
Vapour Diffusion Sitting Drop Method to Induce Nucleation of Calcium Phosphate on Exfoliated Graphene and Graphene Oxide Flakes



 ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of G and GO Suspensions in Presence of L-Ala
2.2. Precipitation Method
2.3. Characterization Methods
3. Results
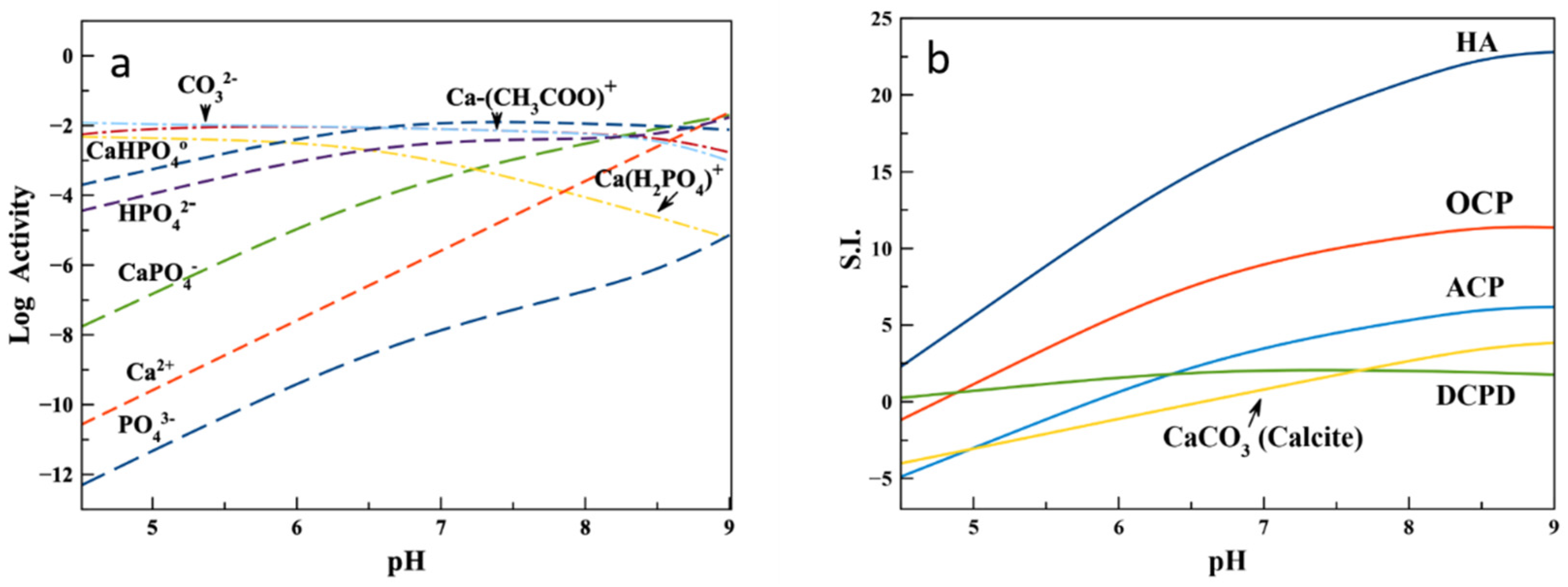
3.1. Ionic Speciation and Evolution of Saturation Indices Respect to Main CaP Phases
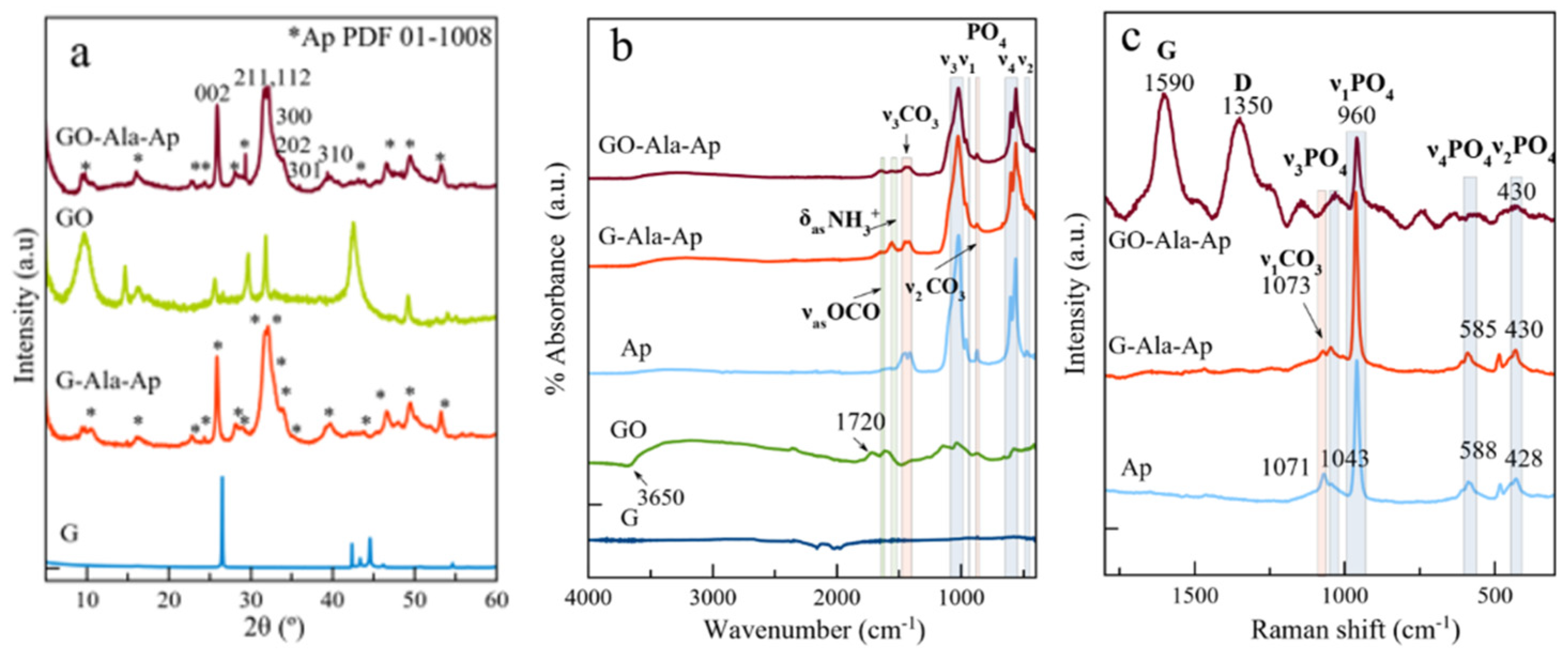
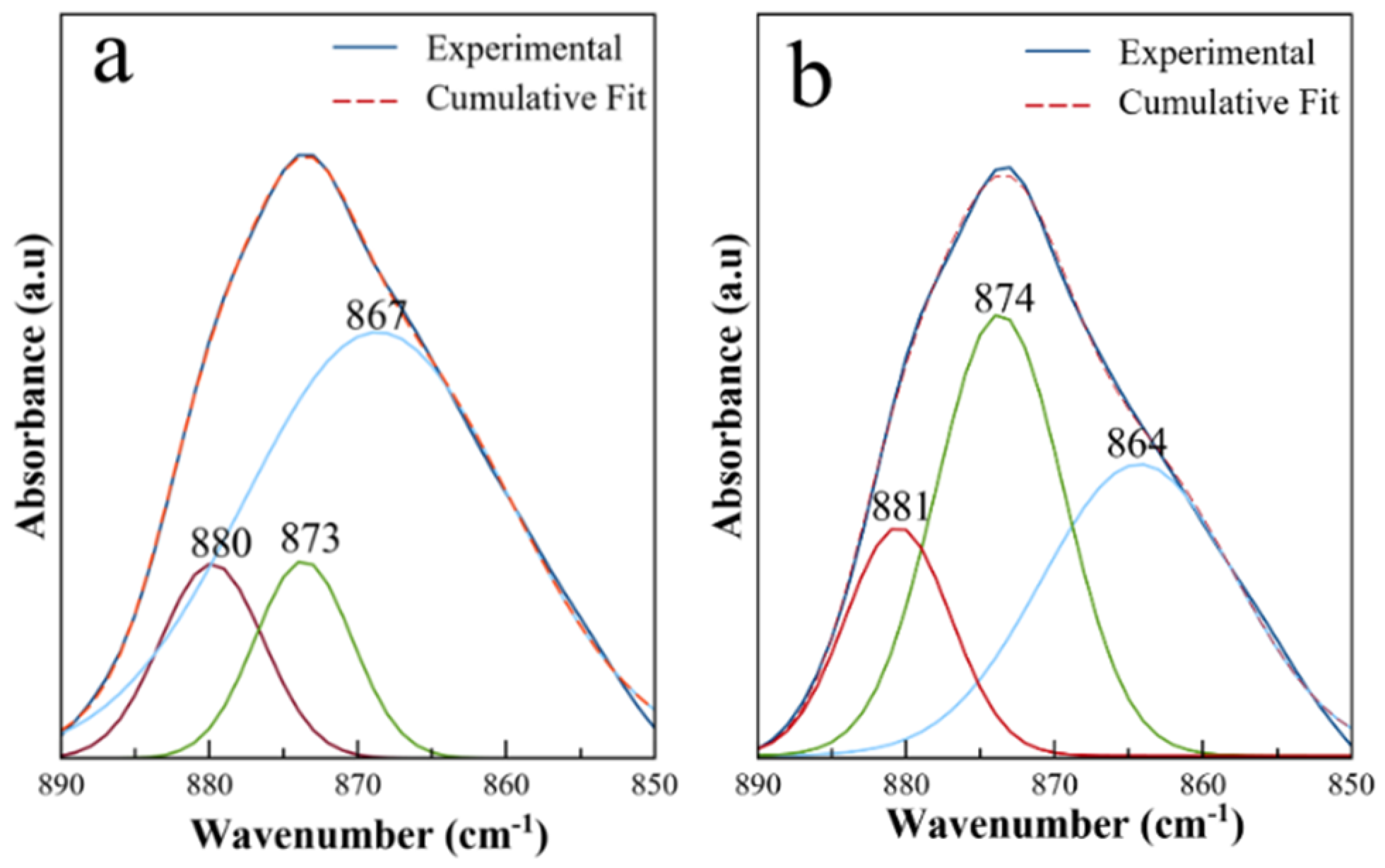
3.2. Structural, Morphological and Spectroscopic Features of Precipitated Solids
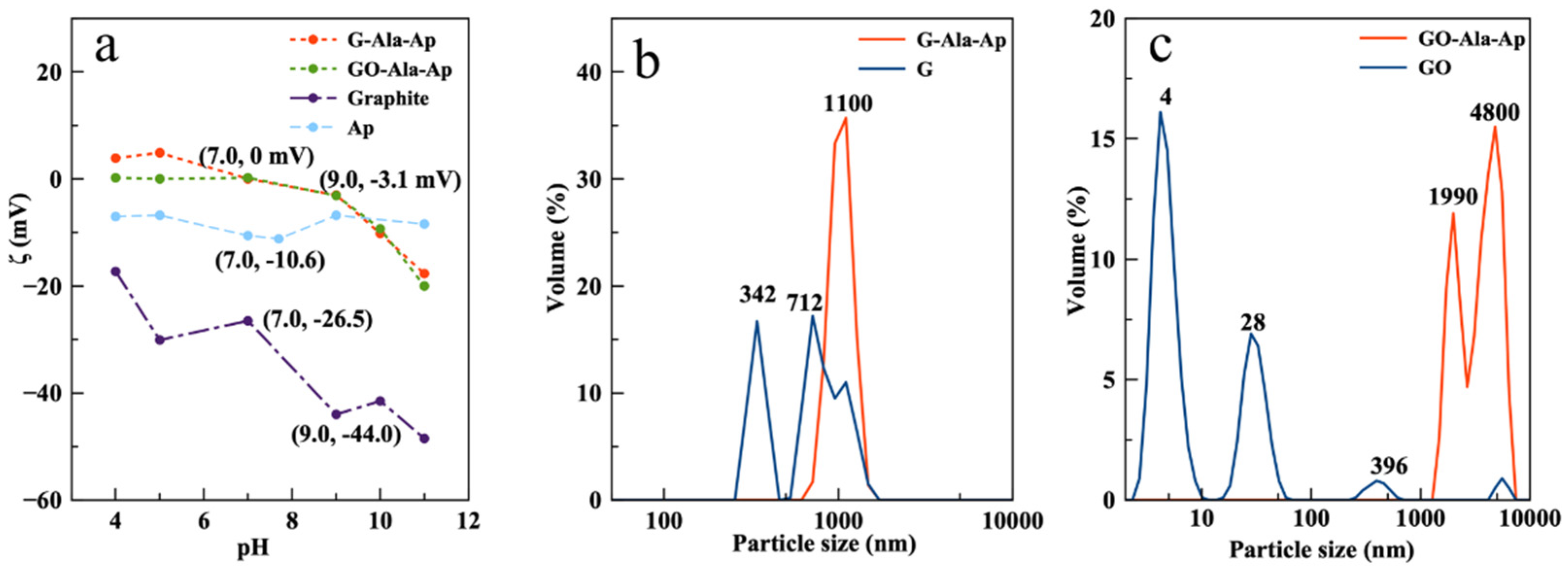
3.3. ζ-Potential and Particle Size Distribution
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Groot, K.; Geesink, R.; Klein, C.P.A.T.; Serekian, P. Plasma sprayed coatings of hydroxylapatite. J. Biomed. Mater. Res. 1987, 21, 1375–1381. [Google Scholar] [CrossRef]
- Lucas, L.C.; Lacefield, W.R.; Ong, J.L.; Whitehead, R.Y. Calcium phosphate coatings for medical and dental implants. Colloids Surf. A Physicochem. Eng. Asp. 1993, 77, 141–147. [Google Scholar] [CrossRef]
