
Tautomeric Equilibrium of an Asymmetric β-Diketone in Halogen-Bonded Cocrystals with Perfluorinated Iodobenzenes


Abstract
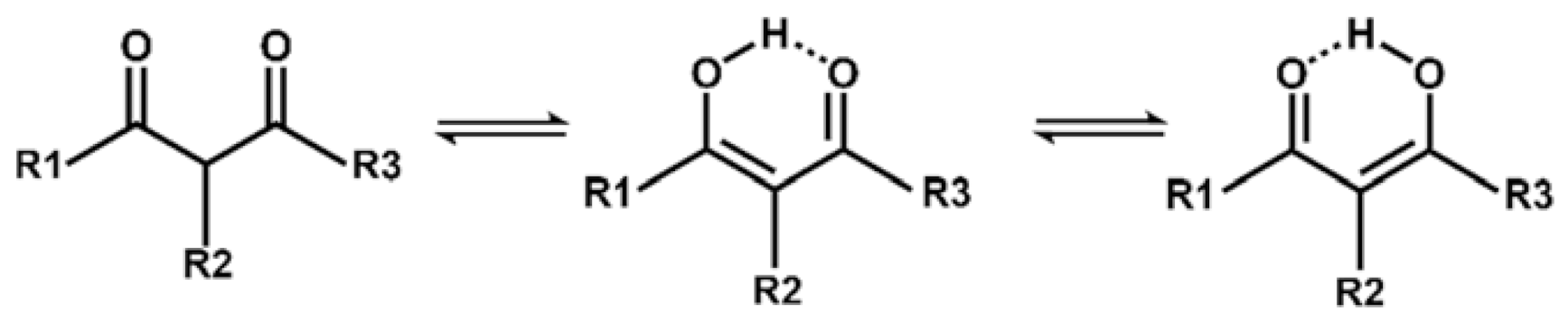
:1. Introduction
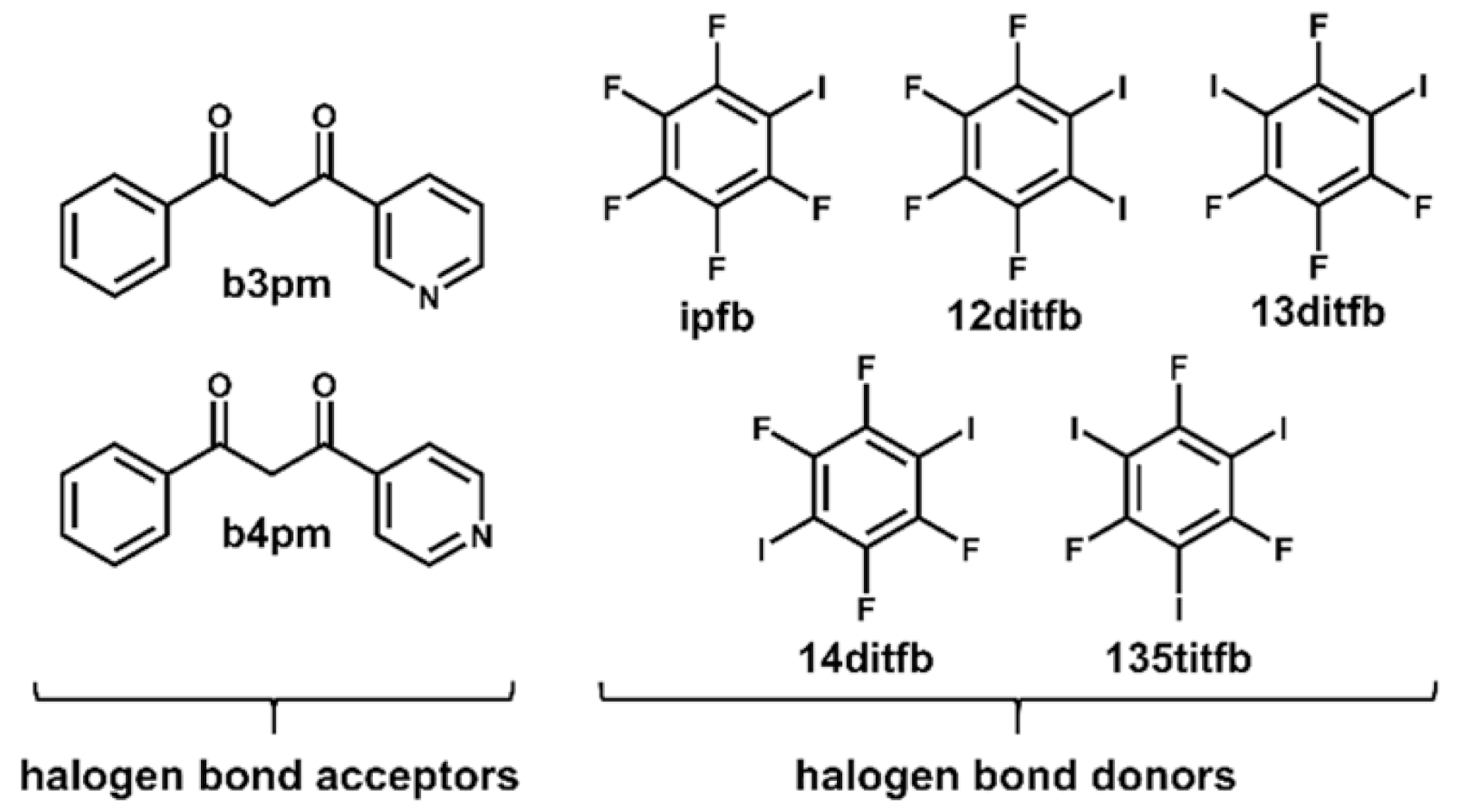
2. Materials and Methods
2.1. Synthesis of Pyridyl Diketones
2.2. Synthesis of the Cocrystals
2.3. X-ray Diffraction Measurements
2.4. Thermal Analysis
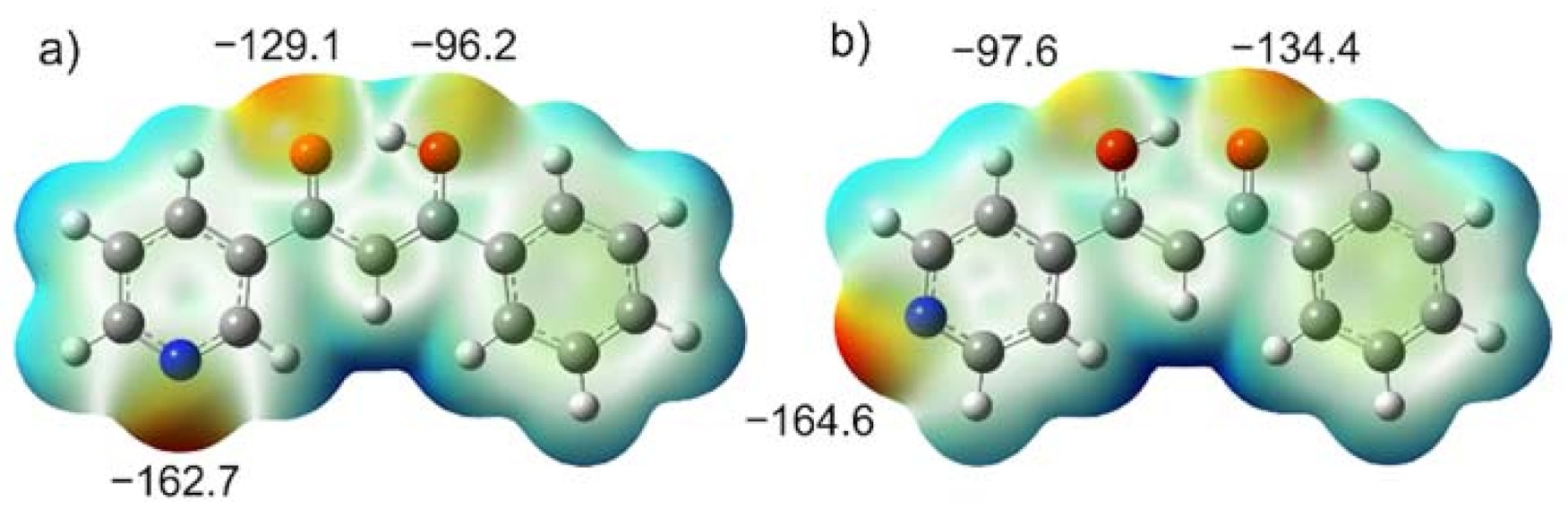
2.5. Computations
3. Results and Discussion
3.1. Preparation of the Cocrystals
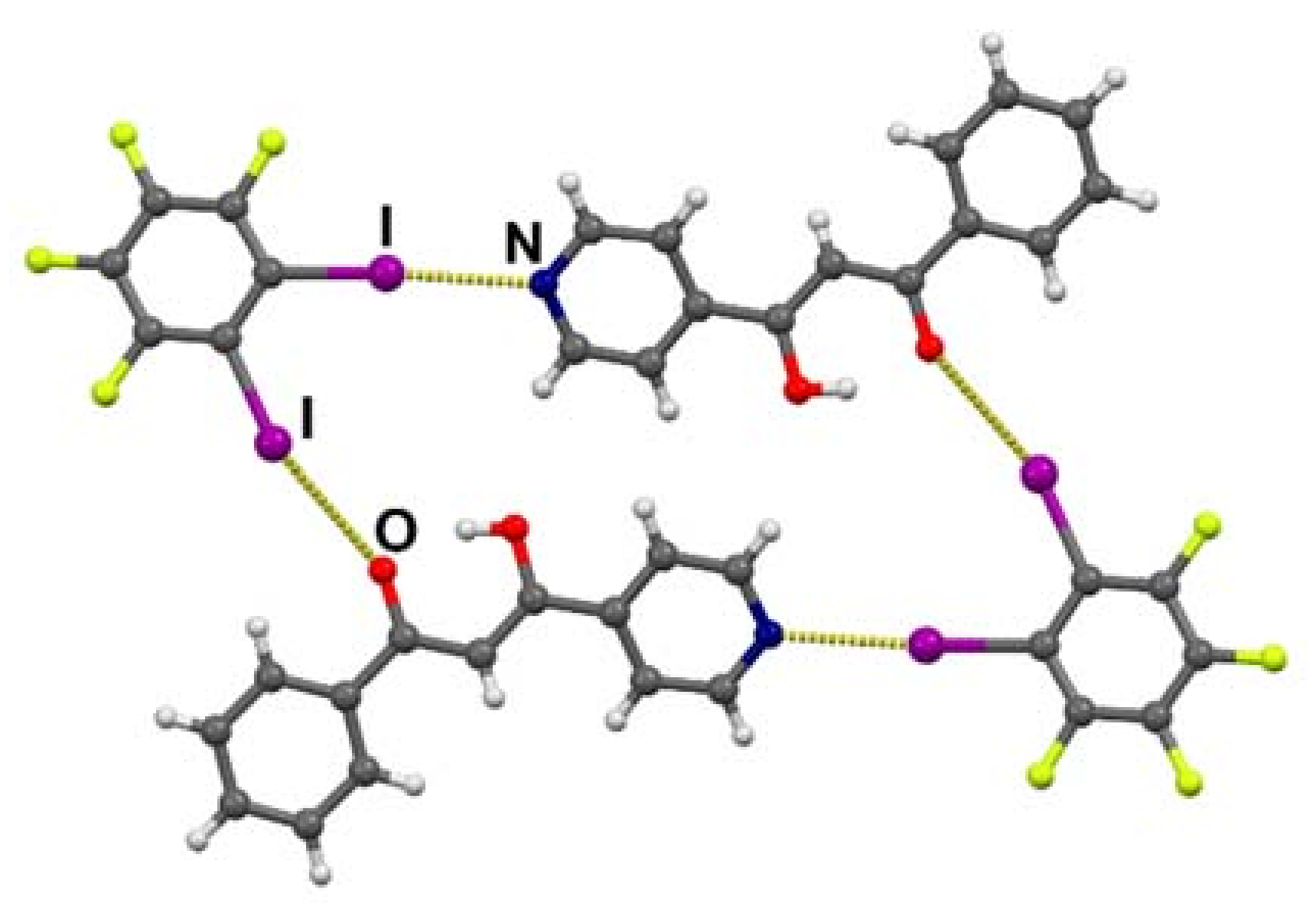
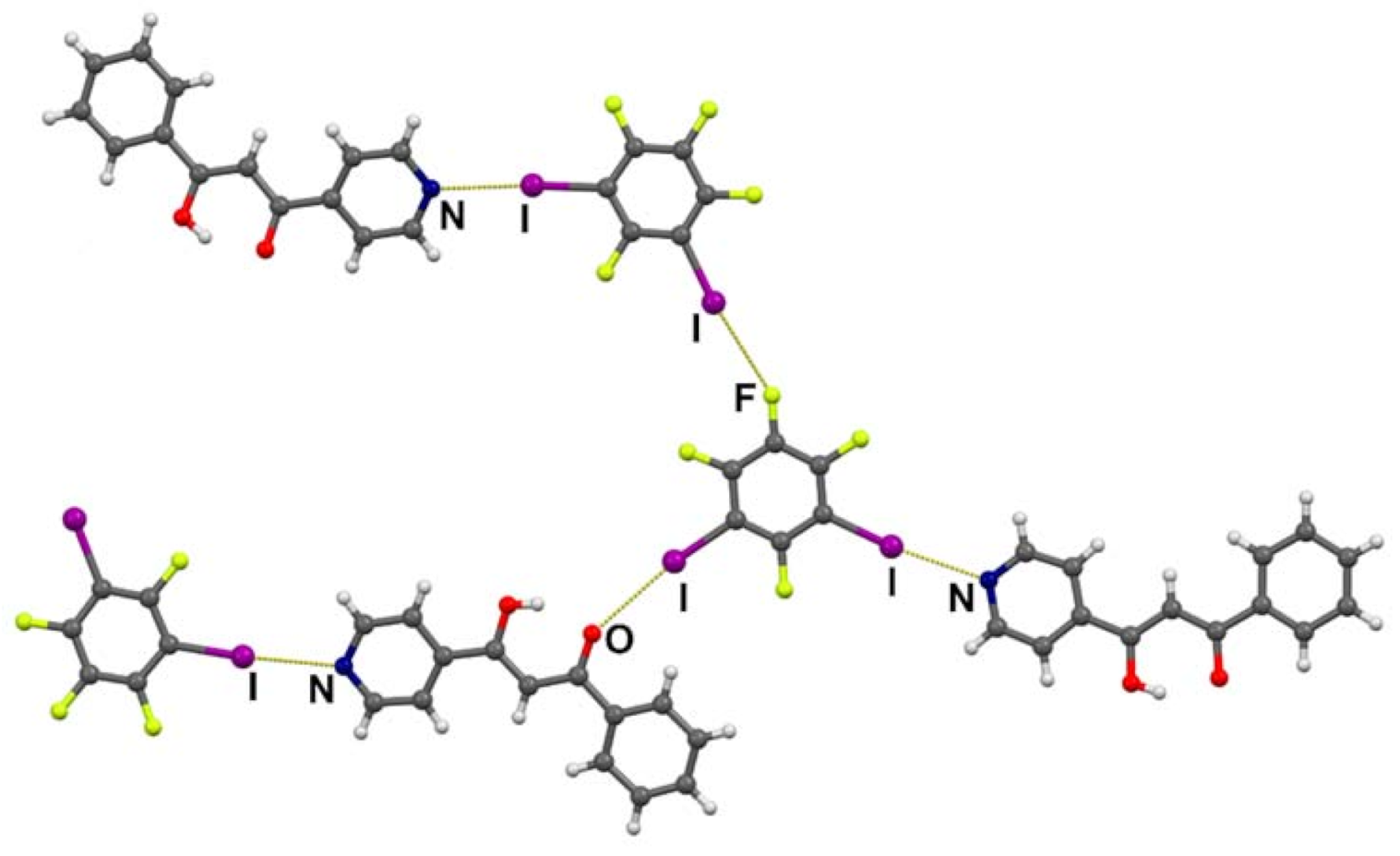
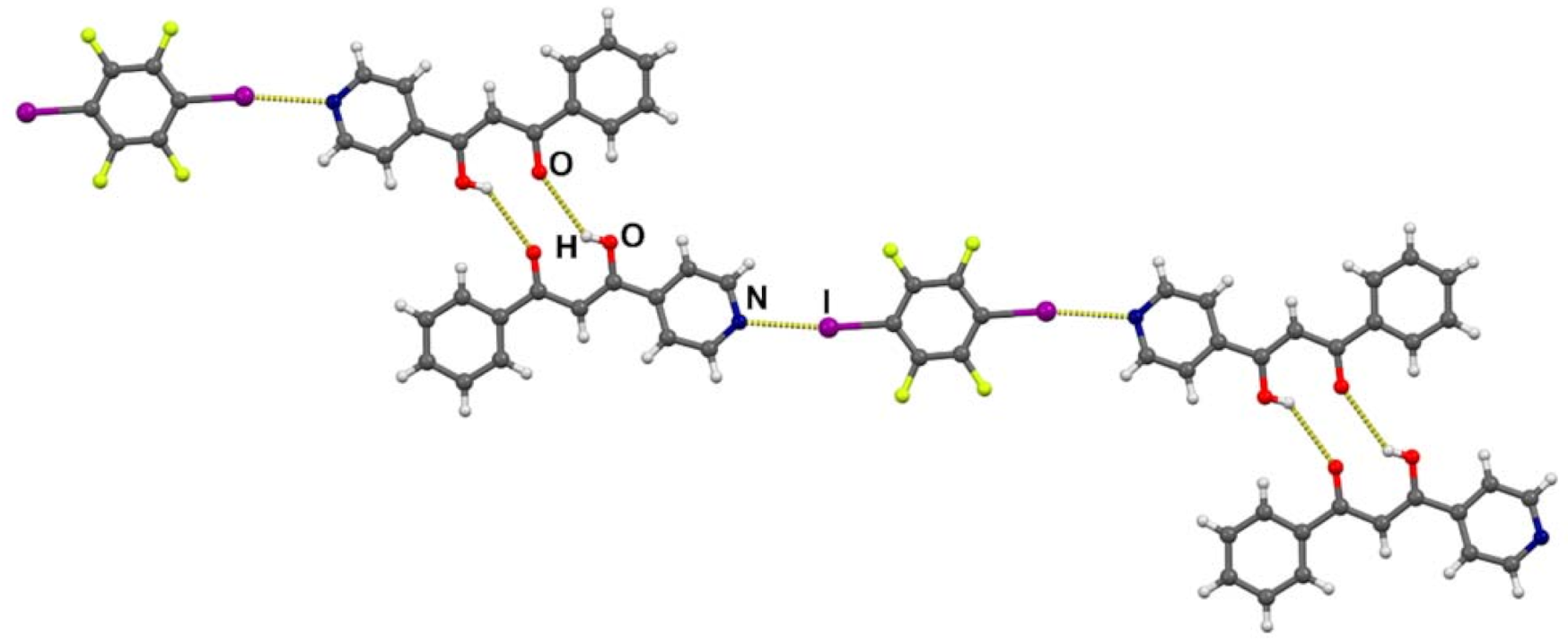
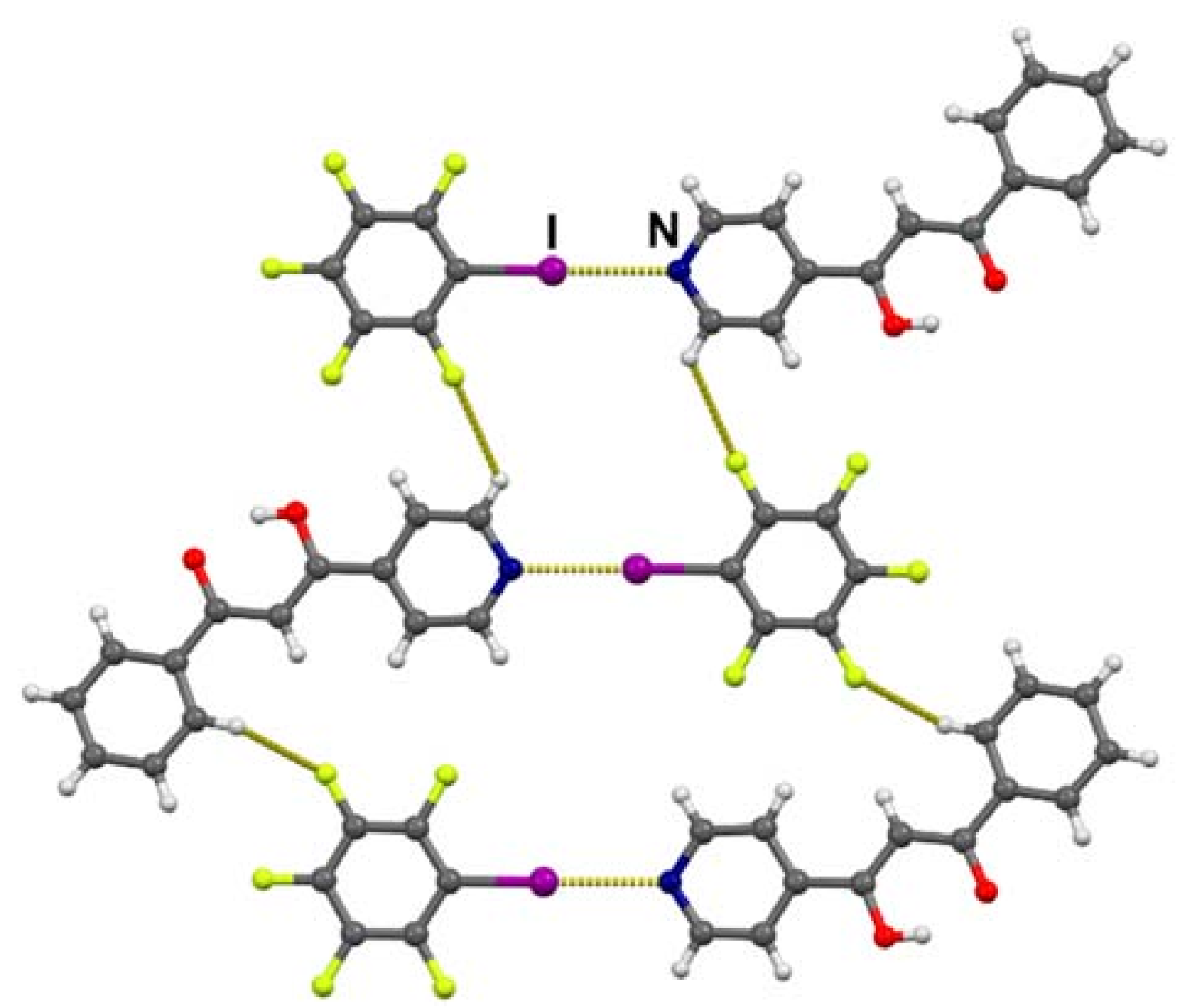
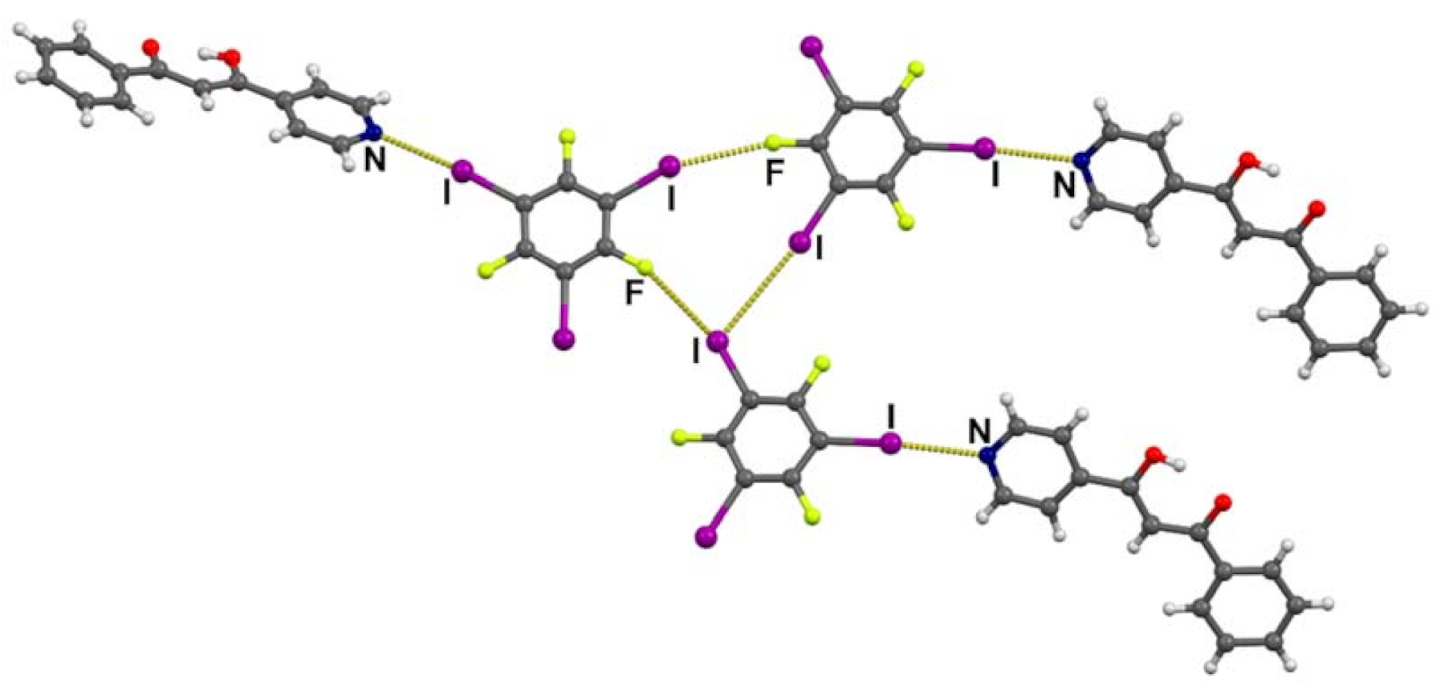
3.2. Crystal Structures of Halogen-Bonded Cocrystals
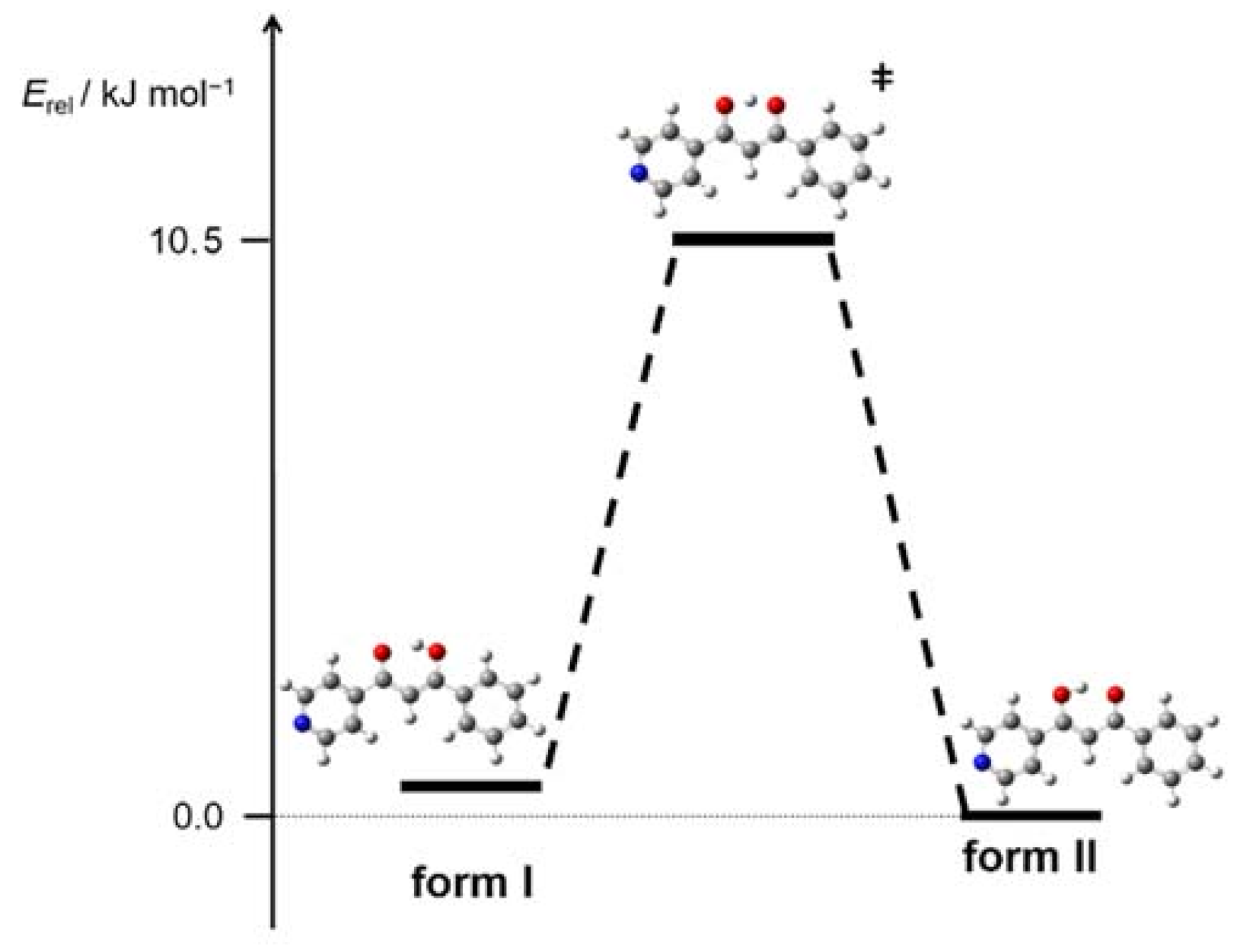
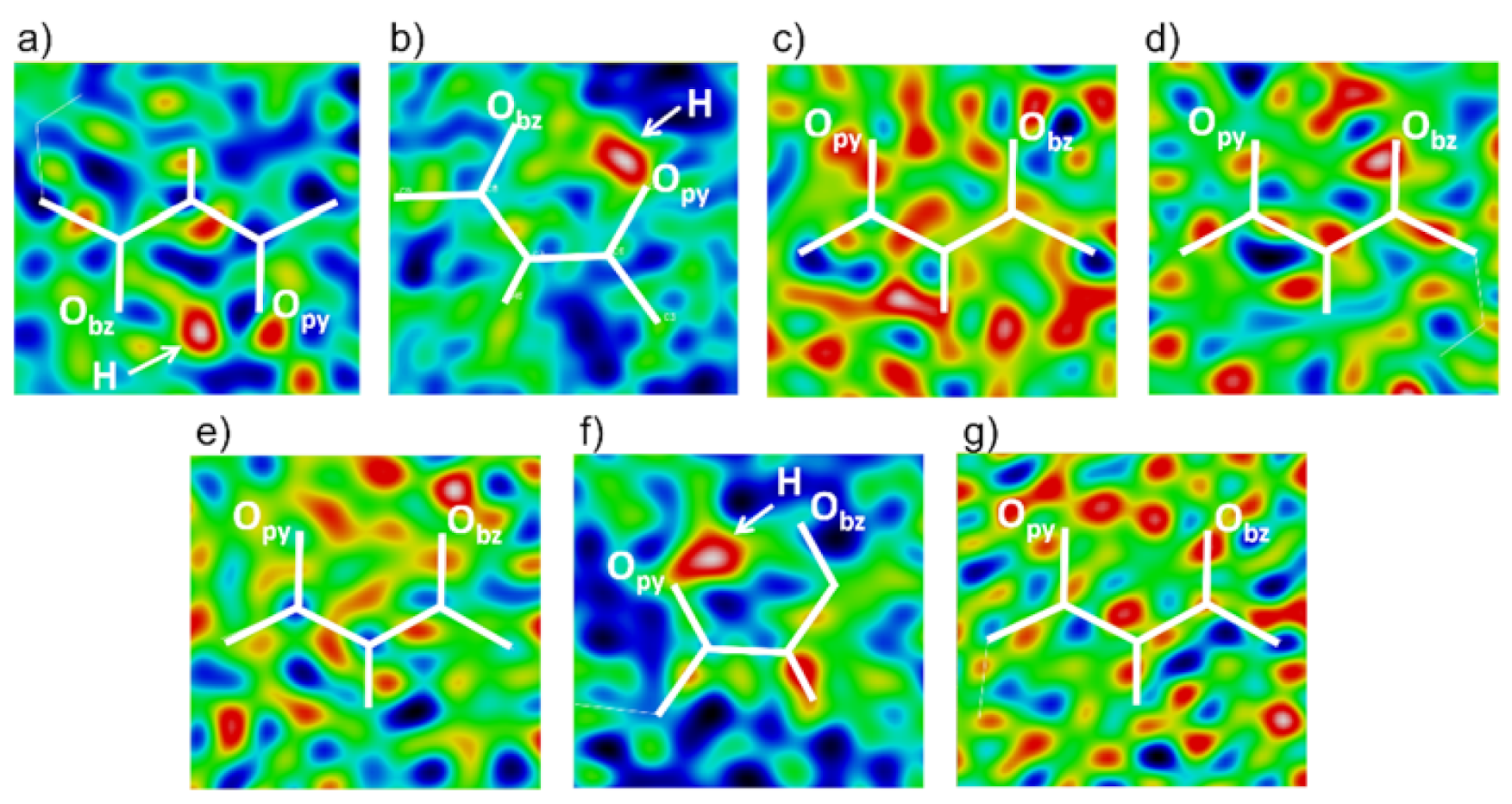
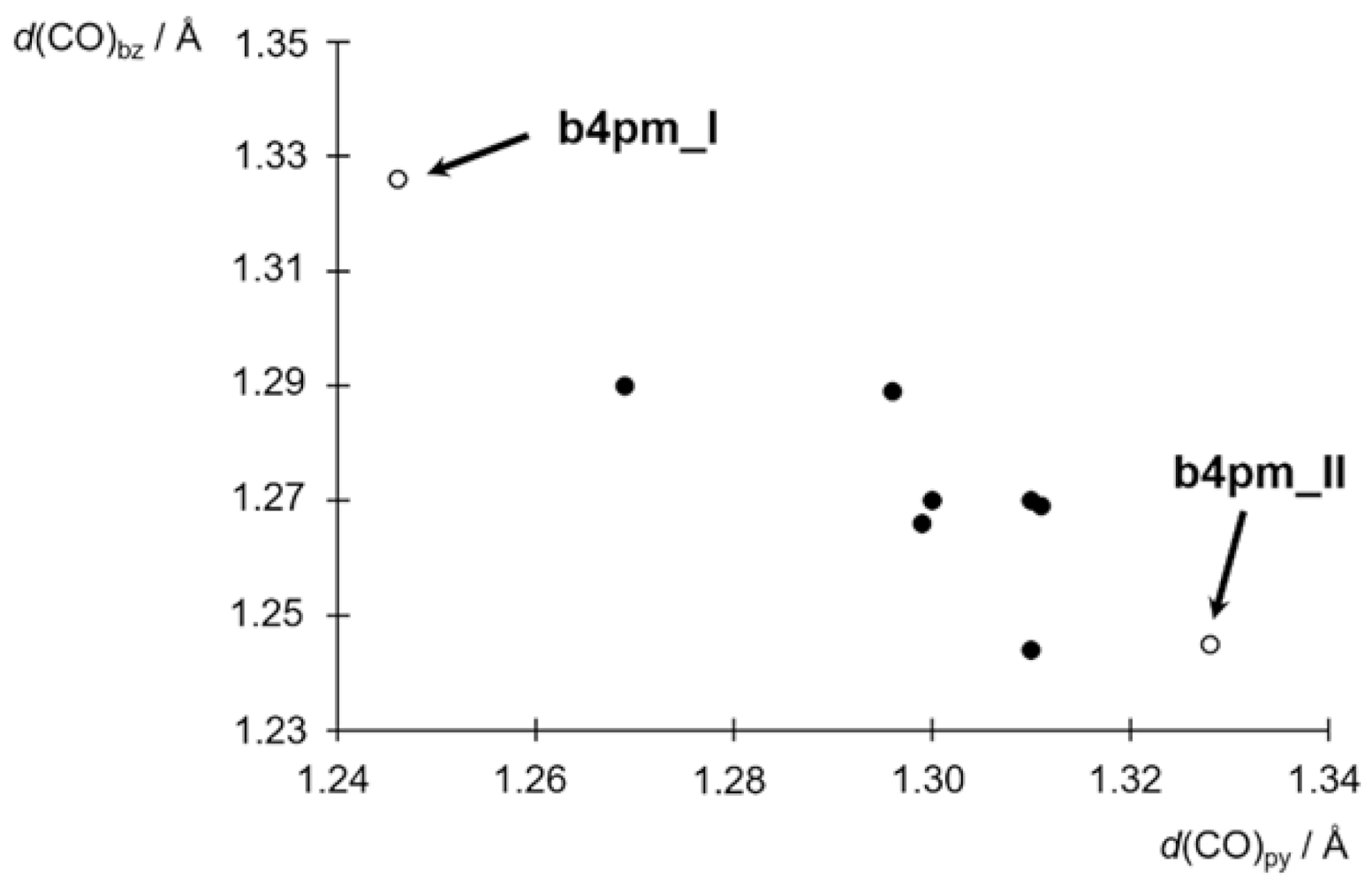
3.3. Tautomerism
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Aakeröy, C.B.; Panikkattu, S.; Chopade, P.D.; Desper, J. Competing hydrogen-bond and halogen-bond donors in crystal engineering. CrystEngComm 2013, 15, 3125–3136. [Google Scholar] [CrossRef] [Green Version]
- Aakeröy, C.B.; Spartz, C.L.; Dembowski, S.; Dwyrea, S.; Despera, J. A systematic structural study of halogen bonding versus hydrogen bonding within competitive supramolecular systems. IUCrJ 2015, 2, 498–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartashevich, E.V.; Tsirelson, V.G. Interplay between non-covalent interactions in complexes and crystals with halogen bonds. Russ. Chem. Rev. 2014, 83, 1181–1203. [Google Scholar] [CrossRef]
- Awwadi, F.F.; Taher, D.; Haddad, S.F.; Turnbull, M.M. Competition between Hydrogen and Halogen Bonding Interactions: Theoretical and Crystallographic Studies. Cryst. Growth Des. 2014, 14, 1961–1971. [Google Scholar] [CrossRef]
- Jing, B.; Li, Q.; Li, R.; Gong, B.; Liu, Z.; Li, W.; Cheng, J.; Sun, J. Competition and cooperativity between hydrogen bond and halogen bond in HNC⋯(HOBr)n and (HNC)n⋯HOBr (n = 1 and 2) systems. Comput. Theor. Chem. 2011, 963, 417–421. [Google Scholar] [CrossRef]
