Influence of MoS2 on Activity and Stability of Carbon Nitride in Photocatalytic Hydrogen Production

 , , , , ,
, , , , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Catalytic Activity
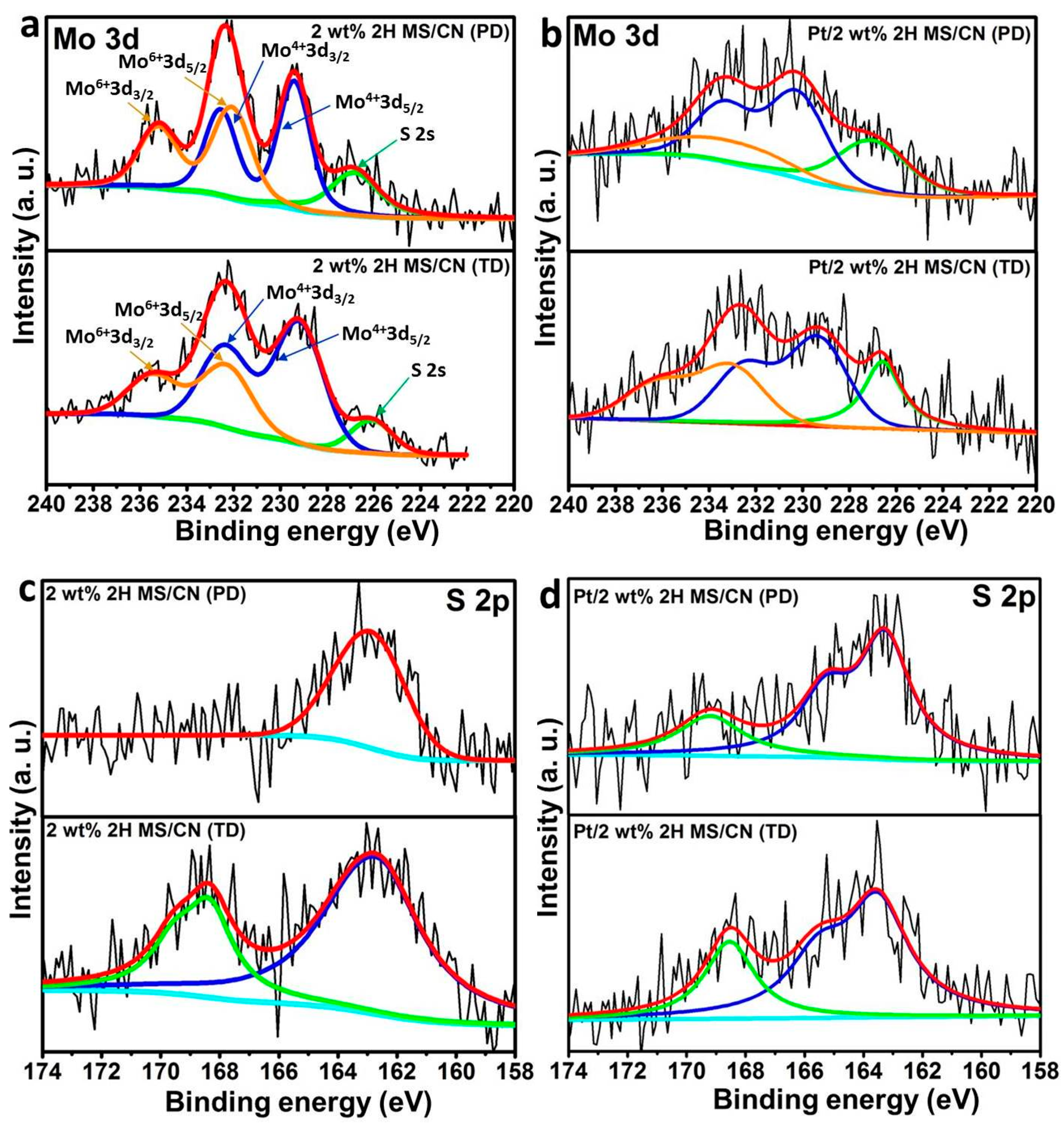
2.2. Characterization of Fresh and Used Catalysts
2.3. Monitoring Charge Separation and Transfer
3. Materials and Methods
3.1. Materials
3.2. Synthesis of MoS2/C3N4 Composites
3.2.1. Synthesis of 2 wt% 2H MoS2/C3N4 Composite by In Situ-Photodeposition Method (2H MS-CN (PD))
3.2.2. Synthesis of 2 wt% 2H MoS2/C3N4 Composite by Sonochemical Method (2H MS-CN (SC))
3.2.3. Synthesis of 2 wt% 1T MoS2/C3N4 Composite by Sonochemical Method (1T MS-CN (SC))
3.2.4. Synthesis of 2H MoS2 and 2 wt% 2H MoS2/C3N4 Composite by Thermal Decomposition (Impregnation) Method (2H MS-CN (TD))
3.3. Catalyst Characterization
3.4. In Situ Electron Paramagnetic Resonance Spectroscopy Studies
3.5. Photocatalytic Hydrogen Evolution Tests
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Armaroli, N.; Balzani, V. The future of energy supply: Challenges and opportunities. Angew. Chem. Int. Ed. 2007, 46, 52–66. [Google Scholar] [CrossRef] [PubMed]
- Takanabe, K.; Domen, K. Preparation of Inorganic Photocatalytic Materials for Overall Water Splitting. ChemCatChem 2012, 4, 1485–1497. [Google Scholar] [CrossRef]
- Lewis, N.S.; Nocera, D.G. Powering the planet: Chemical challenges in solar energy utilization. Proc. Natl. Acad. Sci. USA 2006, 103, 15729–15735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, J.A. Sustainable hydrogen production. Science 2004, 305, 972–974. [Google Scholar] [CrossRef] [PubMed]
- Colón, G. Towards the hydrogen production by photocatalysis. Appl. Catal. A Gen. 2016, 518, 48–59. [Google Scholar] [CrossRef]
- Li, X.; Yu, J.; Low, J.; Fang, Y.; Xiao, J.; Chen, X. Engineering heterogeneous semiconductors for solar water splitting. J. Mater. Chem. A 2015, 3, 2485–2534. [Google Scholar] [CrossRef]
- Kudo, A.; Miseki, Y. Heterogeneous photocatalyst materials for water splitting. Chem. Soc. Rev. 2009, 38, 253–278. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shen, S.; Guo, L.; Mao, S.S. Semiconductor-based Photocatalytic Hydrogen Generation. Chem. Rev. 2010, 110, 6503–6570. [Google Scholar] [CrossRef]
- Wang, R.; Gu, L.; Zhou, J.; Liu, X.; Teng, F.; Li, C.; Shen, Y.; Yuan, Y. Quasi-Polymeric Metal-Organic Framework UiO-66/g-C3N4 Heterojunctions for Enhanced Photocatalytic Hydrogen Evolution under Visible Light Irradiation. Adv. Mater. Interfaces 2015, 2, 1500037. [Google Scholar] [CrossRef]
- Han, X.B.; Li, Y.G.; Zhang, Z.M.; Tan, H.Q.; Lu, Y.; Wang, E.B. Polyoxometalate-based nickel clusters as visible light-driven water oxidation catalysts. J. Am. Chem. Soc. 2015, 137, 5486–5493. [Google Scholar] [CrossRef]