- De Grootl, K.; Wolke, J.G.C.; Jansen, J.A. Calcium phosphate coatings for medical implants. Proc. Inst. Mech. Eng. Part H J. Eng. Med. 1998, 212, 137–147. [Google Scholar] [CrossRef]
- Gómez-Morales, J.; Rodríguez-Clemente, R.; Armas, B.; Combescure, C.; Berjoan, R.; Cubo, J.; Martínez, E.; García-Carmona, J.; Garelik, S.; Murtra, J. Controlled nucleation and growth of thin hydroxyapatite layers on titanium implants by using induction heating technique. Langmuir 2004, 20, 5174–5178. [Google Scholar] [CrossRef]
- Surmenev, R.A.; Surmeneva, M.A.; Ivanova, A.A. Significance of calcium phosphate coatings for the enhancement of new bone osteogenesis—A review. Acta Biomater. 2014, 10, 557–579. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Xiong, P.; Yan, F.; Li, S.; Ren, C.; Yin, Z.; Li, A.; Li, H.; Ji, X.; Zheng, Y.; et al. An overview of graphene-based hydroxyapatite composites for orthopedic applications. Bioact. Mater. 2018, 3, 1–18. [Google Scholar] [CrossRef]
- Gómez-Morales, J.; Iafisco, M.; Delgado-López, J.M.; Sarda, S.; Drouet, C. Progress on the preparation of nanocrystalline apatites and surface characterization: Overview of fundamental and applied aspects. Prog. Cryst. Growth Charact. Mater. 2013, 59, 1–46. [Google Scholar] [CrossRef] [Green Version]
