Stability and Phase Transitions of Nontoxic γ-Cyclodextrin-K+ Metal-Organic Framework in Various Solvents

Abstract
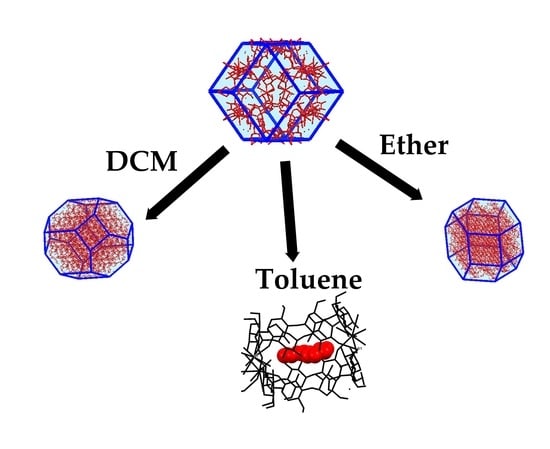
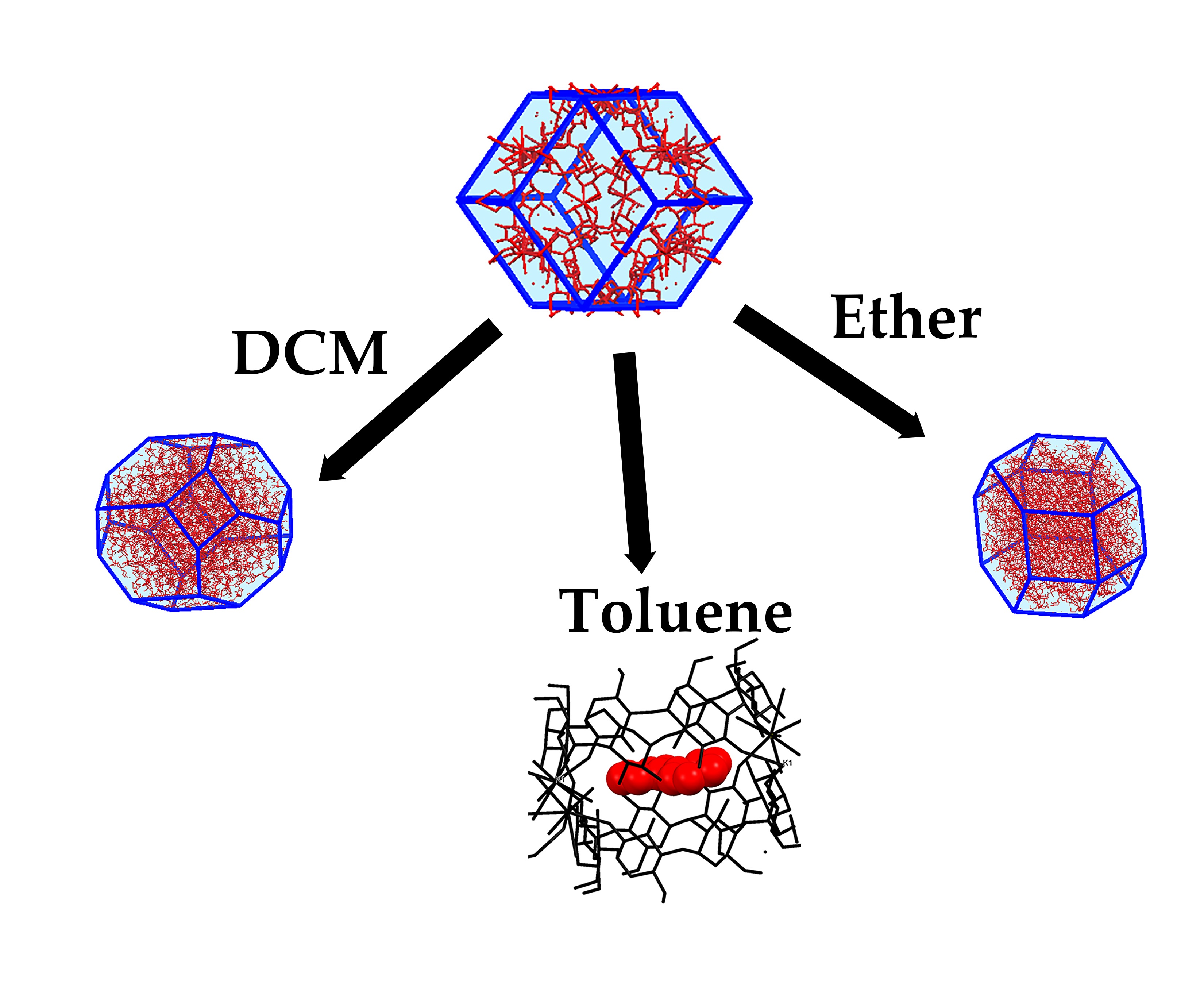
:
1. Introduction
2. Materials and Methods
2.1. Materials and Measurements
2.2. Synthesis of α-Phase
2.3. Synthesis of New Phases
2.4. Evaluation of Stability of α-Phase in Various Solvents
2.5. Crystal Structure Determination
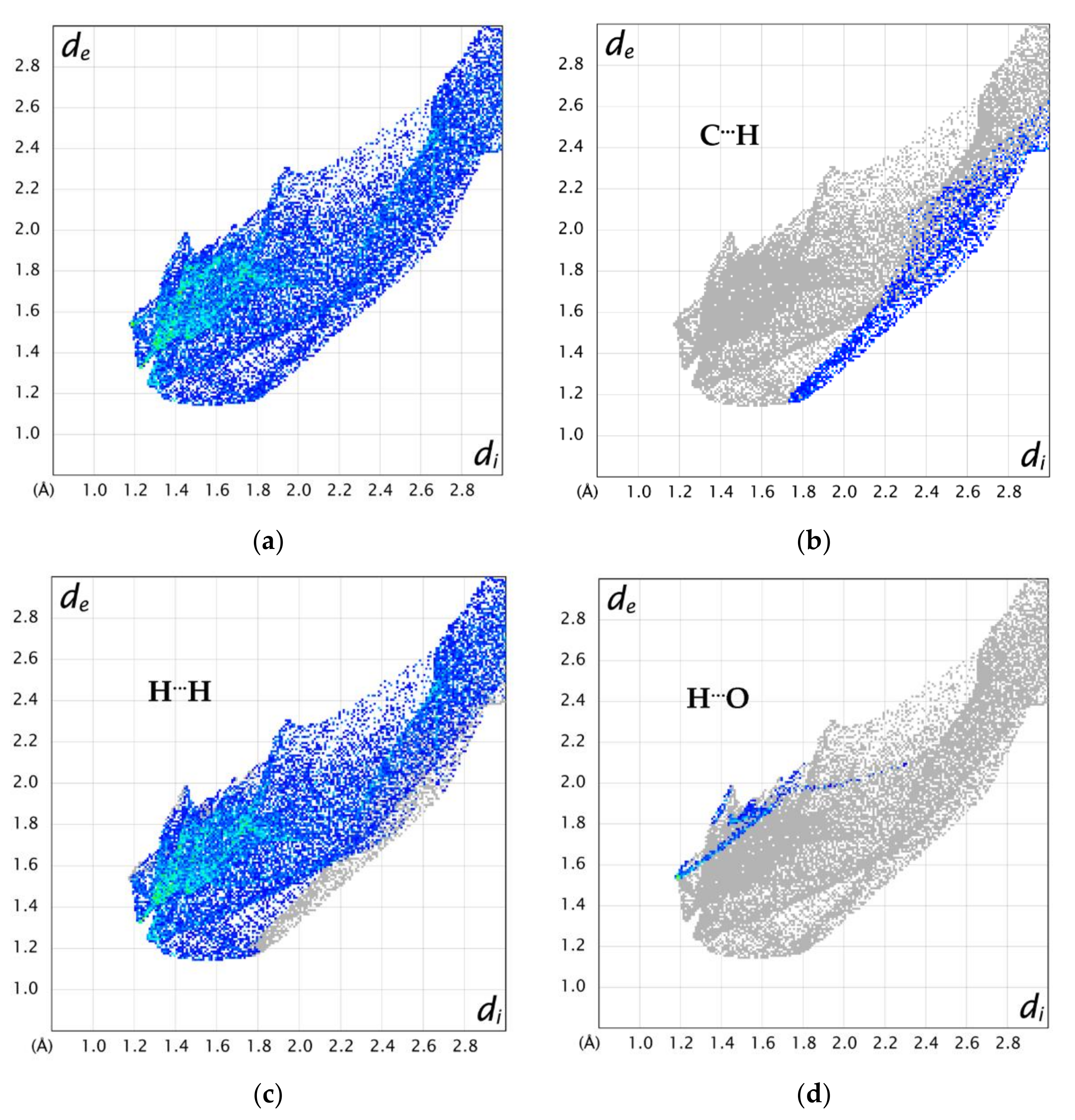
2.6. Hirshfeld Surface Analysis
2.7. Thermal Stability Measurements
3. Results
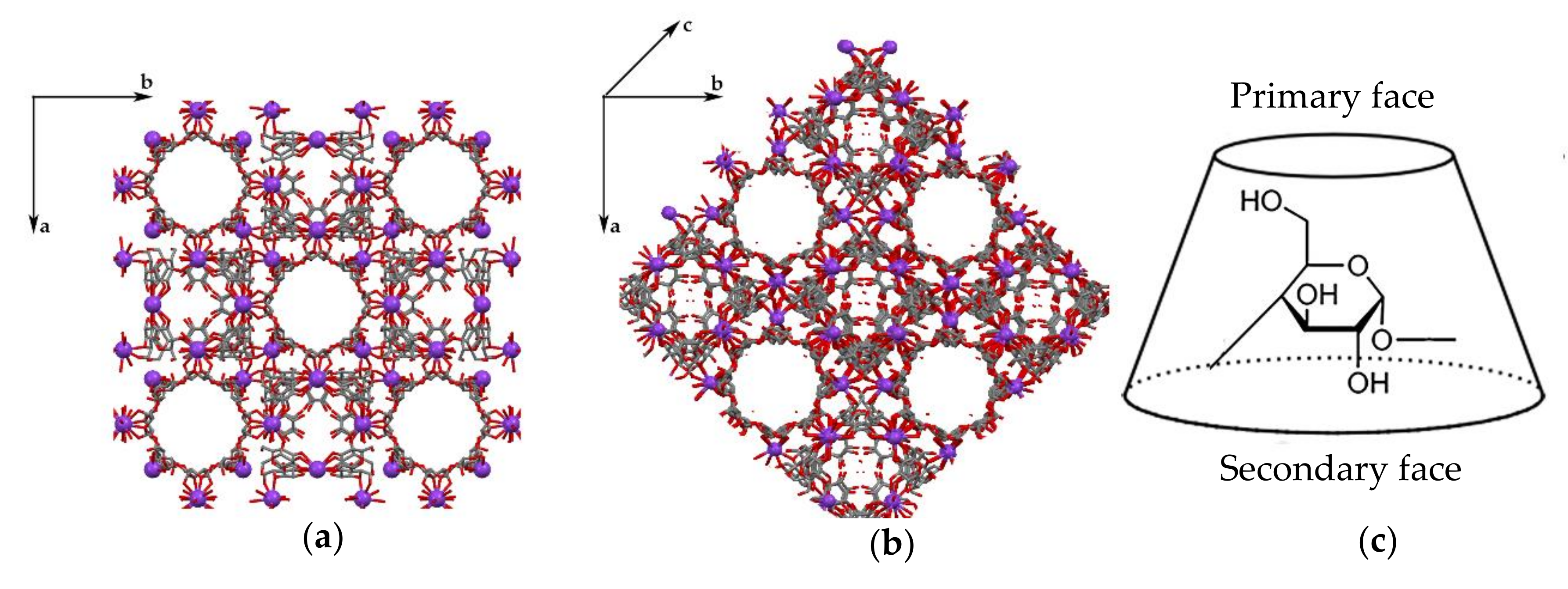
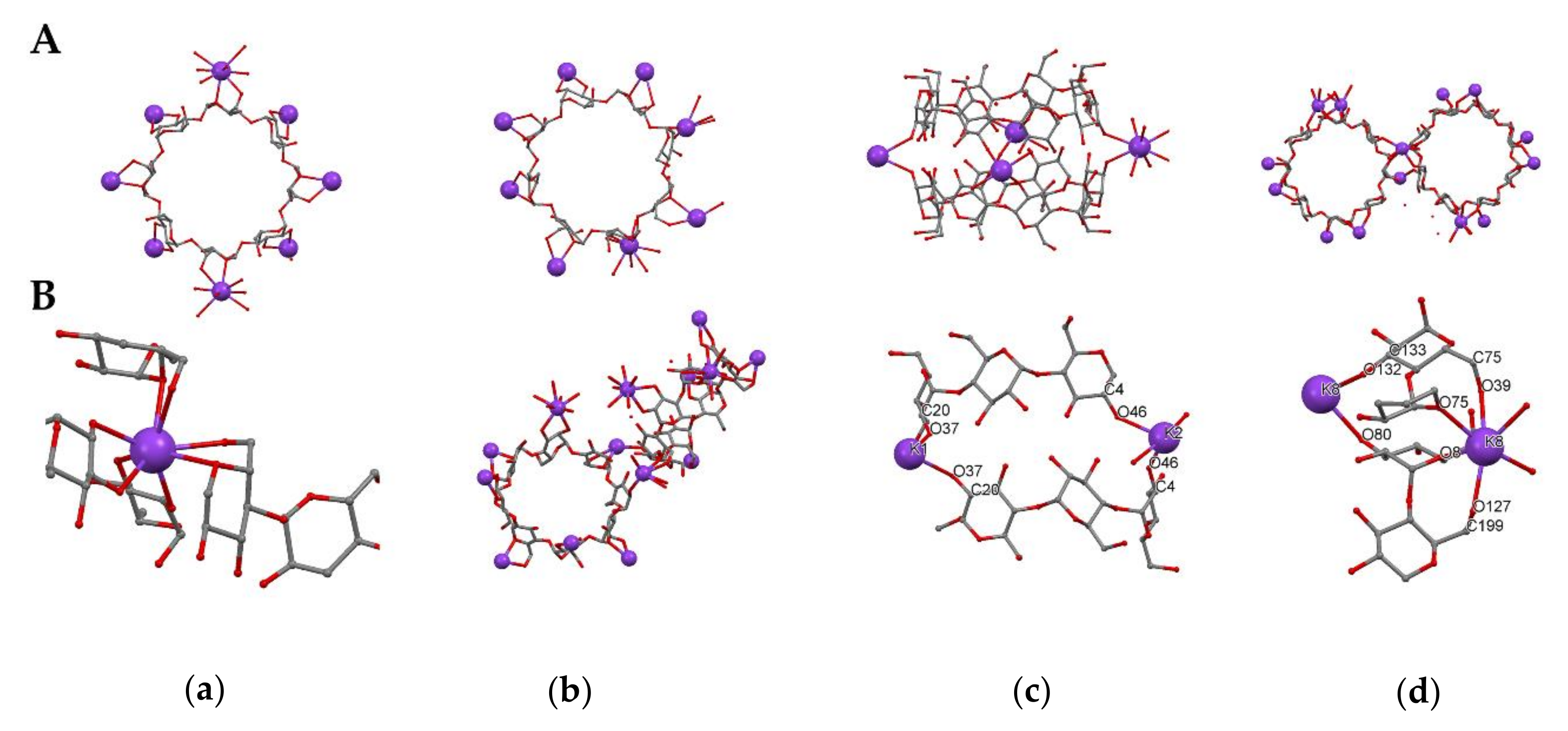
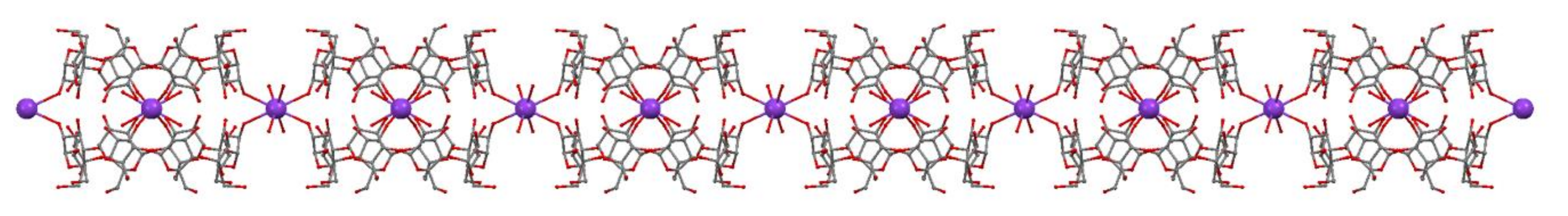
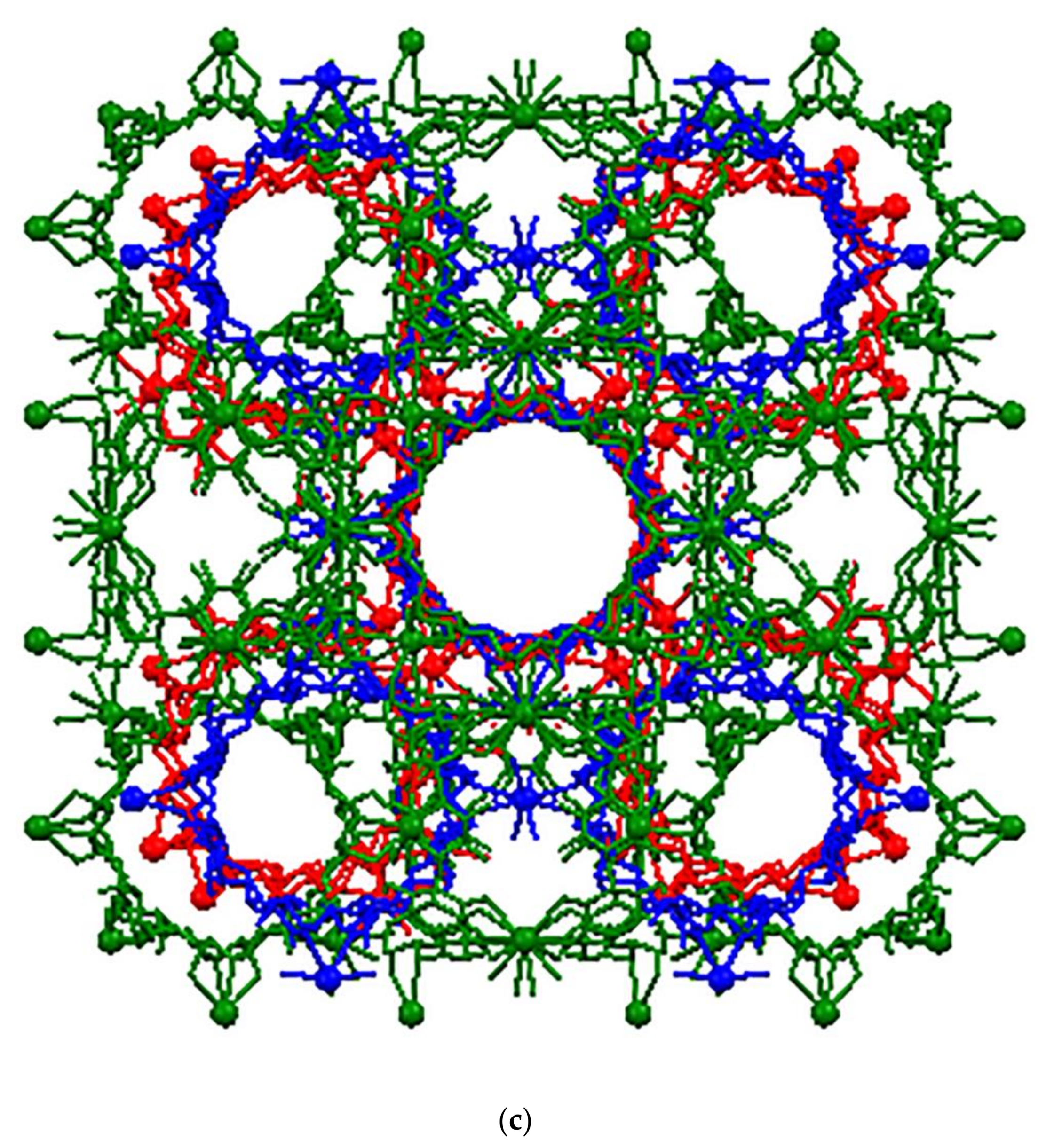
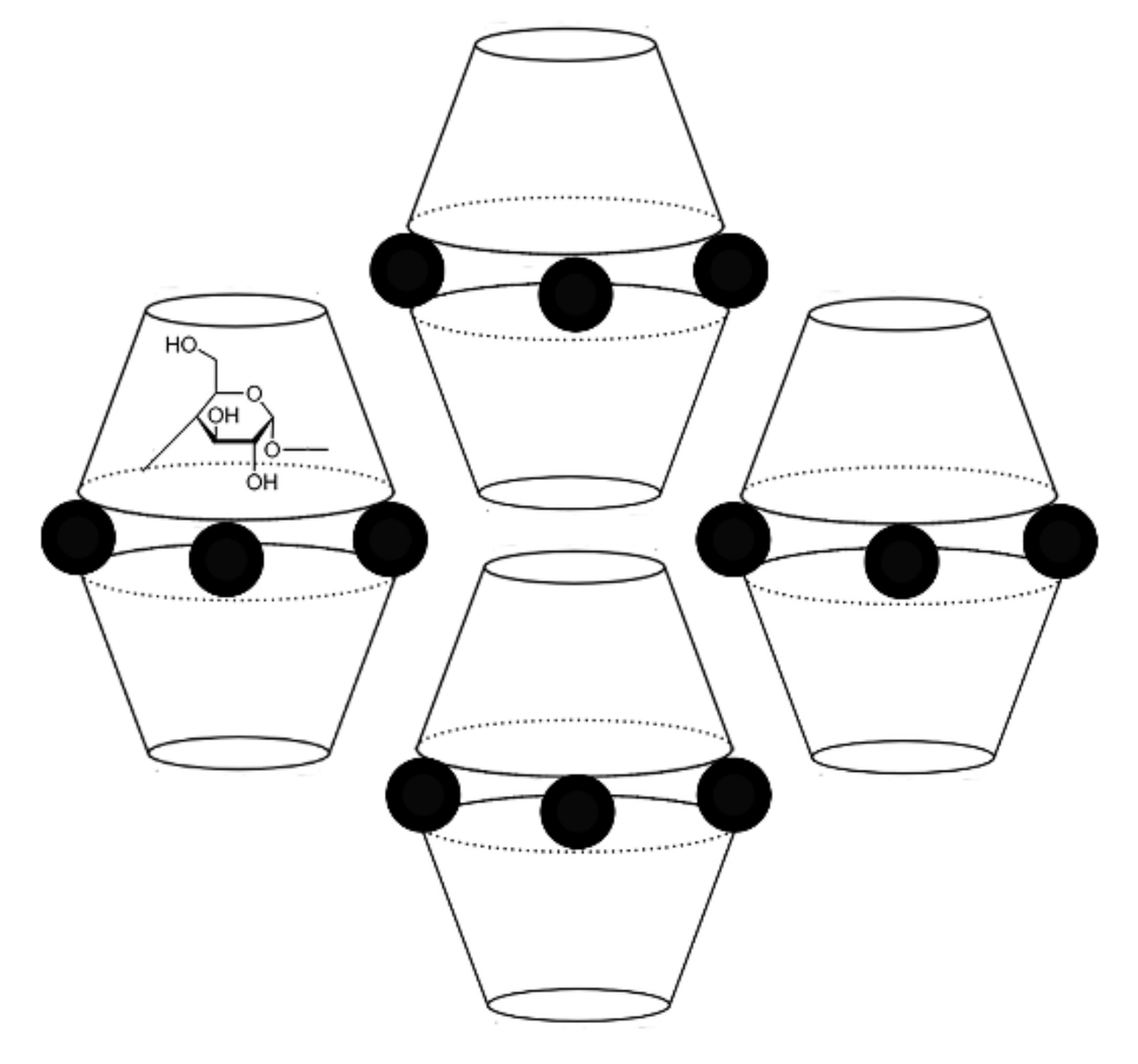
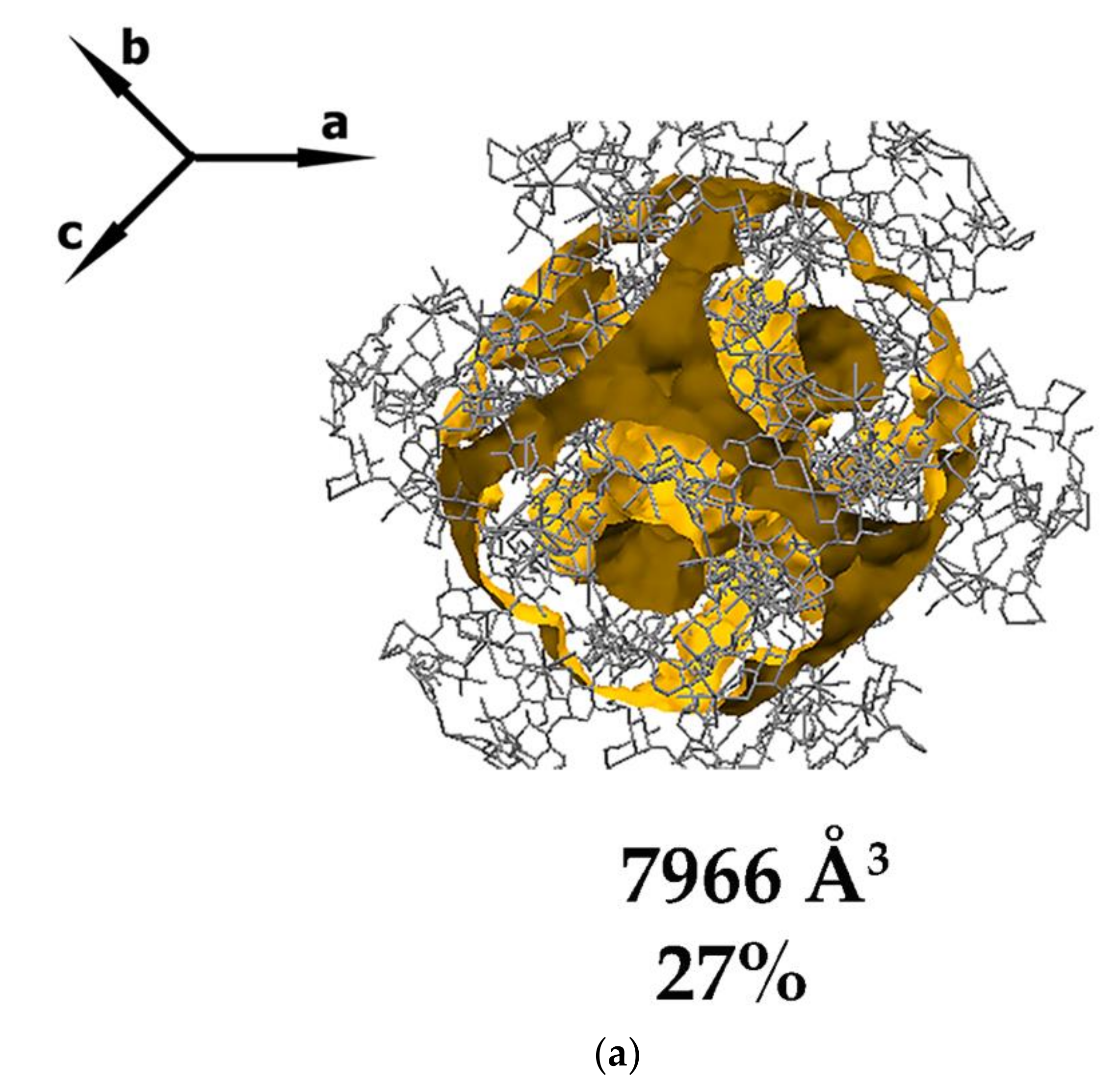
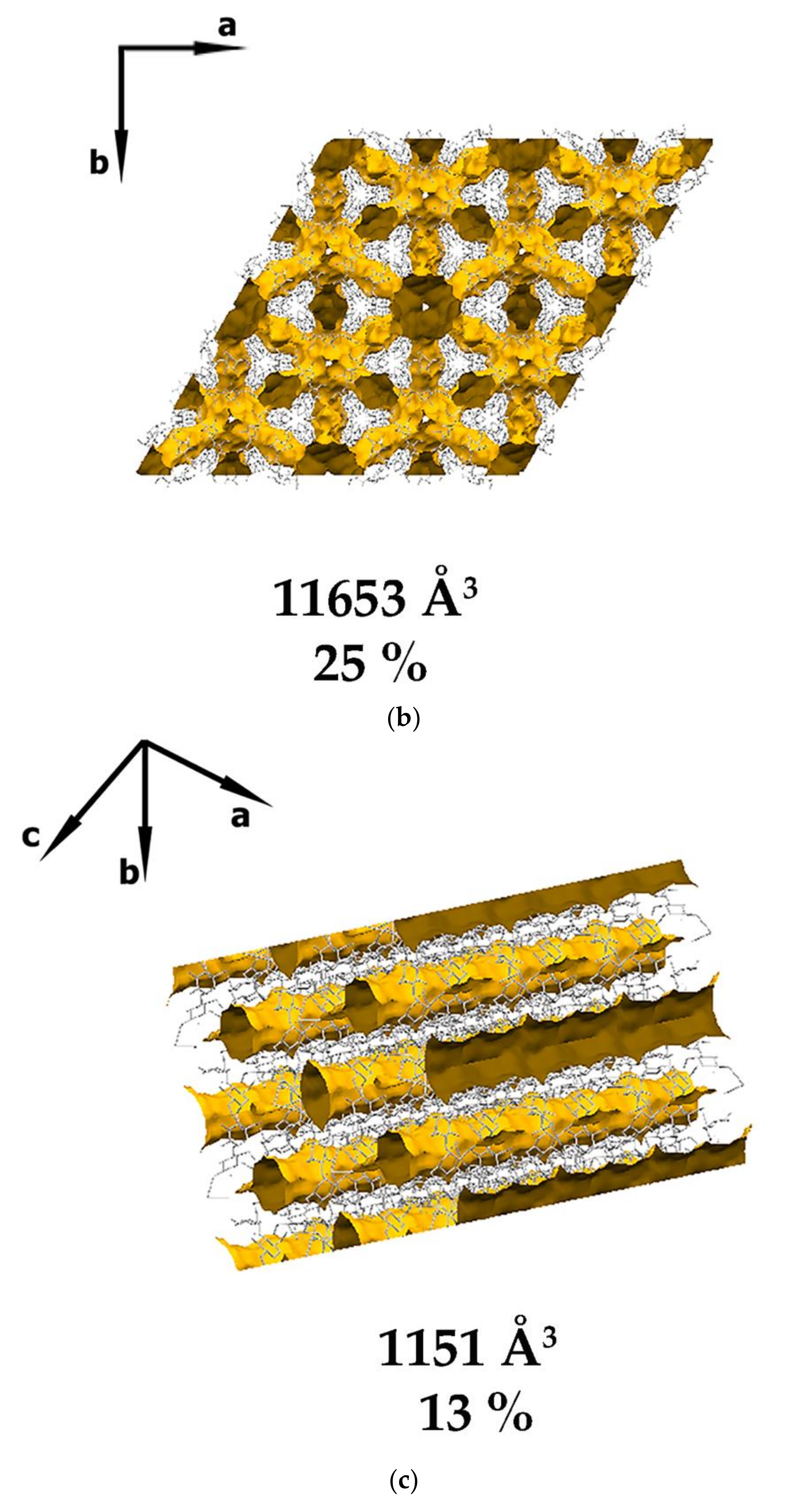
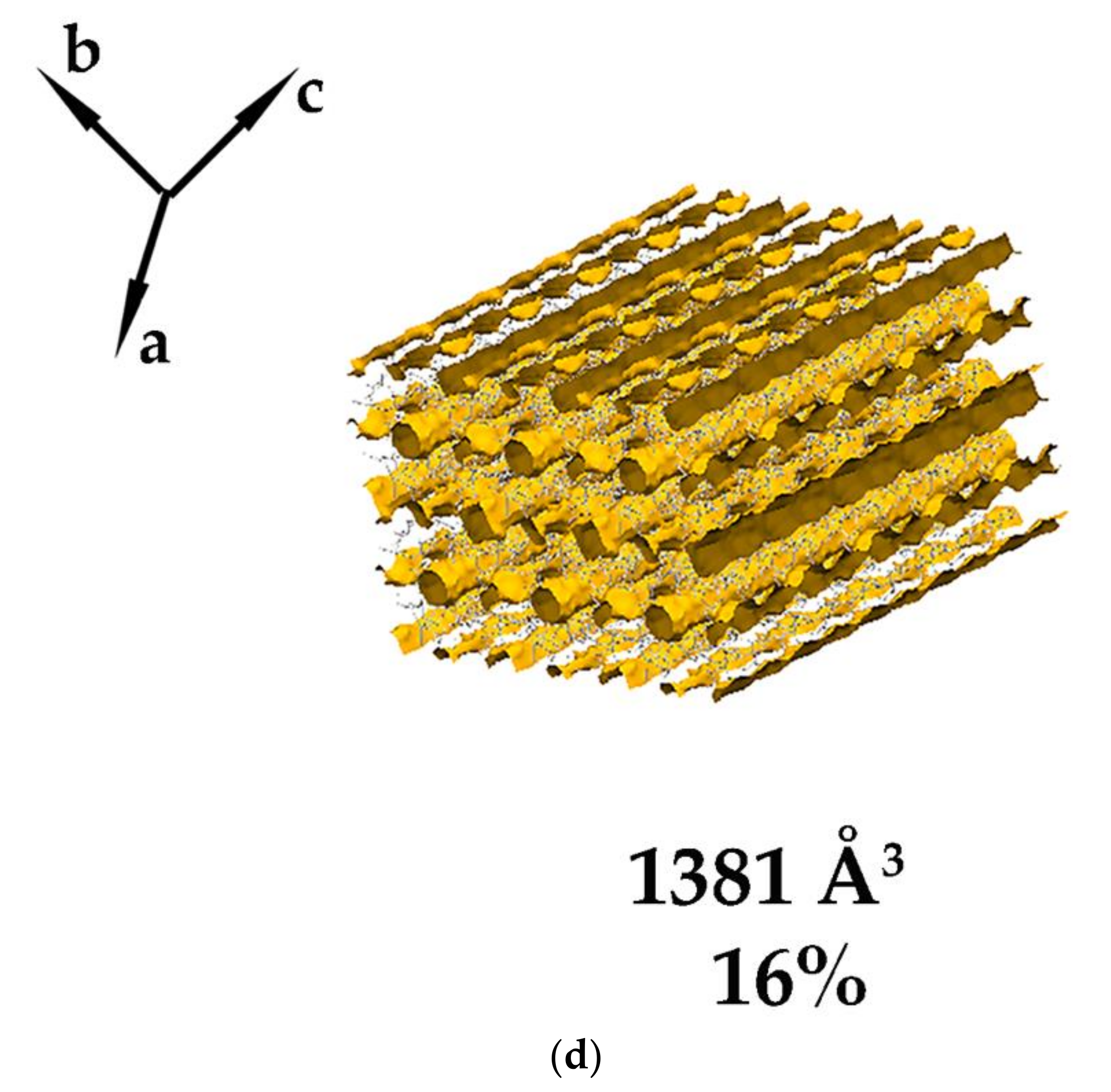
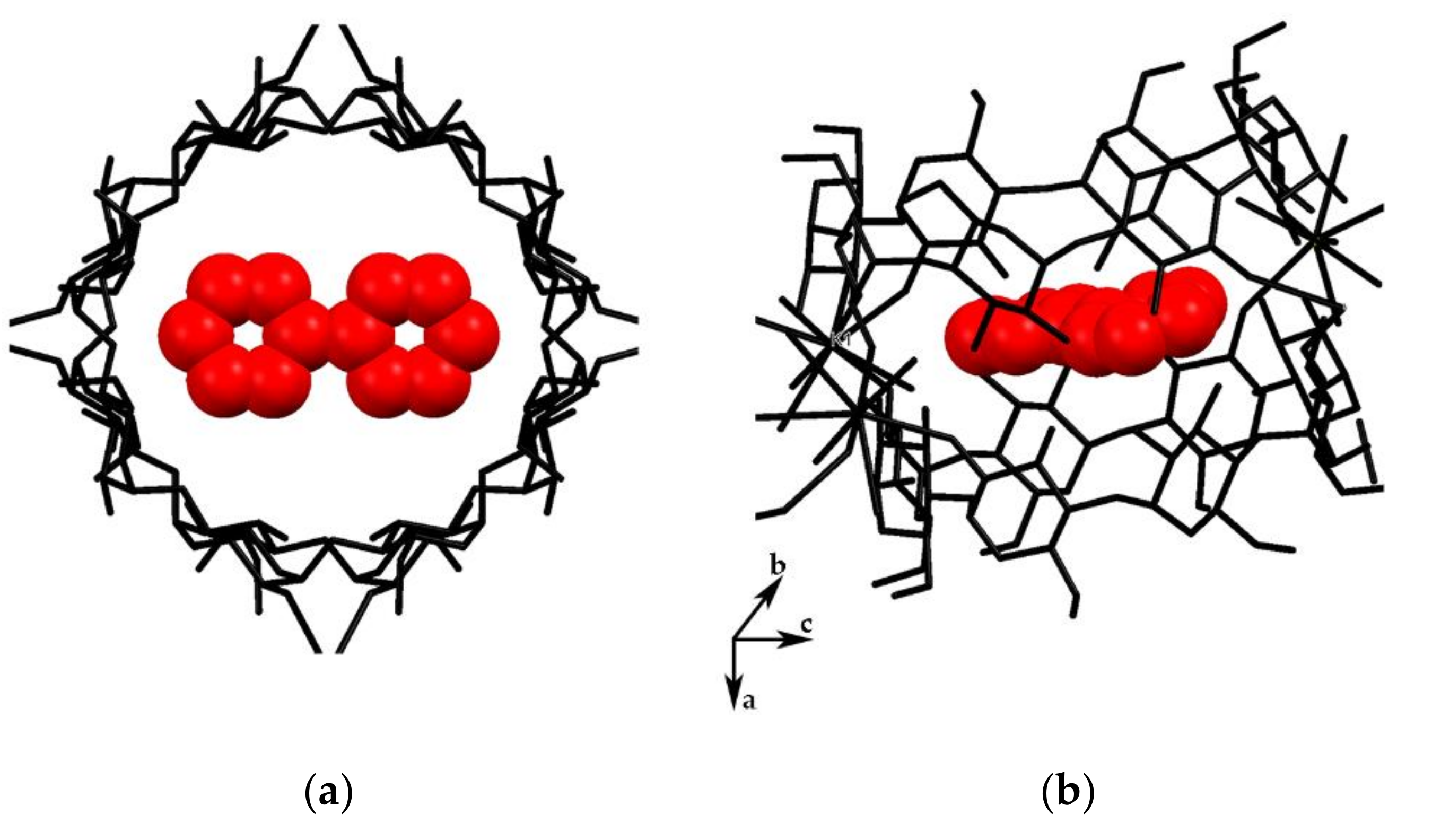
3.1. Crystal Structure Analysis of the New Phases of γ-CD-MOF-1
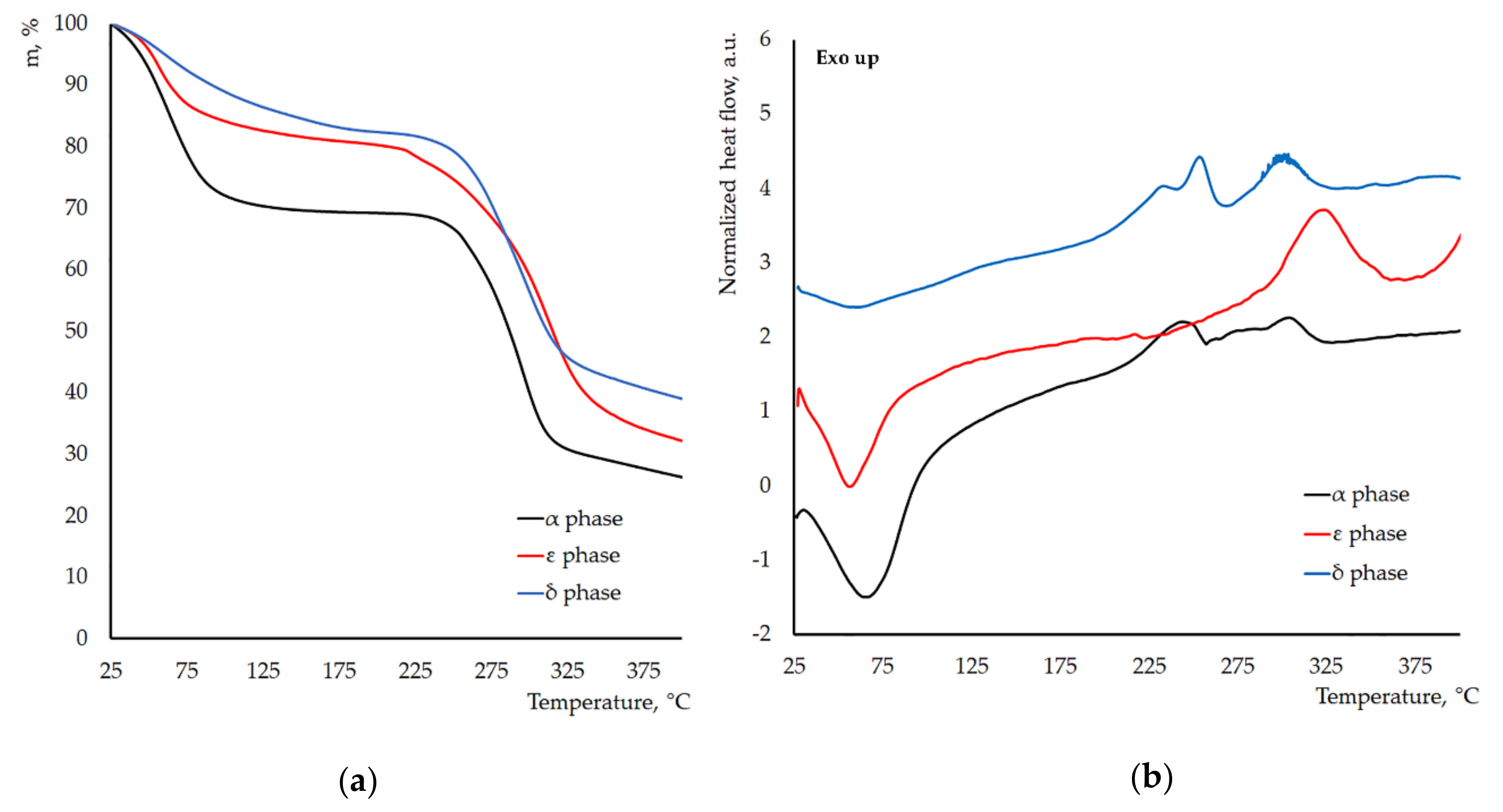
3.2. Thermal Stability of the New Crystal Phases of γ-CD-MOF-1
