2.1. Sorbent Materials
The raw material of the purified clinoptilolite tuff (PCT) originates from the open-pit mine in Nižný Hrabovec (eastern Slovak Republic). It was purified according to a patented treatment, involving several ion exchange steps, micronization, and terminal heat treatment [
25]. The heavy-metal-reduced, extremely fine-grained, and dry-sterilized product is available as a dietary supplement on the US market under the trade name G-PUR
®. The two non-purified tuffs are commercially available medical devices on the European market.
A detailed investigation of the deposit at Nižný Hrabovec as well as the raw material’s characteristics and its evolution were described by Tschegg et al. [
18,
25].
For characterization of the sorbent materials, the chemical and mineralogical compositions were determined using ICP-MS (NexION 350 D, Perkin Elmer, Waltham, MA, USA) and XRD (D-8 Advance, Bruker, Billerica, MA, USA). For observing the heavy metal content in the zeolite samples of 100 mg from the raw material of the PCT, the PCT and the two NPCTs were taken and primarily digested using an Anton Paar microwave 3000. As digestion reagent, 1% (w/w) hydrogen fluoride (HF) and 69% (w/w) sub-boiled HNO3 were mixed. After digestion, the samples were measured with the ICP-MS (mentioned above). Indium (Merck, Kenilworth, NJ, USA) was added as internal standard to the solutions. For calibration, the multi-element standard VI (Merck) was utilized.
For the mineralogical analyses of the powder samples, XRD (X-Ray Diffractometry) was performed. A Bruker D-8 Advance diffractometer (at Glock Health, Science and Research GmbH) equipped with a Cu source was used. Tube conditions were 40-kV voltage and 40-mA current. Measurements were performed on powdered specimens in top-loaded sample holders using a θ–2θ configuration, scanning angles from 6° to 70°, and 0.01° step size at 0.5 s/step. For the evaluation of the phases, the Bruker software DIFFRAC.EVA (release 2016) was used. The reference sample NIST SRM1976a (sintered alumina) and an intralaboratory clinoptilolite tuff reference sample were analyzed before each batch was run through quality control. XRD analysis is depicted in
Figure 1.
2.2. Determination of the Ion Exchange Capacity and Particle Size Distribution
For the purified clinoptilolite tuff and the non-purified tuffs, the ion exchange capacity (mol·kg−1) was determined using the 930 Compact IC Flex Chamber/SeS/PP/Deg (Metrohm, Herisau, Switzerland).
In total, 0.8 g of the tuff and 20 mL of a 0.15 mol·L−1 NH4Cl-solution were mixed by continuous movement (orbital shaker Heidolph Promax 1020) for two hours. In this way, metal cations bound to the tuff were exchanged with the ammonia from NH4Cl.
Afterwards, the solution was centrifuged for 15 min at 1923 g (Mega Star 1.6R, VWR) to obtain the fluid fraction. The supernatant was filtered through a 0.45 µm PVDF membrane filter, diluted (1:100) with HNO
3/dipicolinic acid 34 mM/14 mM (Sigma Aldrich, St. Louis, MO, USA), and analyzed. The ion exchange capacity was calculated over the difference in ammonia content between the blank solution (without sorbent) and the solution after being contacted with the tuff. The samples were measured four times. Results are presented in
Table 2.
The particle size of the PCT, its raw material, and the NPCTs were measured with a Mastersizer 2000 (Malvern Panalytical, Malvern, UK). The reference sample NIST SRM 1021 was run with the tuffs to ensure the accuracy and precision of the method. Measurements were carried out in triplicates, and results are given in
Table 2.