- Duplock, E.J.; Scheffler, M.; Lindan, P.J.D. Hallmark of perfect graphene. Phys. Rev. Lett. 2004, 92, 225502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Wei, X.; Kysar, J.W.; Hone, J. Measurement of the elastic properties and intrinsic strength of monolayer graphene. Science 2008, 321, 385–388. [Google Scholar] [CrossRef]
- Yang, K.; Gong, H.; Shi, X.; Wan, J.; Zhang, Y.; Liu, Z. In vivo biodistribution and toxicology of functionalized nano-graphene oxide in mice after oral and intraperitoneal administration. Biomaterials 2013, 34, 2787–2795. [Google Scholar] [CrossRef]
- Sheehy, D.E.; Schmalian, J. Optical transparency of graphene as determined by the fine-structure constant. Phys. Rev. B Condens. Matter Mater. Phys. 2009, 80, 193411. [Google Scholar] [CrossRef] [Green Version]
- Iafisco, M.; Gómez-Morales, J.; Hernández-Hernández, M.A.; García-Ruiz, J.M.; Roveri, N. Biomimetic carbonate-hydroxyapatite nanocrystals prepared by vapor diffusion. Adv. Eng. Mater. 2010, 12, 218–223. [Google Scholar] [CrossRef]
- Gómez-Morales, J.; Verdugo-Escamilla, C.; Gavira, J.A. Bioinspired Calcium Phosphate Coated Mica Sheets by Vapor Diffusion and Its Effects on Lysozyme Assembly and Crystallization. Cryst. Growth Des. 2016, 16, 5150–5158. [Google Scholar] [CrossRef]
- Gómez-Morales, J.; González-Ramírez, L.A.; Verdugo-Escamilla, C.; Penas, R.F.; Oltolina, F.; Prat, M.; Falini, G. Induced nucleation of biomimetic nanoapatites on exfoliated graphene biomolecule flakes by vapor diffusion in microdroplets. Crystals 2019, 9, 341. [Google Scholar] [CrossRef] [Green Version]
- Ciesielski, A.; Samorì, P. Graphene via sonication assisted liquid-phase exfoliation. Chem. Soc. Rev. 2014, 43, 381–398. [Google Scholar] [CrossRef]
- Dai, Z.; Li, Y.; Lu, W.; Jiang, D.; Li, H.; Yan, Y.; Lv, G.; Yang, A. In vivo biocompatibility of new nano-calcium-deficient hydroxyapatite/poly-amino acid complex biomaterials. Int. J. Nanomed. 2015, 10, 6303–6316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafsson, J.P. Visual MINTEQ 3.1, Freeware Chemical Equilibrium Model. Available online: https://vminteq.lwr.kth.se/ (accessed on 23 May 2021).
