Immobilization of a Novel ESTBAS Esterase from Bacillus altitudinis onto an Epoxy Resin: Characterization and Regioselective Synthesis of Chloramphenicol Palmitate

Abstract
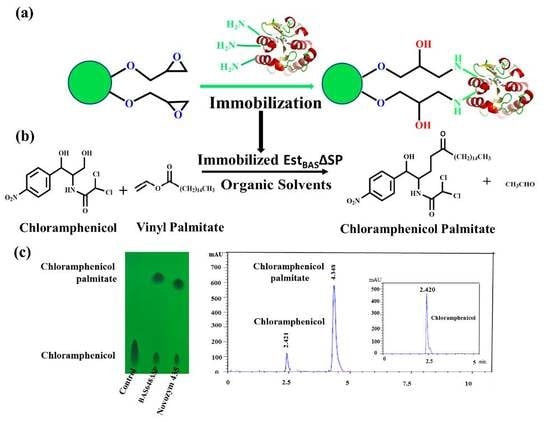
:
1. Introduction
2. Results and Discussion
2.1. Cloning of the EstBAS Gene and Sequence Analysis of EstBAS
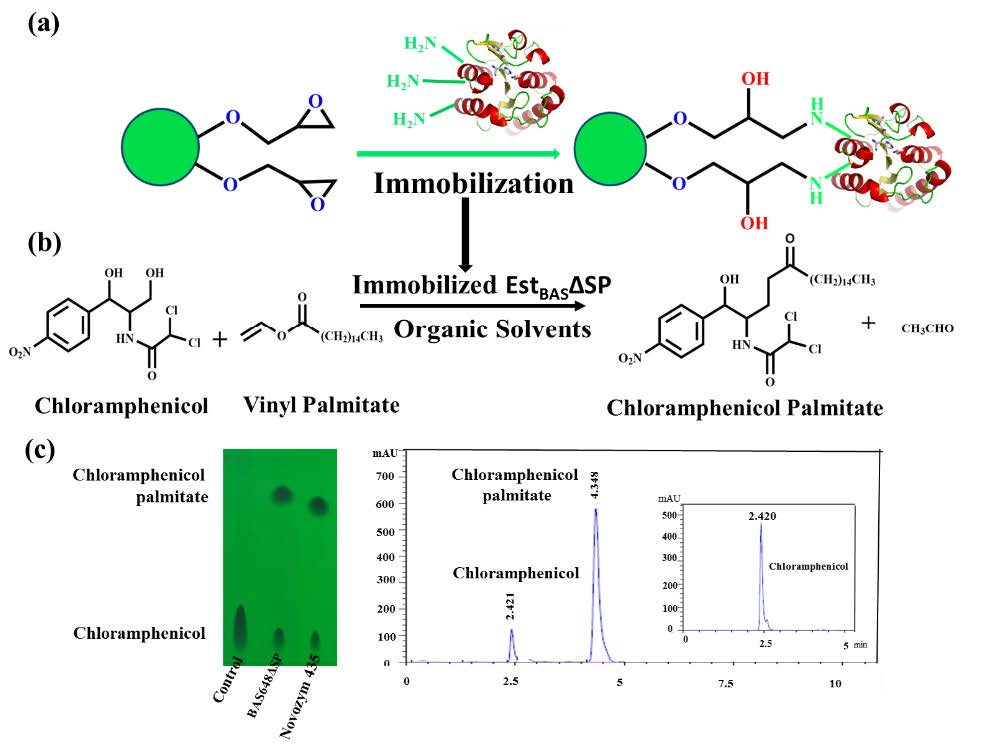
2.2. Expression and Purification of Recombinant EstBASΔSP
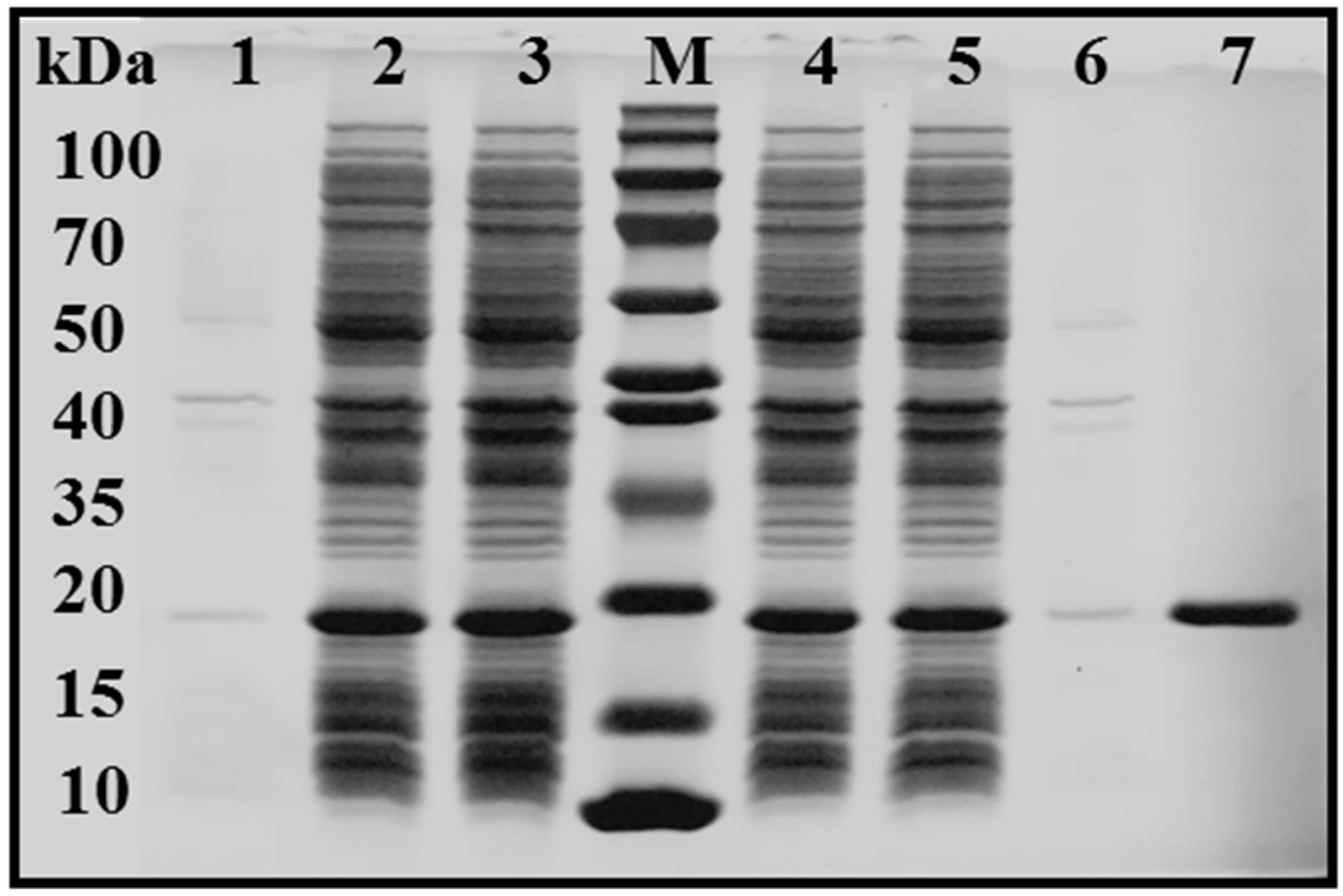
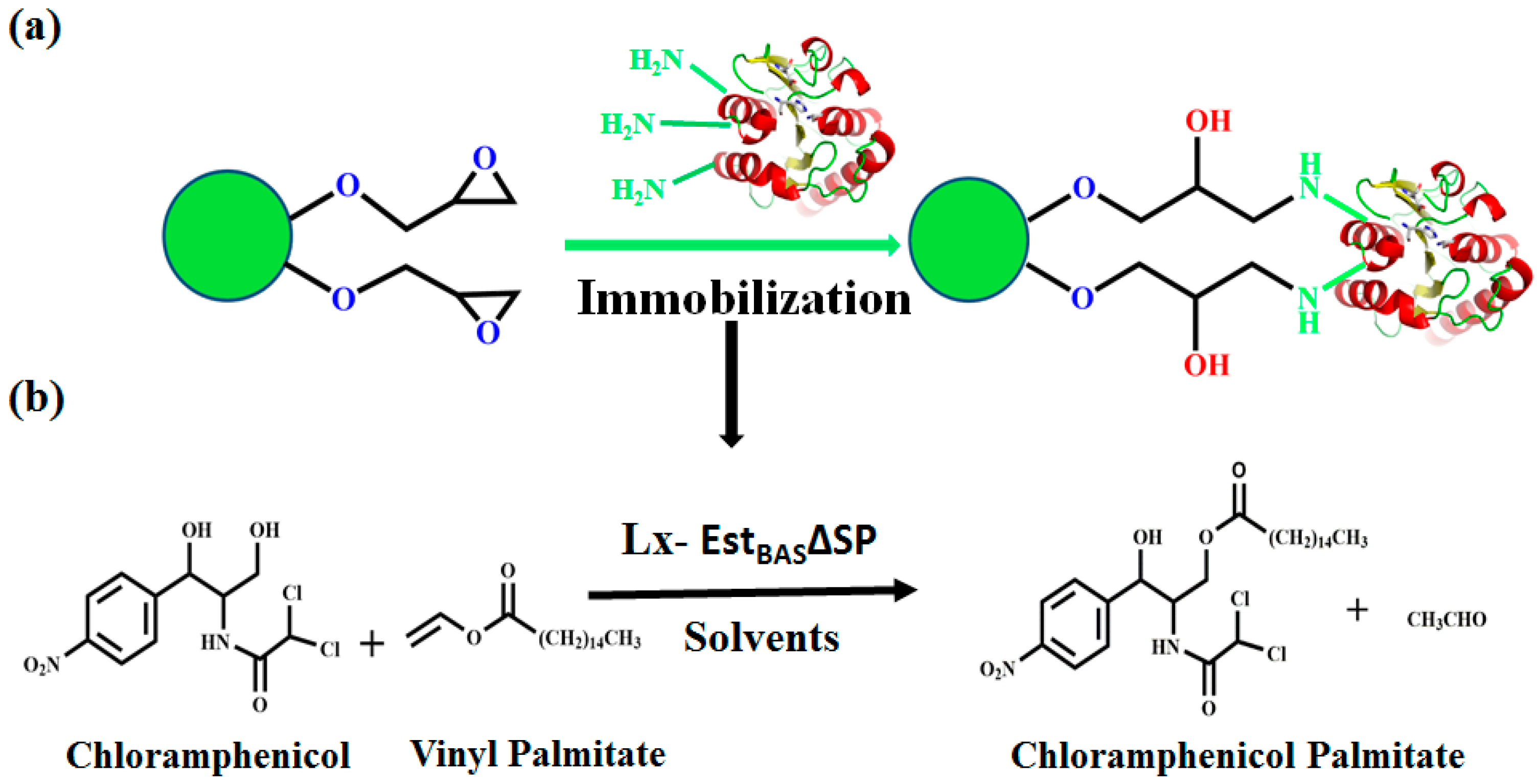