2.3. Single-Ion Adsorption Experiments
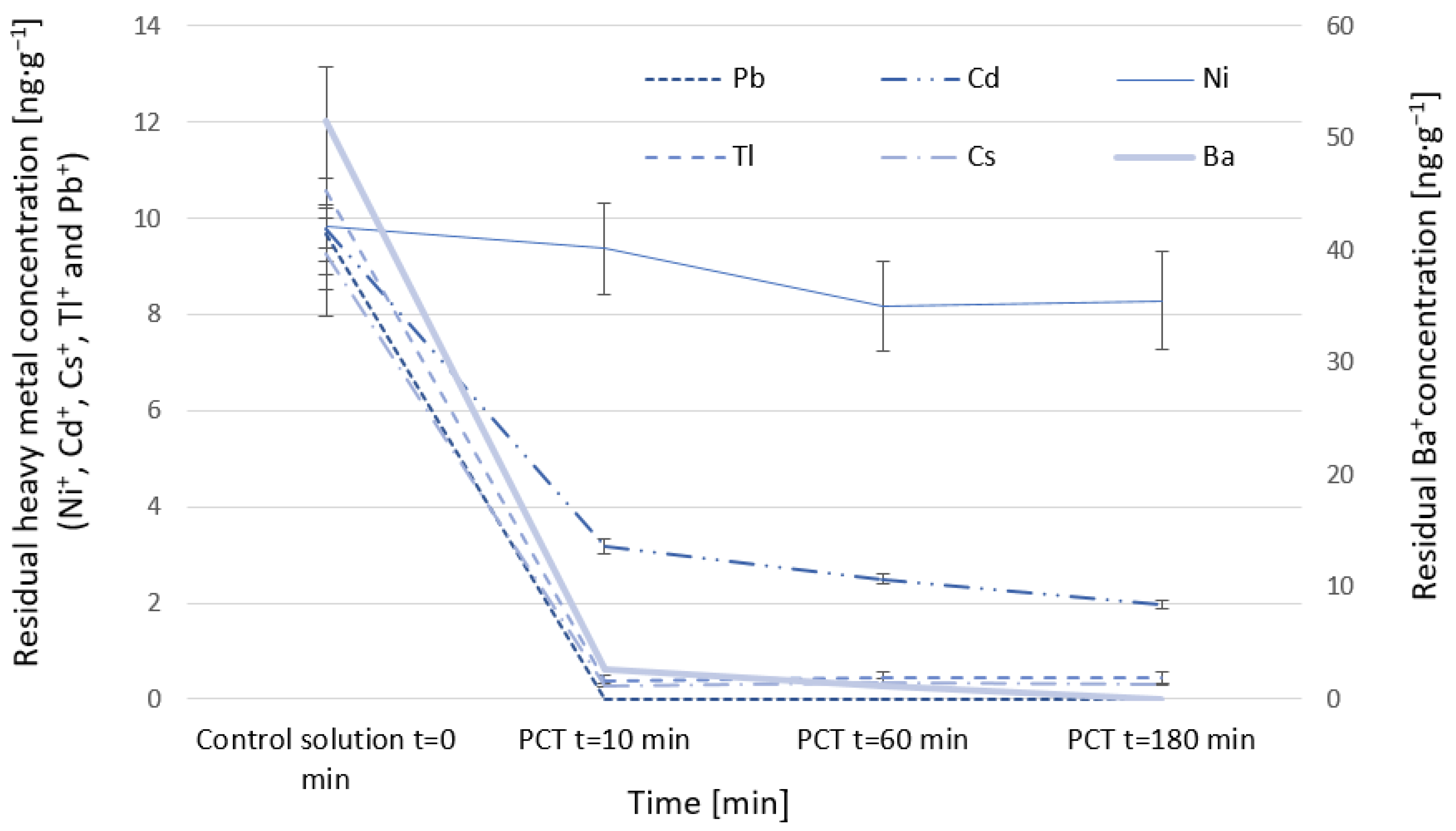
In the following section, the adsorption of heavy metal cations Ni+, Cd+, Cs+, Ba+, Tl+, and Pb+ by the PCT is investigated in single-ion solutions. The batch method was employed, using concentrations from 10 to 1000 ng·g−1 on the basis of (w/w) in non-carbonated mineral water.