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cook, T.R.; Zheng, Y.R.; Stang, P.J. Metal-organic frameworks and self-assembled supramolecular coordination complexes: Comparing and contrasting the design, synthesis, and functionality of metal-organic materials. Chem. Rev. 2013, 113, 734–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKinlay, A.C.; Morris, R.E.; Horcajada, P.; Férey, G.; Gref, R.; Couvreur, P.; Serre, C. BioMOFs: Metal-organic frameworks for biological and medical applications. Angew. Chem. Int. Ed. 2010, 49, 6260–6266. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Chen, Q.W.; Zhuang, C.; Tang, P.P.; Lin, N.; Wei, L.Q. Controlled release of drug molecules in metal–organic framework material HKUST-1. Inorg. Chem. Commun. 2017, 79, 78–81. [Google Scholar] [CrossRef]
- Li, H.; Lv, N.; Li, X.; Liu, B.; Feng, J.; Ren, X.; Guo, T.; Chen, D.; Fraser Stoddart, J.; Gref, R.; et al. Composite CD-MOF nanocrystals-containing microspheres for sustained drug delivery. Nanoscale 2017, 9, 7454–7463. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Xi, H.; Chen, S.; Ediger, M.D.; Yu, L. Crystallization near glass transition: Transition from diffusion-controlled to diffusionless crystal growth studied with seven polymorphs. J. Phys. Chem. B 2008, 112, 5594–5601. [Google Scholar] [CrossRef]
- Han, Y.; Liu, W.; Huang, J.; Qiu, S.; Zhong, H.; Liu, D.; Liu, J. Cyclodextrin-Based Metal-Organic Frameworks (CD-MOFs) in Pharmaceutics and Biomedicine. Pharmaceutics 2018, 10, 271. [Google Scholar] [CrossRef] [Green Version]