- Lazić, S. Microcrystalline hydroxyapatite formation from alkaline solutions. J. Cryst. Growth 1995, 147, 147–154. [Google Scholar] [CrossRef]
- Betts, F.; Posner, A.S. An X-ray radial distribution study of amorphous calcium phosphate. Mater. Res. Bull. 1974, 9, 353–360. [Google Scholar] [CrossRef]
- Boskey, A.L.; Posner, A.S. Conversion of amorphous calcium phosphate to microcrystalline hydroxyapatite. A pH-dependent, solution-mediated, solid-solid conversion. J. Phys. Chem. 1973, 77, 2313–2317. [Google Scholar] [CrossRef]
- Habraken, W.J.E.M.; Tao, J.; Brylka, L.J.; Friedrich, H.; Bertinetti, L.; Schenk, A.S.; Verch, A.; Dmitrovic, V.; Bomans, P.H.H.; Frederik, P.M.; et al. Ion-association complexes unite classical and non-classical theories for the biomimetic nucleation of calcium phosphate. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef] [Green Version]
- Iafisco, M.; Degli Esposti, L.; Ramírez-Rodríguez, G.B.; Carella, F.; Gómez-Morales, J.; Ionescu, A.C.; Brambilla, E.; Tampieri, A.; Delgado-López, J.M. Fluoride-doped amorphous calcium phosphate nanoparticles as a promising biomimetic material for dental remineralization. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Martínez-Casado, F.J.; Iafisco, M.; Delgado-López, J.M.; Martínez-Benito, C.; Ruiz-Pérez, C.; Colangelo, D.; Oltolina, F.; Prat, M.; Gómez-Morales, J. Bioinspired Citrate−Apatite Nanocrystals Doped with Divalent Transition Metal Ions. Cryst. Growth Des. 2016, 16, 145–153. [Google Scholar] [CrossRef]
- Grunenwald, A.; Keyser, C.; Sautereau, A.M.; Crubézy, E.; Ludes, B.; Drouet, C. Revisiting carbonate quantification in apatite (bio)minerals: A validated FTIR methodology. J. Archaeol. Sci. 2014, 49, 134–141. [Google Scholar] [CrossRef] [Green Version]
- Garcia, A.R.; De Barros, R.B.; Lourenço, J.P.; Ilharco, L.M. The infrared spectrum of solid L-alanine: Influence of pH-induced structural changes. J. Phys. Chem. A 2008, 112, 8280–8287. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Rodríguez, G.B.; Delgado-López, J.M.; Gómez-Morales, J. Evolution of calcium phosphate precipitation in hanging drop vapor diffusion by in situ Raman microspectroscopy. CrystEngComm 2013, 15, 2206–2212. [Google Scholar] [CrossRef]
- Hao, Y.; Wang, Y.; Wang, L.; Ni, Z.; Wang, Z.; Wang, R.; Koo, C.K.; Shen, Z.; Thong, J.T.L. Probing layer number and stacking order of few-layer graphene by Raman Spectroscopy. Small 2010, 6, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Rey, C.; Combes, C.; Drouet, C.; Cazalbou, S.; Grossin, D.; Brouillet, F.; Sarda, S. Surface properties of biomimetic nanocrystalline apatites; applications in biomaterials. Prog. Cryst. Growth Charact. Mater. 2014, 60, 63–73. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Morales, J.; Verdugo-Escamilla, C.; Fernández-Penas, R.; Parra-Milla, C.M.; Drouet, C.; Maue-Bosc, F.; Oltolina, F.; Prat, M.; Fernández-Sánchez, J.F. Luminescent biomimetic citrate-coated europium-doped carbonated apatite nanoparticles for use in bioimaging: Physico-chemistry and cytocompatibility. RSC Adv. 2018, 8, 2385–2397. [Google Scholar] [CrossRef] [Green Version]