2.3. Optimization of EstBASΔSP Immobilization
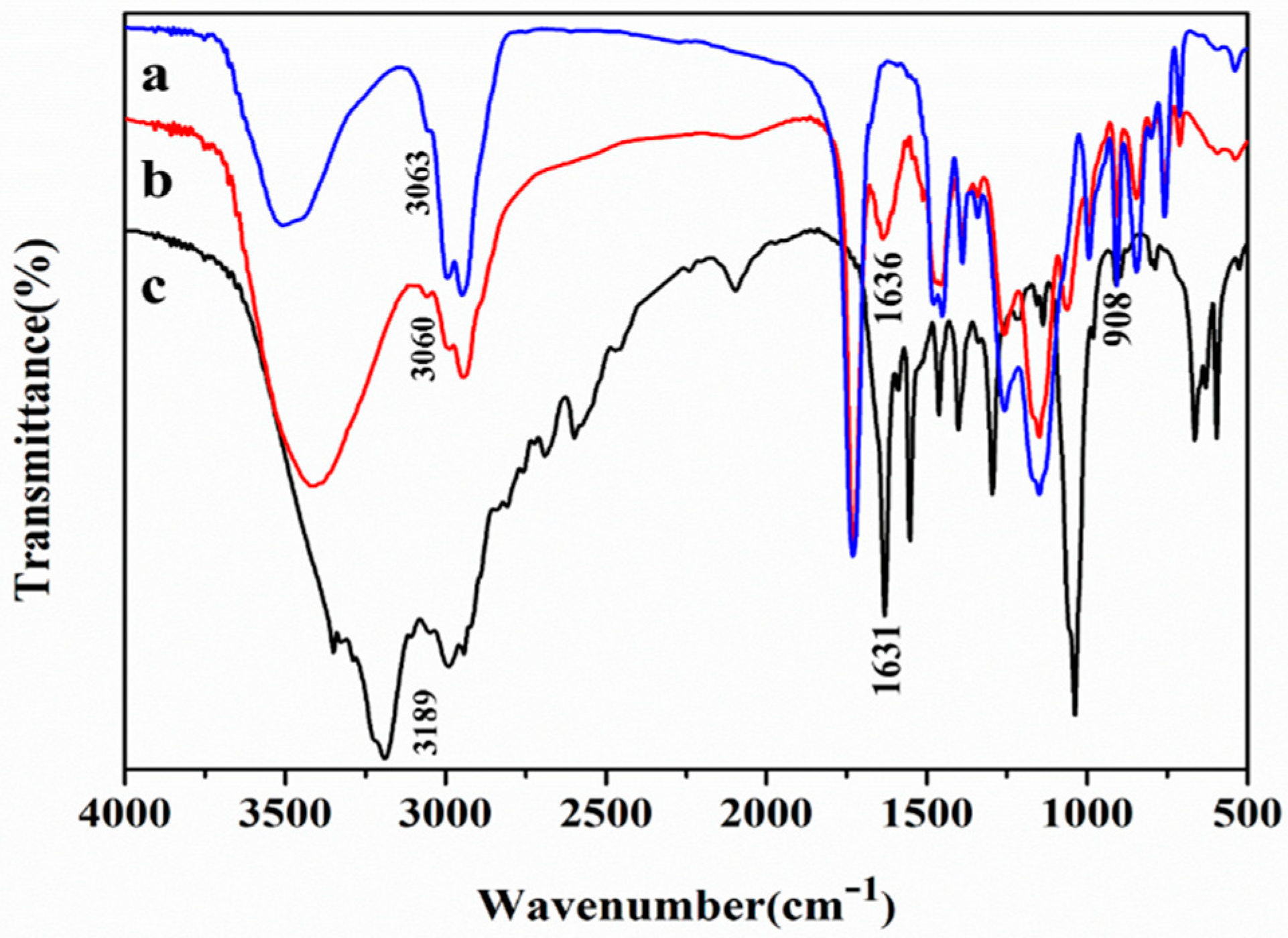
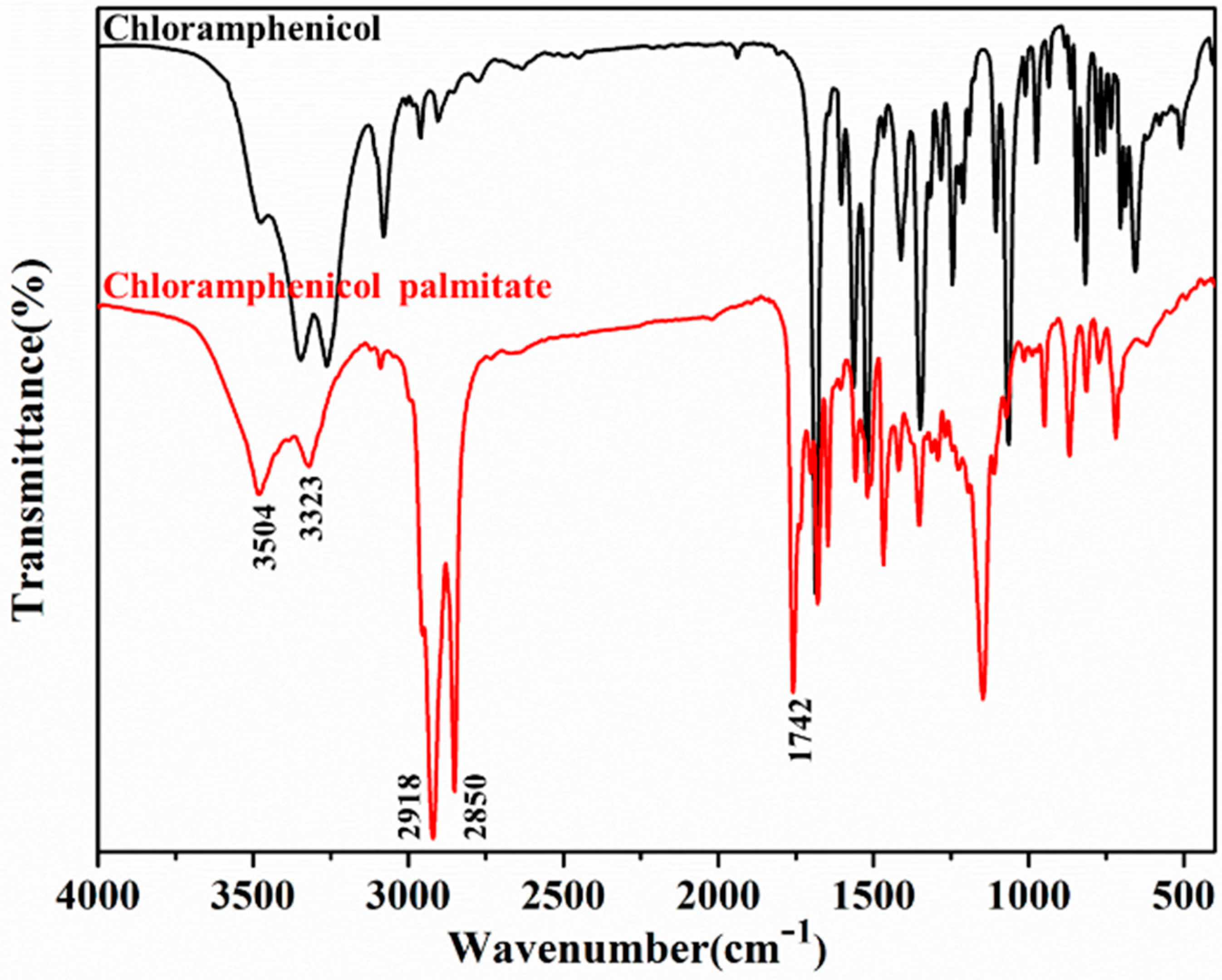
2.4. Characterization of Lx-EstBASΔSP by FTIR
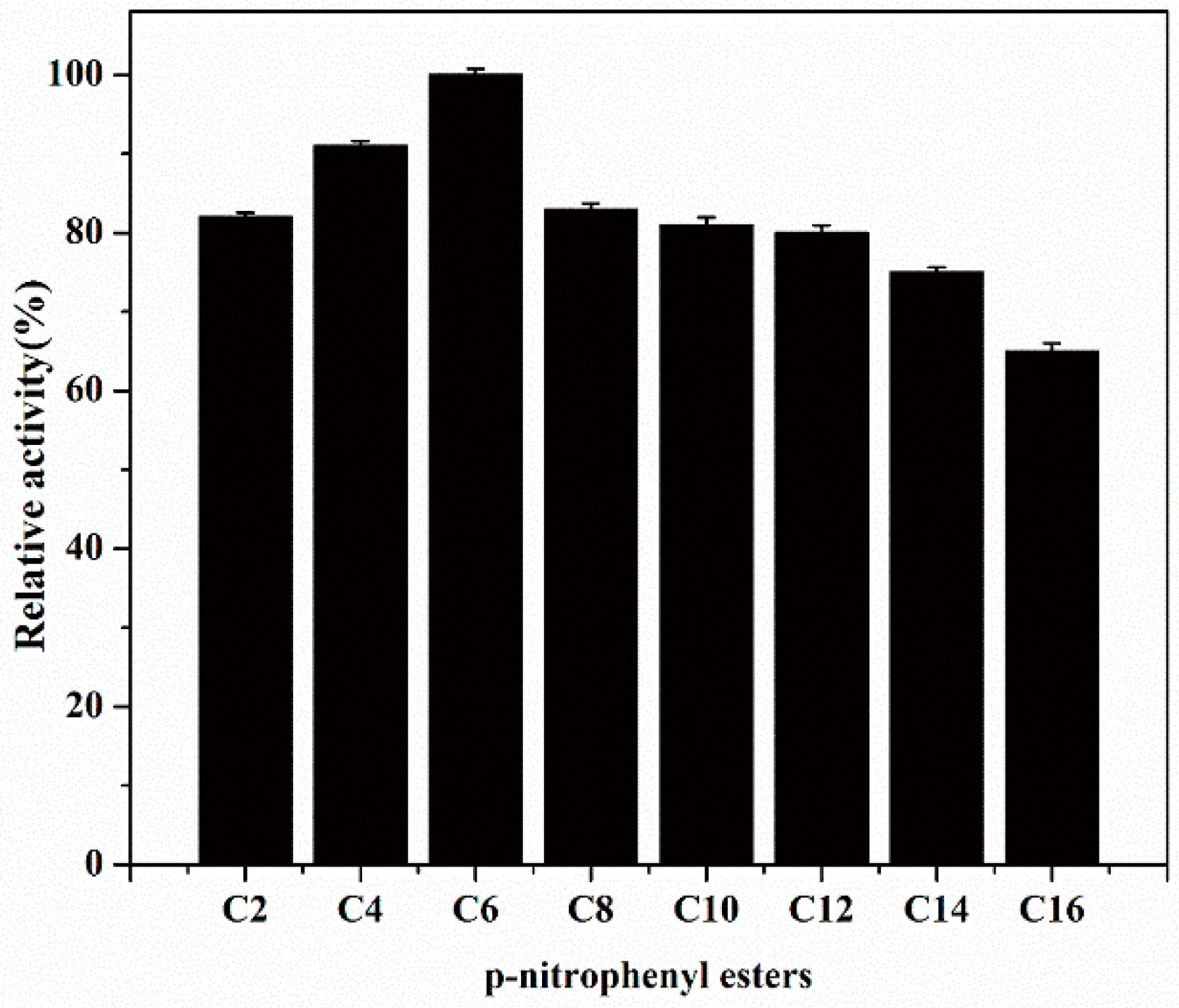