- Bauzá, A.; Frontera, A. Competition between lone pair-π, halogen-π and triel bonding interactions involving BX3 (X = F, Cl, Br and I) compounds: An ab initio study. Theor. Chem. Acc. 2017, 136, 37. [Google Scholar] [CrossRef]
- Ji, B.; Wang, W.; Deng, D.; Zhang, Y.; Cao, L.; Zhou, L.; Ruana, C.; Li, T. Structural competition between π⋯π interactions and halogen bonds: A crystallographic study. CrystEngComm 2013, 15, 769–774. [Google Scholar] [CrossRef]
- Nemec, V.; Lisac, K.; Bedeković, N.; Fotović, L.; Stilinović, V.; Cinčić, D. Crystal engineering strategies towards halogen-bonded metal–organic multi-component solids: Salts, cocrystals and salt cocrystals. CrystEngComm 2021, 23, 3063–3083. [Google Scholar] [CrossRef]
- Sumi, Y.; Sasaki, K.; Tsuzuki, S.; Shibata, N. Studies of Halogen Bonding Induced by Pentafluorosulfanyl Aryl Iodides: A Potential Group of Halogen Bond Donors in a Rational Drug Design. Molecules 2019, 24, 3610–3625. [Google Scholar] [CrossRef] [Green Version]
- Hutchins, K.M.; Kummer, K.A.; Groeneman, R.H.; Reinheimer, E.W.; Sinnwell, M.A.; Swensona, D.C.; MacGillivray, L.R. Thermal expansion properties of three isostructural co-crystals composed of isosteric components: Interplay between halogen and hydrogen bonds. CrystEngComm 2016, 18, 8354–8357. [Google Scholar] [CrossRef]
- Riel, A.M.S.; Rowe, R.K.; Ho, E.N.; Carlsson, A.C.C.; Rappé, A.K.; Berryman, O.B.; Ho, P.S. Hydrogen Bond Enhanced Halogen Bonds: A Synergistic Interaction in Chemistry and Biochemistry. Acc. Chem. Res. 2019, 52, 2870–2880. [Google Scholar] [CrossRef]
- Li, Q.; Lin, Q.; Li, W.; Cheng, J.; Gong, B.; Sun, J. Cooperativity between the Halogen Bond and the Hydrogen Bond in H3N⋅⋅⋅XY⋅⋅⋅HF Complexes (X, Y = F, Cl, Br). ChemPhysChem 2008, 9, 2265–2269. [Google Scholar] [CrossRef]
- Ciancaleoni, G. Cooperativity between hydrogen- and halogen bonds: The case of selenourea. Phys. Chem. Chem. Phys. 2018, 20, 8506–8514. [Google Scholar] [CrossRef]