- Huxford, R.C.; Della Rocca, J.; Lin, W. Metal-organic frameworks as potential drug carriers. Curr. Opin. Chem. Biol. 2010, 14, 262–268. [Google Scholar] [CrossRef] [Green Version]
- Almáši, M.; Zeleňák, V.; Palotai, P.; Beňová, E.; Zeleňáková, A. Metal-organic framework MIL-101(Fe)-NH2 functionalized with different long-chain polyamines as drug delivery system. Inorg. Chem. Commun. 2018, 93, 115–120. [Google Scholar] [CrossRef]
- Rojas, S.; Colinet, I.; Cunha, D.; Hidalgo, T.; Salles, F.; Serre, C.; Guillou, N.; Horcajada, P. Toward Understanding Drug Incorporation and Delivery from Biocompatible Metal-Organic Frameworks in View of Cutaneous Administration. ACS Omega 2018, 3, 2994–3003. [Google Scholar] [CrossRef]
- Rojas, S.; Devic, T.; Horcajada, P. Metal organic frameworks based on bioactive components. J. Mater. Chem. B 2017, 5, 2560–2573. [Google Scholar] [CrossRef]
- Smaldone, R.A.; Forgan, R.S.; Furukawa, H.; Gassensmith, J.J.; Slawin, A.M.Z.; Yaghi, O.M.; Stoddart, J.F. Metalorganic frameworks from edible natural products. Angew. Chem. Int. Ed. 2010, 49, 8630–8634. [Google Scholar] [CrossRef] [PubMed]