- Ong, W.-J.; Tan, L.-L.; Ng, Y.H.; Yong, S.-T.; Chai, S.-P. Graphitic Carbon Nitride (g-C3N4)-Based Photocatalysts for Artificial Photosynthesis and Environmental Remediation: Are We a Step Closer to Achieving Sustainability? Chem. Rev. 2016, 116, 7159–7329. [Google Scholar] [CrossRef] [PubMed]
- Reza Gholipour, M.; Dinh, C.T.; Beland, F.; Do, T.O. Nanocomposite heterojunctions as sunlight-driven photocatalysts for hydrogen production from water splitting. Nanoscale 2015, 7, 8187–8208. [Google Scholar] [CrossRef] [PubMed]
- Moniz, S.J.A.; Shevlin, S.A.; Martin, D.J.; Guo, Z.-X.; Tang, J. Visible-light driven heterojunction photocatalysts for water splitting a critical review. Energy Environ. Sci. 2015, 8, 731–759. [Google Scholar] [CrossRef]
- Marschall, R. Semiconductor Composites: Strategies for Enhancing Charge Carrier Separation to Improve Photocatalytic Activity. Adv. Funct. Mater. 2014, 24, 2421–2440. [Google Scholar] [CrossRef]
- Hollmann, D.; Karnahl, M.; Tschierlei, S.; Kailasam, K.; Schneider, M.; Radnik, J.; Grabow, K.; Bentrup, U.; Junge, H.; Beller, M.; et al. Structure-Activity Relationships in Bulk Polymeric and Sol–Gel-Derived Carbon Nitrides during Photocatalytic Hydrogen Production. Chem. Mater. 2014, 26, 1727–1733. [Google Scholar] [CrossRef]
- Wang, X.; Blechert, S.; Antonietti, M. Polymeric Graphitic Carbon Nitride for Heterogeneous Photocatalysis. ACS Catal. 2012, 2, 1596–1606. [Google Scholar] [CrossRef]
- Wang, X.; Maeda, K.; Thomas, A.; Takanabe, K.; Xin, G.; Carlsson, J.M.; Domen, K.; Antonietti, M. A metal-free polymeric photocatalyst for hydrogen production from water under visible light. Nat. Mater. 2009, 8, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, J.; Wang, X.; Huang, Y.; Zeng, M.; Xu, J. Rationally designed 1D Ag@AgVO3 nanowire/graphene/protonated g-C3N4 nanosheet heterojunctions for enhanced photocatalysis via electrostatic self-assembly and photochemical reduction methods. J. Mater. Chem. A 2015, 3, 10119–10126. [Google Scholar] [CrossRef]
- Yin, S.; Han, J.; Zhou, T.; Xu, R. Recent progress in g-C3N4 based low cost photocatalytic system: Activity enhancement and emerging applications. Catal. Sci. Technol. 2015, 5, 5048–5061. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, L.; Chen, Z.; Hu, J.; Li, S.; Wang, Z.; Liu, J.; Wang, X. Semiconductor heterojunction photocatalysts: Design, construction, and photocatalytic performances. Chem. Soc. Rev. 2014, 43, 5234–5244. [Google Scholar] [CrossRef]