- Guardia, L.; Fernández-Merino, M.J.; Paredes, J.I.; Solís-Fernández, P.; Villar-Rodil, S.; Martínez-Alonso, A.; Tascón, J.M.D. High-throughput production of pristine graphene in an aqueous dispersion assisted by non-ionic surfactants. Carbon 2011, 49, 1653–1662. [Google Scholar] [CrossRef]
- Butyrskaya, E.; Zapryagaev, S.; Izmailova, E.; Nechaeva, L. Sorption Interactions between l/d-Alanine and Carbon Nanotubes in Aqueous Solutions. J. Phys. Chem. C 2017, 121, 20524–20531. [Google Scholar] [CrossRef]
- Aliyev, E.; Filiz, V.; Khan, M.M.; Lee, Y.J.; Abetz, C.; Abetz, V. Structural characterization of graphene oxide: Surface functional groups and fractionated oxidative debris. Nanomaterials 2019, 9, 1180. [Google Scholar] [CrossRef] [Green Version]
- Shih, C.J.; Lin, S.; Sharma, R.; Strano, M.S.; Blankschtein, D. Understanding the pH-dependent behavior of graphene oxide aqueous solutions: A comparative experimental and molecular dynamics simulation study. Langmuir 2012, 28, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Pandit, S.; De, M. Interaction of amino acids and graphene oxide: Trends in thermodynamic properties. J. Phys. Chem. C 2017, 121, 600–608. [Google Scholar] [CrossRef]
- Liu, Y.; Huang, J.; Li, H. Synthesis of hydroxyapatite-reduced graphite oxide nanocomposites for biomedical applications: Oriented nucleation and epitaxial growth of hydroxyapatite. J. Mater. Chem. B 2013, 1, 1826–1834. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FTIR Band | Wavenumber |
|---|---|
| Graphene Oxide | |
| O−H stretching | 3650 cm−1 |
| C=O stretching | 1720 cm−1 |
| PO43− | |
| υ3PO4 asymmetric stretching | 1000–1100 cm−1 |
| υ1PO4 symmetric stretching | 958–960 cm−1 |
| υ2PO4 symmetric bending | 470 cm−1 |
| υ4PO4 asymmetric bending | 608 and 564 cm−1 |
| CO32− | |
| υ3CO3 | 1414 cm−1 and 1473 cm−1 |
| υ2CO3 | 875 cm−1 |
| L-Ala | |
| -OCO- antisymmetric stretch | 1620 cm−1 |
| -NH3+ antisymmetric deformation | 1590 cm−1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Acebedo-Martínez, F.J.; Fernández-Penas, R.; Verdugo-Escamilla, C.; Choquesillo-Lazarte, D.; Gómez-Morales, J. Vapour Diffusion Sitting Drop Method to Induce Nucleation of Calcium Phosphate on Exfoliated Graphene and Graphene Oxide Flakes. Crystals 2021, 11, 767. https://doi.org/10.3390/cryst11070767
Acebedo-Martínez FJ, Fernández-Penas R, Verdugo-Escamilla C, Choquesillo-Lazarte D, Gómez-Morales J. Vapour Diffusion Sitting Drop Method to Induce Nucleation of Calcium Phosphate on Exfoliated Graphene and Graphene Oxide Flakes. Crystals. 2021; 11(7):767. https://doi.org/10.3390/cryst11070767
Chicago/Turabian StyleAcebedo-Martínez, Francisco Javier, Raquel Fernández-Penas, Cristóbal Verdugo-Escamilla, Duane Choquesillo-Lazarte, and Jaime Gómez-Morales. 2021. "Vapour Diffusion Sitting Drop Method to Induce Nucleation of Calcium Phosphate on Exfoliated Graphene and Graphene Oxide Flakes" Crystals 11, no. 7: 767. https://doi.org/10.3390/cryst11070767