- Patyk-Kaźmierczak, E.; Warren, M.R.; Allan, D.R.; Katrusiak, A. Pressure inverse solubility and polymorphism of an edible γ-cyclodextrin-based metal-organic framework. Phys. Chem. Chem. Phys. 2017, 19, 9086–9091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chem, A.; Ed, I.; Biradha, K.; Hongo, Y.; Fujita, M. for Crystal-to-Crystal Sliding of Two-Dimensional Coordination Layers Triggered by Guest Exchange. Angew. Chem. Int. Ed. 2002, 41, 3395–3398. [Google Scholar]
- Suh, M.P.; Ko, J.W.; Choi, H.J. A Metal−Organic Bilayer Open Framework with a Dynamic Component: Single-Crystal-to-Single-Crystal Transformations. J. Am. Chem. Soc. 2002, 124, 10976–10977. [Google Scholar] [CrossRef]
- Suh, M.P.; Moon, H.R.; Lee, E.Y.; Jang, S.Y. A Redox-Active Two-Dimensional Coordination Polymer: Preparation of Silver and Gold Nanoparticles and Crystal Dynamics on Guest Removal. J. Am. Chem. Soc. 2006, 128, 4710–4718. [Google Scholar] [CrossRef]
- Park, H.J.; Lim, D.; Yang, W.S.; Oh, T.; Suh, M.P. A Highly Porous Metal–Organic Framework: Structural Transformations of a Guest-Free MOF Depending on Activation Method and Temperature. Chem. A Eur. J. 2011, 17, 7251–7260. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, T.K.; Suh, M.P.; Moon, H.R. Solvent-induced single-crystal to single-crystal transformation of a Zn 4 O-containing doubly interpenetrated metal–organic framework with a pcu net. Cryst. Eng. Commun. 2015, 17, 8807–8811. [Google Scholar] [CrossRef]