- Jiang, D.; Li, J.; Xing, C.; Zhang, Z.; Meng, S.; Chen, M. Two-Dimensional CaIn2S4/g-C3N4 Heterojunction Nanocomposite with Enhanced Visible-Light Photocatalytic Activities: Interfacial Engineering and Mechanism Insight. ACS Appl. Mater. Interfaces 2015, 7, 19234–19242. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Huang, J.; Zhang, M.; Yuan, Q.; Dong, B. Ultrathin hexagonal SnS2 nanosheets coupled with g-C3N4 nanosheets as 2D/2D heterojunction photocatalysts toward high photocatalytic activity. Appl. Catal. B Environ. 2015, 163, 298–305. [Google Scholar] [CrossRef]
- Hou, Y.; Wen, Z.; Cui, S.; Guo, X.; Chen, J. Constructing 2D porous graphitic C3N4 nanosheets/nitrogen-doped graphene/layered MoS2 ternary nanojunction with enhanced photoelectrochemical activity. Adv. Mater. 2013, 25, 6291–6297. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Fang, Q.; Liu, Q.; Liu, D.; Wang, C.; Xiang, T.; Khalil, A.; Chen, S.; Song, L. Engineering interfacial charge-transfer by phase transition realizing enhanced photocatalytic hydrogen evolution activity. Inorg. Chem. Front. 2017, 4, 663–667. [Google Scholar] [CrossRef]
- Lei, Z.; Zhan, J.; Tang, L.; Zhang, Y.; Wang, Y. Recent Development of Metallic (1T) Phase of Molybdenum Disulfide for Energy Conversion and Storage. Adv. Energy Mater. 2018, 8, 1703482. [Google Scholar] [CrossRef]
- Liu, Q.; Li, X.; He, Q.; Khalil, A.; Liu, D.; Xiang, T.; Wu, X.; Song, L. Gram-Scale Aqueous Synthesis of Stable Few-Layered 1T-MoS2: Applications for Visible-Light-Driven Photocatalytic Hydrogen Evolution. Small 2015, 11, 5556–5564. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.; Li, Y. Recent advances in heterogeneous electrocatalysts for the hydrogen evolution reaction. J. Mater. Chem. A 2015, 3, 14942–14962. [Google Scholar] [CrossRef]
- Sivasankaran, R.; Rockstroh, N.; Hollmann, D.; Kreyenschulte, C.; Agostini, G.; Lund, H.; Acharjya, A.; Rabeah, J.; Bentrup, U.; Junge, H.; et al. Relations between Structure, Activity and Stability in C3N4 Based Photocatalysts Used for Solar Hydrogen Production. Catalysts 2018, 8, 52. [Google Scholar] [CrossRef]
- Wu, M.; Zhan, J.; Wu, K.; Li, Z.; Wang, L.; Geng, B.; Wang, L.; Pan, D. Metallic 1T MoS2 nanosheet arrays vertically grown on activated carbon fiber cloth for enhanced Li-ion storage performance. J. Mater. Chem. A 2017, 5, 14061–14069. [Google Scholar] [CrossRef]
- Tyborski, T.; Merschjann, C.; Orthmann, S.; Yang, F.; Lux-Steiner, M.C.; Schedel-Niedrig, T. Crystal structure of polymeric carbon nitride and the determination of its process-temperature-induced modifications. J. Phys. Condens. Matter 2013, 25, 395402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Xia, J.; Zhu, W.; Di, J.; Wang, B.; Yin, S.; Chen, Z.; Li, H. Facile synthesis of few-layered MoS2 modified BiOI with enhanced visible-light photocatalytic activity. Colloids Surf. A Physico. Eng. Asp. 2016, 511, 1–7. [Google Scholar] [CrossRef]