Inorganic chemicals were supplied by Merck as analytical-grade reagents.
Stock solutions were prepared by diluting the standard solutions (Merck) of the heavy metal ions with non-carbonated mineral water to the desired concentrations and were then used for heavy metal cation adsorption and release experiments. The heavy-metal-spiked solutions are referred to as “contaminated” in the following sections.
Experiments were conducted batchwise with volumes of 50 mL: tuff samples with masses of 0.2 g (solid solid/liquid ratio of 1:4) were contacted with the heavy-metal-contaminated non-carbonated mineral water with 10 ng·g
−1 and 1000 ng·g
−1 starting concentrations. The solutions were placed in a heating chamber (Binder 9010-0082) at 37 °C for three hours. Experimental conditions of 37 °C as temperature and a maximum reaction time of 180 min were chosen with reference to the body temperature of humans and digestion time in human beings [
26,
27].
After 10 min, 60 min, and 180 min reaction time, 5 mL of the samples were withdrawn, filtered using a 0.45 µm PVDF filter, and diluted with 1 wt% HNO3. The control (no clinoptilolite) and the test solutions (after being contacted with clinoptilolite) were taken for metal-ion concentration analysis using an inductively coupled plasma mass spectrometer (NexION 350 D, Perkin Elmer) for parts-per-billion (ppb) sample concentrations of Ni+, Cd+, Cs+, Ba+, Tl+, and Pb+, with a limit of quantification of 0.10 ng·g−1 for barium, 0.010 ng·g−1 for lead, 0.011 ng·g−1 for cadmium, 0.031 ng·g−1 for cesium, 0.070 ng·g−1 for nickel, and 0.0021 ng·g−1 for thallium. Every experiment was performed with appropriate controls (no sorbent) and blank solutions (non-carbonated mineral water). Accuracy and precision of the analysis were ensured using equidistant calibration (Multi-element Standard Solution VI, Merck). Indium (Merck) was used as an internal standard and added before the measurements. This was to ensure that the difference in heavy metal concentrations before and after the experiment was due to adsorption processes and not due to, for example, precipitation of heavy metals at high pH. All control solutions underwent the same experimental conditions as the test samples.