- Saha, S.; Sastry, G.N. Cooperative or Anticooperative: How Noncovalent Interactions Influence Each Other. J. Phys. Chem. B 2015, 119, 11121–11135. [Google Scholar] [CrossRef]
- Stilinović, V.; Kaitner, B. Salts and Co-Crystals of Gentisic Acid with Pyridine Derivatives: The Effect of Proton Transfer on the Crystal Packing (and Vice Versa). Cryst. Growth Des. 2012, 12, 5763–5772. [Google Scholar] [CrossRef]
- Bedeković, N.; Stilinović, V.; Piteša, T. Aromatic versus Aliphatic Carboxyl Group as a Hydrogen Bond Donor in Salts and Cocrystals of an Asymmetric Diacid and Pyridine Derivatives. Cryst. Growth. Des. 2017, 11, 5732–5743. [Google Scholar] [CrossRef]
- Sarma, B.; Nath, N.K.; Bhogala, B.R.; Nangia, A. Synthon Competition and Cooperation in Molecular Salts of Hydroxybenzoic Acids and Aminopyridines. Cryst. Growth Des. 2009, 9, 1546–1557. [Google Scholar] [CrossRef]
- Lemmerer, A.; Govindraju, S.; Johnston, M.; Motloung, X.; Savig, K.L. Co-crystals and molecular salts of carboxylic acid/pyridine complexes: Can calculated pKa’s predict proton transfer? A case study of nine complexes. CrystEngComm 2015, 17, 3591–3595. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, S.; Tocher, D.A.; Vickers, M.; Karamertzanis, P.G.; Price, S.L. Salt or Cocrystal? A New Series of Crystal Structures Formed from Simple Pyridines and Carboxylic Acids. Cryst. Growth Des. 2009, 9, 2881–2889. [Google Scholar] [CrossRef]
- Cruz-Cabeza, A.J. Acid–base crystalline complexes and the pKa rule. CrystEngComm 2012, 14, 6362–6365. [Google Scholar] [CrossRef]
- Blagus, A.; Cinčić, D.; Friščić, T.; Kaitner, B.; Stilinović, V. Schiff bases derived from hydroxyaryl aldehydes: Molecular and crystal structure, tautomerism, quinoid effect, coordination compounds. Maced. J. Chem. Chem. Eng. 2010, 29, 117–138. [Google Scholar] [CrossRef] [Green Version]
- Rubčić, M.; Užarević, K.; Halasz, I.; Bregović, N.; Mališ, M.; Đilović, I.; Kokan, Z.; Stein, R.S.; Dinnebier, R.E.; Tomišić, V. Desmotropy, Polymorphism, and Solid-State Proton Transfer: Four Solid Forms of an Aromatic o-Hydroxy Schiff Base. Chem. Eur. J. 2012, 18, 5620–5631. [Google Scholar] [CrossRef] [PubMed]