- Mo, Z.; She, X.; Li, Y.; Liu, L.; Huang, L.; Chen, Z.; Zhang, Q.; Xu, H.; Li, H. Synthesis of g-C3N4 at different temperatures for superior visible/UV photocatalytic performance and photoelectrochemical sensing of MB solution. RSC Adv. 2015, 5, 101552–101562. [Google Scholar] [CrossRef]
- Wenderich, K.; Mul, G. Methods, Mechanism, and Applications of Photodeposition in Photocatalysis: A Review. Chem. Rev. 2016, 116, 14587–14619. [Google Scholar] [CrossRef] [PubMed]
- Xiang, T.; Fang, Q.; Xie, H.; Wu, C.; Wang, C.; Zhou, Y.; Liu, D.; Chen, S.; Khalil, A.; Tao, S.; et al. Vertical 1T-MoS2 nanosheets with expanded interlayer spacing edged on a graphene frame for high rate lithium-ion batteries. Nanoscale 2017, 9, 6975–6983. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, I.B.; Swift, P. A study of the tribo-chemical oxidation of molybdenum disulphide using X-ray photo-electron spectroscopy. Wear 1974, 29, 129–133. [Google Scholar] [CrossRef]
- Liu, Q.; Fang, Q.; Chu, W.; Wan, Y.; Li, X.; Xu, W.; Habib, M.; Tao, S.; Zhou, Y.; Liu, D.; et al. Electron-Doped 1T-MoS2 via Interface Engineering for Enhanced Electrocatalytic Hydrogen Evolution. Chem. Mater. 2017, 29, 4738–4744. [Google Scholar] [CrossRef]
- Afanasiev, P.; Lorentz, C. Oxidation of Nanodispersed MoS2 in Ambient Air: The Products and the Mechanistic Steps. J. Phys. Chem. C 2019, 123, 7486–7494. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, L.; Zhang, B.; Guo, X.; Mu, X. Highly selective hydrogenation of furfural to furfuryl alcohol over Pt nanoparticles supported on g-C3N4 nanosheets catalysts in water. Sci. Rep. 2016, 6, 28558. [Google Scholar] [CrossRef]
- Garbarino, S.; Pereira, A.; Hamel, C.; Irissou, É.; Chaker, M.; Guay, D. Effect of Size on the Electrochemical Stability of Pt Nanoparticles Deposited on Gold Substrate. J. Phys. Chem. C 2010, 114, 2980–2988. [Google Scholar] [CrossRef]
- Isaifan, R.J.; Ntais, S.; Baranova, E.A. Particle size effect on catalytic activity of carbon-supported Pt nanoparticles for complete ethylene oxidation. Appl. Catal. A Gen. 2013, 464, 87–94. [Google Scholar] [CrossRef]
- Marcus, P.; Hinnen, C. XPS study of the early stages of deposition of Ni, Cu and Pt on HOPG. Surf. Sci. 1997, 392, 134–142. [Google Scholar] [CrossRef]
- Mason, M.G. Electronic structure of supported small metal clusters. Phys. Rev. B 1983, 27, 748–762. [Google Scholar] [CrossRef]
- Espinós, J.P.; Morales, J.; Barranco, A.; Caballero, A.; Holgado, J.P.; González-Elipe, A.R. Interface Effects for Cu, CuO, and Cu2O Deposited on SiO2 and ZrO2. XPS Determination of the Valence State of Copper in Cu/SiO2 and Cu/ZrO2 Catalysts. J. Phys. Chem. B 2002, 106, 6921–6929. [Google Scholar] [CrossRef]