Quality assurance checks were run at intervals.
All batch experiments were performed in triplicate, and the averaged values and standard deviations were reported.
A Metrohm pH meter (WTW pH 3210) was used for the pH measurements.
Highest-purity reagents were used throughout this study.
From the results obtained, it was decided to further compare the PCT with NPCTs in terms of adsorption behavior towards heavy metal cations from multi-element solutions.
2.3.1. Batch Adsorption and Release Experiments in Contaminated Non-Carbonated Mineral Water
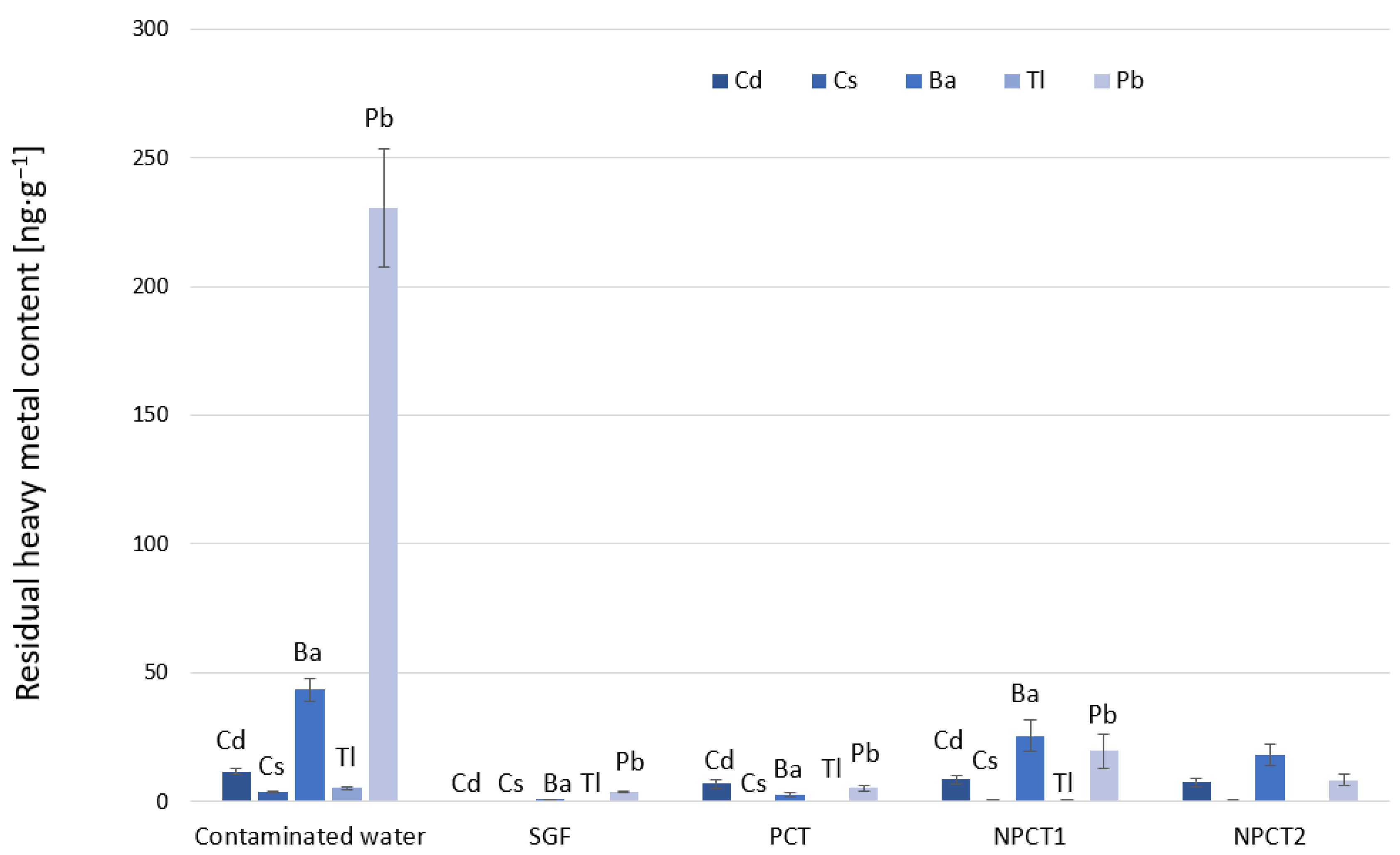
Release experiments were conducted in order to evaluate how strongly heavy metal ions were immobilized by the three different clinoptilolite tuffs. Adsorption behavior was investigated in synthetically prepared multi-ion solutions.
Together with 0.2 g sorbent in a 1:4 solid/liquid ratio, the synthetically treated non-carbonated mineral water spiked with a mix of the heavy metal cations as described above was incubated at 37 °C for three hours. Samples were taken after 10 min, 60 min, and 180 min. The suspension was then filtered through a 0.45 µm PVDF filter in order to remove the sorbent from the solution. The samples of the liquid supernatant were kept in 3 wt% HNO3, and the concentrations of the heavy metals in the supernatant were determined by an inductively coupled plasma mass spectrometer. Quality assurance checks with concentrations of 1 ng·g−1 and 10 ng·g−1 were used.
Measurements were carried out in triplicates.
2.3.2. Batch Adsorption and Release Experiments in Non-Contaminated Non-Carbonated Mineral Water
The experiment was conducted as described above (see
Section 2.3.1). However, no heavy-metal-spiked water was used; instead, adsorption/release of heavy metals in naturally occurring amounts was investigated in the non-carbonated mineral water.