- Juribašić, M.; Bregović, N.; Stilinović, V.; Tomišić, V.; Cindrić, M.; Šket, P.; Plavec, J.; Rubčić, M.; Užarević, K. Supramolecular Stabilization of Metastable Tautomers in Solution and the Solid State. Chem. Eur. J. 2014, 20, 17333–17345. [Google Scholar] [CrossRef] [PubMed]
- Užarević, K.; Rubčić, M.; Stilinović, V.; Kaitner, B.; Cindrić, M. Keto–enol tautomerism in asymmetric Schiff bases derived from p-phenylenediamine. J. Mol. Struct. 2010, 984, 232–239. [Google Scholar] [CrossRef]
- Užarević, K.; Stilinović, V.; Rubčić, M. Supramolecular Control over Tautomerism in Organic Solids in Tautomerism: Concepts and Applications in Science and Technology; Antonov, L., Ed.; John Wiley & Sons: Weinheim, Germany, 2016; pp. 295–328. [Google Scholar]
- Hadjoudis, E.; Mavridis, I.M. Photochromism and thermochromism of Schiff bases in the solid state: Structural aspects. Chem. Soc. Rev. 2004, 33, 579–588. [Google Scholar] [CrossRef]
- Metrangolo, P.; Resnati, G. Halogen Bonding: A Paradigm in Supramolecular Chemistry. Chem. Eur. J. 2001, 7, 2511–2519. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [Green Version]
- Stilinović, V.; Horvat, G.; Hrenar, T.; Nemec, V.; Cinčić, D. Halogen and Hydrogen Bonding between (N-Halogeno)-succinimides and Pyridine Derivatives in Solution, the Solid State and In Silico. Chem. Eur. J. 2017, 23, 5244–5257. [Google Scholar] [CrossRef]
- Eraković, M.; Cinčić, D.; Molčanov, K.; Stilinović, V. A Crystallographic Charge Density Study of the Partial Covalent Nature of Strong N⋅⋅⋅ Br Halogen Bonds. Angew. Chem. 2019, 131, 15849–15853. [Google Scholar] [CrossRef]
- Zbačnik, M.; Vitković, M.; Vulić, V.; Nogalo, I.; Cinčić, D. Competition between Halogen Bonds in Cocrystals of Imines Derived from o-Vanillin. Cryst. Growth Des. 2016, 16, 6381–6389. [Google Scholar] [CrossRef]
- Carletta, A.; Zbačnik, M.; Van Gysel, M.; Vitković, M.; Tumanov, N.; Stilinović, V.; Wouters, J.; Cinčić, D. Playing with Isomerism: Cocrystallization of Isomeric N-Salicylideneaminopyridines with Perfluorinated Compounds as Halogen Bond Donors and Its Impact on Photochromism. Cryst. Growth Des. 2018, 18, 6833–6842. [Google Scholar] [CrossRef]