- Morales, J.; Espinos, J.P.; Caballero, A.; Gonzalez-Elipe, A.R.; Mejias, J.A. XPS Study of Interface and Ligand Effects in Supported Cu2O and CuO Nanometric Particles. J. Phys. Chem. B 2005, 109, 7758–7765. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.S.; Choi, S.H.; Kim, H.G.; Lee, J.S. Location and State of Pt in Platinized CdS/TiO2 Photocatalysts for Hydrogen Production from Water under Visible Light. J. Phys. Chem. C 2008, 112, 17200–17205. [Google Scholar] [CrossRef]
- Zhang, G.; Lan, Z.-A.; Lin, L.; Lin, S.; Wang, X. Overall water splitting by Pt/g-C3N4 photocatalysts without using sacrificial agents. Chem. Sci. 2016, 7, 3062–3066. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Huang, C.; Wang, X. Post-annealing reinforced hollow carbon nitride nanospheres for hydrogen photosynthesis. Nanoscale 2015, 7, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Ma, S.; Zhou, Y.; Zhang, Z.; He, M. Facile Photochemical Synthesis of Au/Pt/g-C3N4 with Plasmon-Enhanced Photocatalytic Activity for Antibiotic Degradation. ACS Appl. Mater. Interfaces 2015, 7, 9630–9637. [Google Scholar] [CrossRef] [PubMed]
- Priebe, J.B.; Radnik, J.; Lennox, A.J.J.; Pohl, M.-M.; Karnahl, M.; Hollmann, D.; Grabow, K.; Bentrup, U.; Junge, H.; Beller, M.; et al. Solar Hydrogen Production by Plasmonic Au-TiO2 Catalysts: Impact of Synthesis Protocol and TiO2 Phase on Charge Transfer Efficiency and H2 Evolution Rates. ACS Catal. 2015, 5, 2137–2148. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, G.; Chen, X.; Lin, S.; Mohlmann, L.; Dolega, G.; Lipner, G.; Antonietti, M.; Blechert, S.; Wang, X. Co-monomer control of carbon nitride semiconductors to optimize hydrogen evolution with visible light. Angew. Chem. 2012, 51, 3183–3187. [Google Scholar] [CrossRef]
- Li, T.; Zhao, L.; He, Y.; Cai, J.; Luo, M.; Lin, J. Synthesis of g-C3N4/SmVO4 composite photocatalyst with improved visible light photocatalytic activities in RhB degradation. Appl. Catal. B Environ. 2013, 129, 255–263. [Google Scholar] [CrossRef]
- Miyashita, K.; Kuroda, S.-I.; Tajima, S.; Takehira, K.; Tobita, S.; Kubota, H. Photoluminescence study of electron–hole recombination dynamics in the vacuum-deposited SiO2/TiO2 multilayer film with photo-catalytic activity. Chem. Phys. Lett. 2003, 369, 225–231. [Google Scholar] [CrossRef]
- Xing, C.; Wu, Z.; Jiang, D.; Chen, M. Hydrothermal synthesis of In2S3/g-C3N4 heterojunctions with enhanced photocatalytic activity. J. Colloid Interface Sci. 2014, 433, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Shen, S.; Guo, P.; Wu, P.; Guo, L. Spatial engineering of photo-active sites on g-C3N4 for efficient solar hydrogen generation. J. Mater. Chem. A 2014, 2, 4605–4612. [Google Scholar] [CrossRef]
- Fan, M.; Hu, B.; Yan, X.; Song, C.; Chen, T.; Feng, Y.; Shi, W. Excellent visible-light-driven photocatalytic performance of Cu2O sensitized NaNbO3 heterostructures. New J. Chem. 2015, 39, 6171–6177. [Google Scholar] [CrossRef]