The concentrations of heavy metal ions in the sample were analyzed. The limit of quantification was 11 ng·g−1 for nickel, 0.05 ng·g−1 for cadmium, 0.012 ng·g−1 for cesium, 0.21 ng·g−1 for barium, 0.08 ng·g−1 for thallium, and 0.44 ng·g−1 for lead.
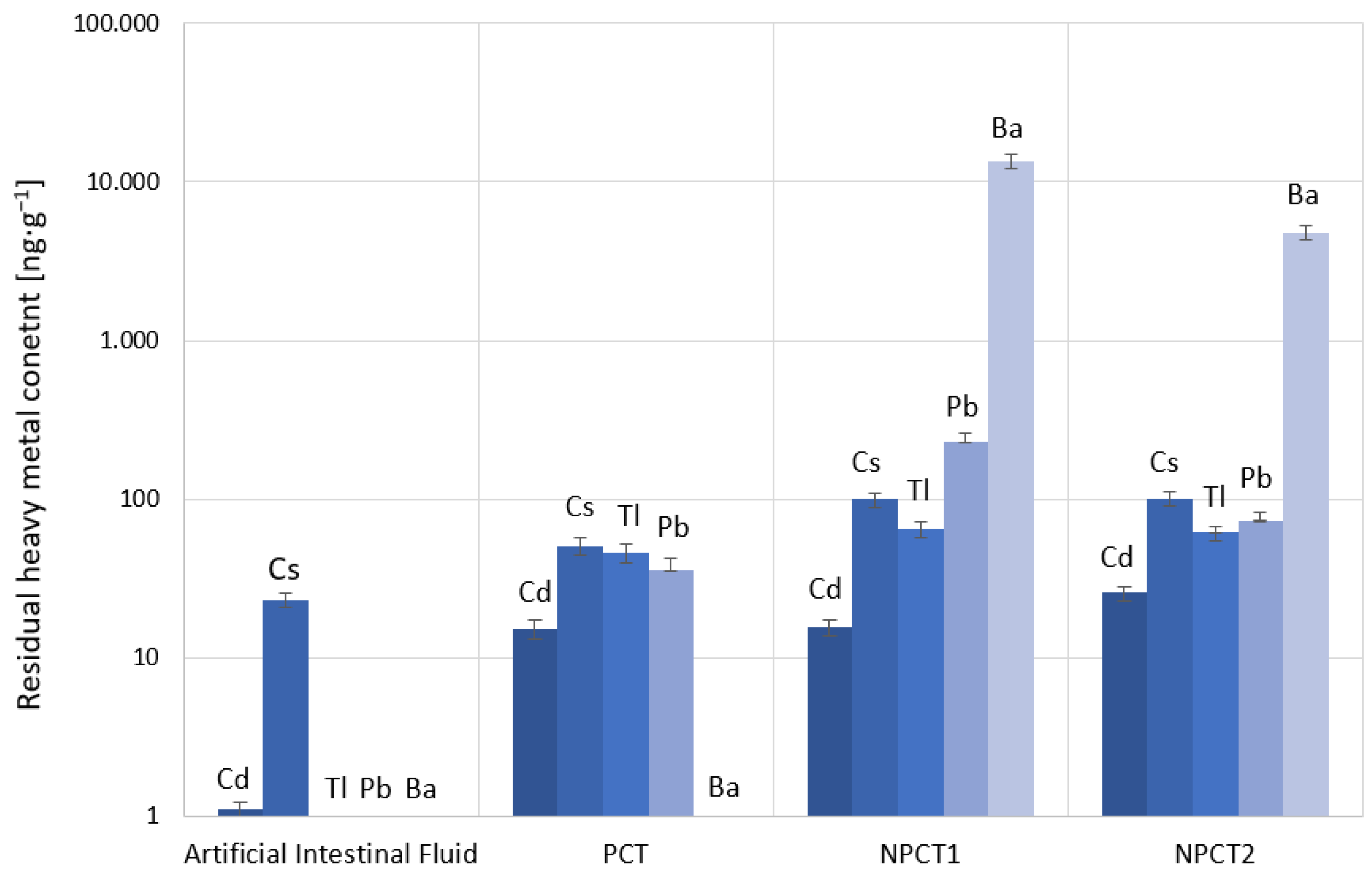
2.3.3. Heavy Metal Adsorption and Release in Artificial Gastric and Intestinal Fluids
Adsorption and release experiments were performed with multi-component solutions investigating the heavy metals stated above in laboratory-synthesized gastric and intestinal fluids. The simulated gastric fluid (SGF) and simulated intestinal fluid (SIF) were prepared following the instructions of the European Pharmacopeia [
28].