- Carletta, A.; Zbačnik, M.; Vitković, M.; Tumanov, N.; Stilinović, V.; Wouters, J.; Cinčić, D. Halogen-bonded cocrystals of N-salicylidene Schiff bases and iodoperfluorinated benzenes: Hydroxyl oxygen as a halogen bond acceptor. CrystEngComm 2018, 20, 5332–5339. [Google Scholar] [CrossRef]
- Carletta, A.; Spinelli, F.; d’Agostino, S.; Ventura, B.; Chierotti, M.R.; Gobetto, R.; Wouters, J.; Grepioni, F. Halogen-Bond Effects on the Thermo- and Photochromic Behaviour of Anil-Based Molecular Co-crystals. Chem. Eur. J. 2017, 23, 5317–5329. [Google Scholar] [CrossRef]
- Zbačnik, M.; Pajski, M.; Stilinović, V.; Vitković, M.; Cinčić, D. The halogen bonding proclivity of the ortho-methoxy–hydroxy group in cocrystals of o-vanillin imines and diiodotetrafluoro-benzenes. CrystEngComm 2017, 19, 5576–5582. [Google Scholar] [CrossRef]
- Chu, M.; Qiu, B.; Zhang, W.; Zhou, Z.; Yang, X.; Yan, Y.; Yao, J.; Li, Y.J.; Zhao, Y.S. Tailoring the Energy Levels and Cavity Structures toward Organic Cocrystal Microlasers. ACS Appl. Mater. Interfaces 2018, 10, 42740–42746. [Google Scholar] [CrossRef]
- Mercier, G.M.; Robeyns, K.; Tumanov, N.; Champagne, B.; Wouters, J.; Leyssens, T. New Insights into Photochromic Properties of N-Salicylideneaniline Derivatives Using a Cocrystal Engineering Approach. Cryst. Growth Des. 2019, 19, 5544–5556. [Google Scholar] [CrossRef]
- Koltsov, A.I. Enol-enol tautomerism in cis-keto-enols. J. Mol. Struct. 1998, 444, 1–11. [Google Scholar] [CrossRef]
- Yoshida, Z.; Ogoshi, H.; Tokumitsu, T. Intramolecular hydrogen bond in enol form of 3-substituted-2,4-pentanedione. Tetrahedron 1970, 26, 5691–5697. [Google Scholar] [CrossRef]
- Yoffe, S.T.; Fedin, E.I.; Petrovskii, P.V.; Kabachnik, M.I. High resolution NMR investigation of enolization of ethyl α-alkylacetoacetates and 3-alkylacetylacetones with branched substituents. Tetrahedron Lett. 1966, 7, 2661–2668. [Google Scholar] [CrossRef]