- Nayak, S.; Mohapatra, L.; Parida, K. Visible light-driven novel g-C3N4/NiFe-LDH composite photocatalyst with enhanced photocatalytic activity towards water oxidation and reduction reaction. J. Mater. Chem. A 2015, 3, 18622–18635. [Google Scholar] [CrossRef]
- Jin, X.; Fan, X.; Tian, J.; Cheng, R.; Li, M.; Zhang, L. MoS2 quantum dot decorated g-C3N4 composite photocatalyst with enhanced hydrogen evolution performance. RSC Adv. 2016, 6, 52611–52619. [Google Scholar] [CrossRef]
- Kailasam, K.; Epping, J.D.; Thomas, A.; Losse, S.; Junge, H. Mesoporous carbon nitride-silica composites by a combined sol-gel/thermal condensation approach and their application as photocatalysts. Energy Environ. Sci. 2011, 4, 4668. [Google Scholar] [CrossRef]
- Nguyen, M.; Tran, P.D.; Pramana, S.S.; Lee, R.L.; Batabyal, S.K.; Mathews, N.; Wong, L.H.; Graetzel, M. In situ photo-assisted deposition of MoS2 electrocatalyst onto zinc cadmium sulphide nanoparticle surfaces to construct an efficient photocatalyst for hydrogen generation. Nanoscale 2013, 5, 1479–1482. [Google Scholar] [CrossRef]
- Li, J.; Liu, E.; Ma, Y.; Hu, X.; Wan, J.; Sun, L.; Fan, J. Synthesis of MoS2/g-C3N4 nanosheets as 2D heterojunction photocatalysts with enhanced visible light activity. Appl. Surf. Sci. 2016, 364, 694–702. [Google Scholar] [CrossRef]
- Ge, L.; Han, C.; Xiao, X.; Guo, L. Synthesis and characterization of composite visible light active photocatalysts MoS2-g-C3N4 with enhanced hydrogen evolution activity. Int. J. Hydrogen Energy 2013, 38, 6960–6969. [Google Scholar] [CrossRef]
- Dai, W.; Yu, J.; Deng, Y.; Hu, X.; Wang, T.; Luo, X. Facile synthesis of MoS2/Bi2WO6 nanocomposites for enhanced CO2 photoreduction activity under visible light irradiation. Appl. Surf. Sci. 2017, 403, 230–239. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sivasankaran, R.P.; Rockstroh, N.; Kreyenschulte, C.R.; Bartling, S.; Lund, H.; Acharjya, A.; Junge, H.; Thomas, A.; Brückner, A. Influence of MoS2 on Activity and Stability of Carbon Nitride in Photocatalytic Hydrogen Production. Catalysts 2019, 9, 695. https://doi.org/10.3390/catal9080695
Sivasankaran RP, Rockstroh N, Kreyenschulte CR, Bartling S, Lund H, Acharjya A, Junge H, Thomas A, Brückner A. Influence of MoS2 on Activity and Stability of Carbon Nitride in Photocatalytic Hydrogen Production. Catalysts. 2019; 9(8):695. https://doi.org/10.3390/catal9080695
Chicago/Turabian StyleSivasankaran, Ramesh P., Nils Rockstroh, Carsten R. Kreyenschulte, Stephan Bartling, Henrik Lund, Amitava Acharjya, Henrik Junge, Arne Thomas, and Angelika Brückner. 2019. "Influence of MoS2 on Activity and Stability of Carbon Nitride in Photocatalytic Hydrogen Production" Catalysts 9, no. 8: 695. https://doi.org/10.3390/catal9080695