- Park, H.J.; Cheon, Y.E.; Suh, M.P. Post-Synthetic Reversible Incorporation of Organic Linkers into Porous Metal-Organic Frameworks through Single-Crystal-to-Single-Crystal Transformations and Modification of Gas-Sorption Properties. Chem. A Eur. J. 2010, 16, 11662–11669. [Google Scholar] [CrossRef]
- Yuan, S.; Feng, L.; Wang, K.; Pang, J.; Bosch, M.; Lollar, C.; Sun, Y.; Qin, J.; Yang, X.; Zhang, P.; et al. Stable Metal–Organic Frameworks: Design, Synthesis, and Applications. Adv. Mater. 2018, 30, 1–35. [Google Scholar] [CrossRef] [Green Version]
- Inokuma, Y.; Yoshioka, S.; Ariyoshi, J.; Arai, T.; Fujita, M. Preparation and guest-uptake protocol for a porous complex useful for “crystal-free” crystallography. Nat. Protoc. 2014, 9, 246–252. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourhis, L.J.; Dolomanov, O. V; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. The anatomy of a comprehensive constrained, restrained refinement program for the modern computing environment – Olex2 dissected. Acta Cryst. Sect. A Found. Adv. 2015, 71, 59–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spek, A.L. Structure validation in chemical crystallography. Acta Cryst. Sect. D Biol. Cryst. 