- Iglesias, E. Substituent effects on enol nitrosation of 1,3-diketones. Int. J. Chem. Kin. 2012, 44, 668–679. [Google Scholar] [CrossRef]
- Iglesias, E. A new method for the nitrosation of 1,3-diketones applied to 3-ethyl-and 3-methyl pentane-2,4-dione. RSC Adv. 2013, 3, 15192–15201. [Google Scholar] [CrossRef]
- Stilinović, V.; Portada, T.; Kaitner, B. Predominance of the triketo tautomer in acyldipivaloylmethanes in solution and the solid state. J. Mol. Struct. 2014, 1063, 123–130. [Google Scholar] [CrossRef]
- Burdett, J.L.; Rogers, M.T. Keto-Enol Tautomerism in β-Dicarbonyls Studied by Nuclear Magnetic Resonance Spectroscopy. I. Proton Chemical Shifts and Equilibrium Constants of Pure Compounds. J. Am. Chem. Soc. 1964, 86, 2105–2109. [Google Scholar] [CrossRef]
- Rogers, M.T.; Burdett, J.L. Keto–enol tautomerism in β-dicarbonyls studied by nuclear magnetic resonance spectroscopy: II. Solvent effects on proton chemical shifts and on equilibrium constants. Can. J. Chem. 1965, 43, 1516–1526. [Google Scholar] [CrossRef] [Green Version]
- Walsh, R.B.; Padgett, C.W.; Metrangolo, P.; Resnati, G.; Hanks, T.W.; Pennington, W.T. Crystal engineering through halogen bonding: Complexes of nitrogen heterocycles with organic iodides. Cryst. Growth Des. 2001, 1, 165–175. [Google Scholar] [CrossRef]
- Ding, X.H.; Ou, C.J.; Wang, S.; Xie, L.H.; Lin, J.Y.; Wang, J.P.; Huang, W. Co-crystallization of 1,3,5-trifluoro-2,4,6-triiodobenzene (1,3,5-TFTIB) with a variety of Lewis bases through halogen-bonding interactions. CrystEngComm 2017, 19, 5504–5521. [Google Scholar] [CrossRef]
- Bedeković, N.; Stilinović, V.; Friščić, T.; Cinčić, D. Comparison of isomeric meta- and para-diiodotetrafluorobenzene as halogen bond donors in crystal engineering. New J. Chem. 2018, 42, 10584–10591. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. 2016, B72, 171–179. [Google Scholar] [CrossRef]
- Brown, J.J.; Brock, A.J.; Pfrunder, M.C.; Sarju, J.P.; Perry, A.Z.; Whitwood, A.C.; Bruce, D.W.; McMurtrie, J.C.; Clegg, J.K. Co-Crystallisation of 1,4-Diiodotetrafluorobenzene with Three Different Symmetric Dipyridylacetylacetone Isomers Produces Four Halogen-Bonded Architectures. Aust. J. Chem. 2017, 70, 594–600. [Google Scholar] [CrossRef]