2.5. Substrate Specificity of EstBASΔSP
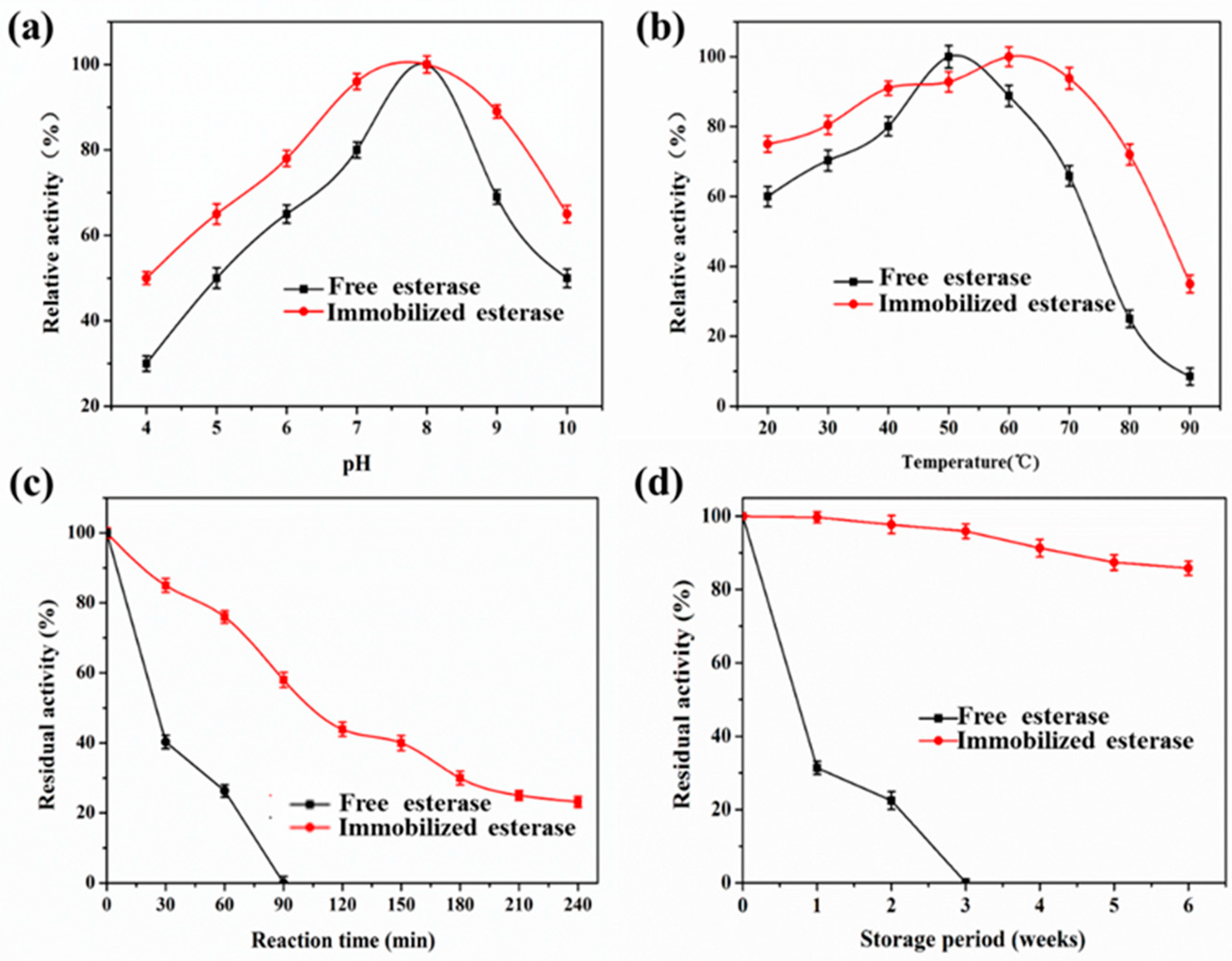
2.6. Effects of pH and Temperature on the Activity of Free EstBASΔSP and Lx-EstBASΔSP
2.7. Effects of Metal Ions and Detergents on Lx-EstBASΔSP and Free EstBASΔSP