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer17; University of Western Australia: Pert, Australia, 2017. [Google Scholar]
- Bučar, D.-K.; Lancaster, R.W.; Bernstein, J. Disappearing Polymorphs Revisited. Angew. Chem. Int. Ed. 2015, 54, 6972–6993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagabas, A.A.; Frasconi, M.; Iehl, J.; Hauser, B.; Farha, O.K.; Hupp, J.T.; Hartlieb, K.J.; Botros, Y.Y.; Stoddart, J.F. γ-Cyclodextrin cuprate sandwich-type complexes. Inorg. Chem. 2013, 52, 2854–2861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, D.W.M. Fast estimation of crystal densities. Acta Cryst. Sect. B Struct. Sci. 2002, 58, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Chen, X. Host-Guest Chemistry in Supramolecular Theranostics. Theranostics 2019, 9, 3041–3074. [Google Scholar] [CrossRef]
- Kathuria, A.; Pauwels, A.K.; Buntinx, M.; Shin, J.; Harding, T. Inclusion of ethanol in a nano-porous, bio-based metal organic framework. J. Incl. Phenom. Macrocycl. Chem. 2019, 95, 91–98. [Google Scholar] [CrossRef]
- Kritskiy, I.; Volkova, T.; Surov, A.; Terekhova, I. γ-Cyclodextrin-metal organic frameworks as efficient microcontainers for encapsulation of leflunomide and acceleration of its transformation into teriflunomide. Carbohydr. Polym. 2019, 216, 224–230. [Google Scholar] [CrossRef]
- Liu, B.; He, Y.; Han, L.; Singh, V.; Xu, X.; Guo, T.; Meng, F.; Xu, X.; York, P.; Liu, Z.; et al. Microwave-Assisted Rapid Synthesis of γ-Cyclodextrin Metal-Organic Frameworks for Size Control and Efficient Drug Loading. Cryst. Growth Des. 2017, 17, 1654–1660. [Google Scholar] [CrossRef]
- Al-Ghamdi, S.; Kathuria, A.; Abiad, M.; Auras, R. Synthesis of nanoporous carbohydrate metal-organic framework and encapsulation of acetaldehyde. J. Cryst. Growth 2016, 451, 72–78. [Google Scholar] [CrossRef] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Known Phases | New Phases | |||
|---|---|---|---|---|---|
| α [11] | β [12] | ε | ε·tol | δ | |
| a/Å | 31.006(8) | 43.6764(14) | 15.7898(1) | 15.8765(1) | 23.80929(17) |
| b/Å | 31.006(8) | 43.6764(14) | 20.2381(1) | 20.1735(1) | 23.80929(17) |
| c/Å | 31.006(8) | 28.1864(9) | 28.1282(2) | 28.1409(2) | 15.26298(14) |
| α = β = γ = 90° | α = β ≠ γ = 120° | α = β = γ = 90° | α = β = γ = 90° | α = β = γ = 90° | |
| V/Å3 | 29807(14) | 46565(3) | 8988.52(10) | 9013.1(1) | 8652.31(15) |
| Space group | I432 | R32 | I222 | I222 | I4 |