- Pfrunder, M.C.; Brock, A.J.; Brown, J.J.; Grosjean, A.; Ward, J.; McMurtrie, J.C.; Clegg, J.K. A three-dimensional cubic halogen-bonded network. Chem. Commun. 2018, 54, 3974–3976. [Google Scholar] [CrossRef]
- CrysAlis CCD V171.34, Oxford Diffraction; Oxford Diffraction Ltd.: Abingdon, UK, 2003.
- CrysAlis RED V171.34, Oxford Diffraction; Oxford Diffraction Ltd.: Abingdon, UK, 2003.
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Cryst. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Cryst. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0-new features for the visualization and investigation of crystal structures. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Philips X’Pert Data Collector 1.3e; Philips Analytical B. V.: Almelo, The Netherlands, 2001.
- Philips X’Pert Graphic & Identify 1.3e Philips; Analytical B. V.: Almelo, The Netherlands, 2001.
- Philips X’Pert Plus 1.0; Philips Analytical B. V.: Almelo, The Netherlands, 1999.
- Degen, T.; Sadki, M.; Bron, E.; König, U.; Nénert, G. The HighScore suite. Powder Diffr. 2014, 29, S13–S18. [Google Scholar] [CrossRef] [Green Version]
- TARe Evaluation Software Version 15.00; Mettler–Toledo GmbH: Greifensee, Switzerland, 2016.
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Godbout, N.; Salahub, D.R.; Andzelm, J.; Wimmer, E. Optimization of Gaussian-type basis sets for local spin density functional calculations. Part I. Boron through neon, optimization technique and validation. Can. J. Chem. 1992, 70, 560–571. [Google Scholar] [CrossRef] [Green Version]
- Sosa, C.; Andzelm, J.; Elkin, B.C.; Wimmer, E.; Dobbs, K.D.; Dixon, D.A. A local density functional study of the structure and vibrational frequencies of molecular transition-metal compounds. J. Phys. Chem. 1992, 96, 6630–6636. [Google Scholar] [CrossRef]