The simulated gastric fluid contained 0.034 mol·L−1 NaCl and 3.2 g·L−1 pepsin. The pH was adjusted to 1.5 by using 0.1 M HCl.
The SIF contained 0.05 M KH2PO4, 19.3 mL 0.2 M NaOH, 40 mL 0.01 M HCl, and 20 g·L−1 pancreatin in a total volume of 250 mL.
The solution’s pH was adjusted to 6.8 using 0.2 M NaOH.
The tuffs were transferred to tubes filled with multi-component heavy metal solution of predetermined concentrations to create a solid/liquid ratio of 1:4 (4 g·L−1). Synthetic gastric fluid was added.
These samples were put in a heating chamber (Binder, 9010-0082) at 37 °C. After 20 min, the reaction mixture was centrifuged at 1923 g (Mega Star 1.6R, VWR, Radnor, PA, USA) and the supernatant decanted. A time of 20 min was chosen because of the time it takes for water to pass through the stomach [
26].
The remaining tuff was mixed with 20 mL SIF and allowed to equilibrate for another three hours at 37 °C. Afterwards, the samples were centrifuged at 1923 g to separate the solid from the fluid, and the supernatant intestinal juice was withdrawn from the remaining adsorbent.
The SIF was digested in nitric acid. Sub-boiled and then concentrated HNO3, concentrated H2O2, and deionized water were added to the SIF, put in a Teflon vessel, capped, and placed in a laboratory microwave (Anton Paar, Multiwave 3000, Tokyo, Japan). After digestion, the volume was brought to 50 mL with purified water.
The SGF and digested SIF samples were kept in 3 wt% HNO3 and analyzed using inductively coupled plasma mass spectrometry (described above). Quality assurance checks with concentrations of 0.5 ng·g−1 and 10 ng·g−1 were run with the samples. Aliquots of synthetic-heavy-metal-spiked water, pure SGF, and pure SIF (without tuffs) were run as blank and control solutions, respectively, and received the same treatment as the extant samples.
2.4. Adsorption Capacity and Removal Efficiency of PCT for Heavy Metal Ions
For the PCT, the heavy metal concentrations in the sample solution before and after the uptake experiments were measured using ICP-MS. The amount of metal adsorbed by the tuff was determined by calculating the difference between the initial metal concentration and the metal concentration after 180 min reaction time.
The adsorption capacity and removal efficiency of PCT for heavy metal ions could be expressed according to following Equations (1) and (2) [
29]:
where AC [ng·g
−1] corresponds to the adsorption capacity of PCT for heavy metal ions, RE (%) describes the removal efficiency for heavy metal ions, C
i [ng∙L
−1] corresponds to the initial heavy metal concentration in the test solution beforehand, and C
f [ng∙L
−1] to the heavy metal concentration after the adsorption experiments. V
f [L] stands for the solution volume of the heavy metal ions and W
c [g] for the dosage of the dry clinoptilolite tuff (0.2 g).
Removal efficiencies were calculated for different times, whereas adsorption capacities were determined only for the last time samples (180 min).







{kind=link}
{kind=link}
{kind=link}
{kind=link}