases Sr3In2As4, Ba3In2As4, Eu3In2As4, Sr5In2As6 and Eu5In2As6. J. Solid State Chem. 2019, 278, 120889. [Google Scholar] [CrossRef]
- Pauling, L. The Nature of the Chemical Bond, 3rd ed.; Cornell University Press: Ithaca, NY, USA, 1960. [Google Scholar]
- Schäfer, H.; Eisenmann, B.; Müller, W. Zintl Phases: Transitions between metallic and ionic bonding. Angew. Chem. Int. Ed. Engl. 1973, 12, 694–712. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formula | Ca3InAs3 |
|---|---|
| Formula weight (g·mol−1) | 459.82 |
| Radiation, λ | Mo Kα, 0.71073 Å |
| Temperature (°C) | –70(2) |
| Crystal system | orthorhombic |
| Space group | Pnma (No. 62) |
| Z | 4 |
| a (Å) | 12.296(2) |
| b (Å) | 4.2553(7) |
| c (Å) | 13.735(2) |
| V (Å3) | 718.7(2) |
| ρcalc (g·cm−3) | 4.25 |
| µMoKα (cm−1) | 190.1 |
| Reflections: parameters | 1151: 44 |
| R1 [I > 2σ(I)] a | 0.0222 |
| R1 (all data) a | 0.0330 |
| wR2 [I > 2σ(I)] a | 0.0463 |
| wR2 (all data) a | 0.0496 |
| Largest peak; deepest hole (e/Å−3) | 0.83; –1.05 |
| Atom | Wyckoff Symbol | Site Symmetry | x | y | z | Ueq (Å2) a |
|---|---|---|---|---|---|---|
| As1 | 4c | .m | 0.04891(4) | 1/4 | 0.36528(4) | 0.0103(1) |
| As2 | 4c | .m | 0.25779(4) | 1/4 | 0.62613(4) | 0.0086(1) |
| As3 | 4c | .m | 0.60837(4) | 1/4 | 0.61098(4) | 0.0085(1) |
| In | 4c | .m | 0.05554(3) | 1/4 | 0.69988(3) | 0.0103(2) |
| Ca1 | 4c | .m | 0.06164(7) | 1/4 | 0.10532(8) | 0.0104(2) |
| Ca2 | 4c | .m | 0.26873(7) | 1/4 | 0.28697(8) | 0.0092(2) |
| Ca3 | 4c | .m | 0.34436(8) | 1/4 | 0.00296(8) | 0.0104(2) |
| Atoms | Distance (Å) | Atoms | Distance (Å) |
|---|---|---|---|
| In–As1 (×2) | 2.6415(5) | As2–In | 2.6853(7) |
| In–As2 | 2.6853(7) | As2–Ca1 (×2) | 3.0885(8) |
| In–As3 | 2.6779(7) | As2–Ca2 (×2) | 3.0845(9) |
| In–Ca1 (×2) | 3.710(1) | As2–Ca3 (×2) | 2.9945(8) |
| In–Ca2 (×2) | 3.2600(8) | As3–In | 2.6779(7) |
| In–Ca3 (×2) | 3.655(1) | As3–Ca1 (×2) | 2.984(8) |
| In–Ca3 | 3.809(1) | As3–Ca1 | 3.026(1) |
| As1–In (×2) | 2.6415(5) | As3–Ca2 (×2) | 2.9623(8) |
| As1–Ca1 | 3.574(1) | As3–Ca3 | 3.297(1) |
| As1–Ca2 | 2.9091(1) | Ca1–Ca2 | 3.565(1) |
| As1–Ca3 | 3.099(1) | Ca1–Ca3 | 3.750(1) |
| As1–Ca3 (×2) | 3.1345(9) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, W.; Baranets, S.; Bobev, S. Synthesis, Crystal and Electronic Structure of the New Ternary Compound Ca3InAs3. Crystals 2022, 12, 1467. https://doi.org/10.3390/cryst12101467
Peng W, Baranets S, Bobev S. Synthesis, Crystal and Electronic Structure of the New Ternary Compound Ca3InAs3. Crystals. 2022; 12(10):1467. https://doi.org/10.3390/cryst12101467
Chicago/Turabian StylePeng, Wanyue, Sviatoslav Baranets, and Svilen Bobev. 2022. "Synthesis, Crystal and Electronic Structure of the New Ternary Compound Ca3InAs3" Crystals 12, no. 10: 1467. https://doi.org/10.3390/cryst12101467