- Siiskonen, A.; Priimagi, A. Benchmarking DFT methods with small basis sets for the calculation of halogen-bond strengths. J. Mol. Model. 2017, 23, 50. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods. V. Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173–1213. [Google Scholar] [CrossRef] [Green Version]
- Curtiss, L.A.; McGrath, M.P.; Blaudeau, J.P.; Davis, N.E.; Binning, R.C.; Radom, L. Extension of Gaussian-2 theory to molecules containing third-row atoms Ga-Kr. J. Chem. Phys. 1995, 103, 6104–6113. [Google Scholar] [CrossRef]
- GaussView, Version 5.1; Dennington, R.; Keith, T.A.; Millam, J.M. (Eds.) Semichem Inc.: Shawnee, KS, USA, 2008. [Google Scholar]
- Dudek, M.; Clegg, J.K.; Glasson, C.R.K.; Kelly, N.; Gloe, K.; Gloe, K.; Kelling, A.; Buschmann, H.J.; Jolliffe, K.A.; Lindoy, L.F.; et al. Interaction of Copper(II) with ditopic Pyridyl-β-diketone Ligands: Dimeric, Framework and Metallogel Structures. Cryst. Growth Des. 2011, 11, 1697–1704. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Halogen Bond Donor | b4pm | tm/°C | ΔfusH/kJ mol−1 |
|---|---|---|---|
| ipfb | (b4pm)(ipfb) | 80.4 | 20.3 |
| 12tfib | (b4pm)(12tfib) | 86.3 | 21.6 |
| 13tfib | (b4pm)(13tfib) | 99.8 | 26.9 |
| 14tfib | (b4pm)2(14tfib) | 101.5 | 26.7 |
| 135tfib | (b4pm)(135tfib) | 108.6 | 37.5 |
| Complex | Ebind/kJ mol−1 |
|---|---|
| (b4pm)(ipfb)_N | −27.9 |
| (b4pm)(ipfb)_O | −23.6 |
| (b4pm)(ipfb)_OH | −19.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinez, V.; Bedeković, N.; Stilinović, V.; Cinčić, D. Tautomeric Equilibrium of an Asymmetric β-Diketone in Halogen-Bonded Cocrystals with Perfluorinated Iodobenzenes. Crystals 2021, 11, 699. https://doi.org/10.3390/cryst11060699
Martinez V, Bedeković N, Stilinović V, Cinčić D. Tautomeric Equilibrium of an Asymmetric β-Diketone in Halogen-Bonded Cocrystals with Perfluorinated Iodobenzenes. Crystals. 2021; 11(6):699. https://doi.org/10.3390/cryst11060699
Chicago/Turabian StyleMartinez, Valentina, Nikola Bedeković, Vladimir Stilinović, and Dominik Cinčić. 2021. "Tautomeric Equilibrium of an Asymmetric β-Diketone in Halogen-Bonded Cocrystals with Perfluorinated Iodobenzenes" Crystals 11, no. 6: 699. https://doi.org/10.3390/cryst11060699