| Dcalc/g·cm−3 | 0.962 | 0.903 | 1.122 | 1.245 | 1.112 |
| Packing Index/% | 45 | 43 | 54 | 63 | 53 |
| Time/Days | 3 | 6 | 9 | 14 |
|---|---|---|---|---|
| Solvent | ||||
| MTBE-1,2 | α | α | α | α |
| DCM-1 | ε | ε | ε | ε |
| DCM-2 1 | α | ε | ε | ε |
| Toluene-1,2 | α | x 2 | ε·tol | ε·tol |
| Diethyl ether-1,2 | α | β | β | δ |
| THF-1 | α | α | β | α |
| THF-2 1 | α | β | β | α |
| Chloroform-1 | α | α | β | α |
| Chloroform-2 1 | α | β | β | β |
| Cyclohexane-1,2 | α | β | β | β |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krūkle-Bērziņa, K.; Belyakov, S.; Mishnev, A.; Shubin, K. Stability and Phase Transitions of Nontoxic γ-Cyclodextrin-K+ Metal-Organic Framework in Various Solvents. Crystals 2020, 10, 37. https://doi.org/10.3390/cryst10010037
Krūkle-Bērziņa K, Belyakov S, Mishnev A, Shubin K. Stability and Phase Transitions of Nontoxic γ-Cyclodextrin-K+ Metal-Organic Framework in Various Solvents. Crystals. 2020; 10(1):37. https://doi.org/10.3390/cryst10010037
Chicago/Turabian StyleKrūkle-Bērziņa, Kristīne, Sergey Belyakov, Anatoly Mishnev, and Kirill Shubin. 2020. "Stability and Phase Transitions of Nontoxic γ-Cyclodextrin-K+ Metal-Organic Framework in Various Solvents" Crystals 10, no. 1: 37. https://doi.org/10.3390/cryst10010037