2.8. Stability of Lx-EstBASΔSP and Free EstBASΔSP
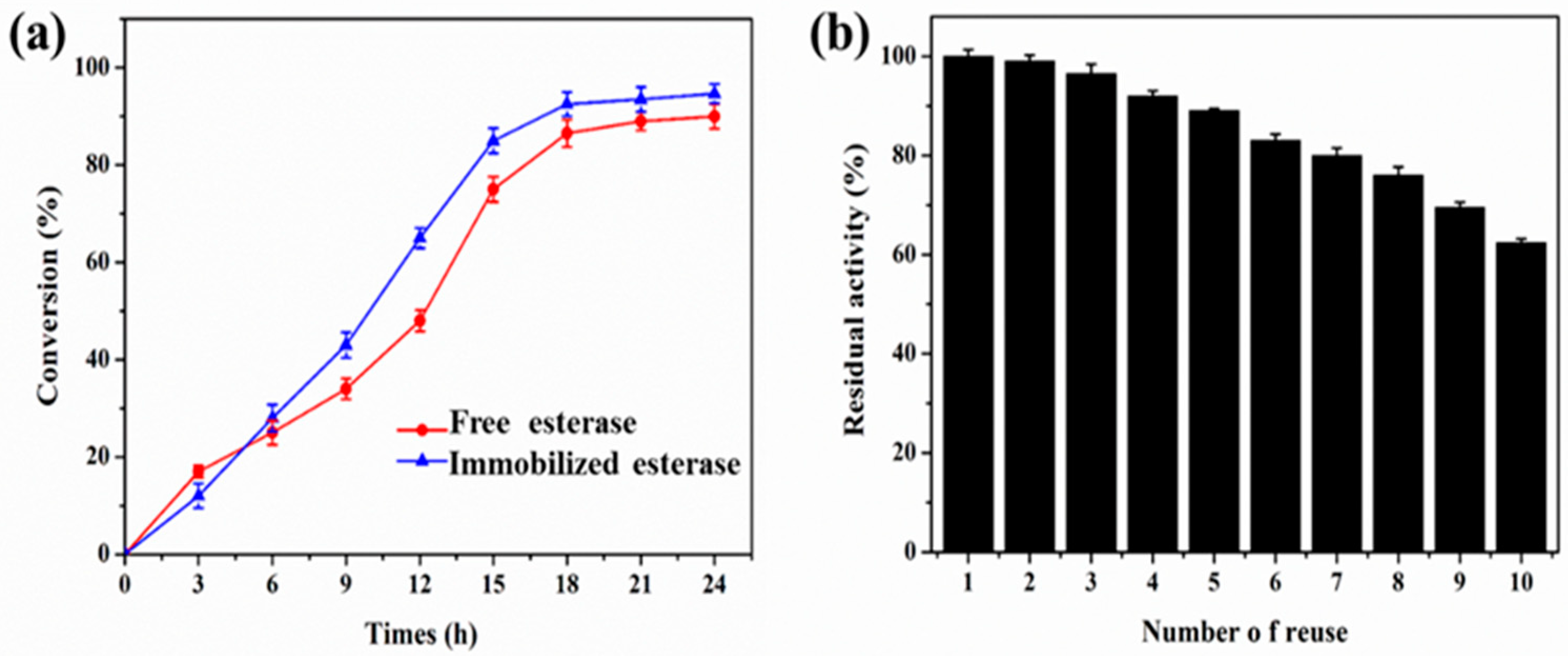
2.9. Regioselective Synthesis of Chloramphenicol Esters by Lx-EstBASΔSP and Free EstBASΔSP
2.10. Product Identification as Chloramphenicol Palmitate
3. Materials and Methods
3.1. Materials
3.2. Triacylglycerol Esterase and Gene Analysis
3.3. Cloning of the Esterase Gene from B. altitudinis
3.4. Effects of the SP on Overexpression and Purification of Triacylglycerol Esterase
3.5. Immobilization of Purified EstBASΔSP
3.6. Biochemical Characterization of Lx-EstBASΔSP and Free EstBASΔSP
3.7. Effects of Metal Ions, Detergents, and Organic Solvents on EstBASΔSP Activity
3.8. Characterization of Lx-EstBASΔSP by Scanning Electron Microscopy (SEM) and FTIR
3.9. Regioselective Synthesis of Chloramphenicol Esters by Lx-EstBASΔSP and Free EstBASΔSP
3.10. Reusability of Lx-EstBASΔSP
3.11. Chloramphenicol Palmitate Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| FTIR | Fourier transform infrared spectroscopy |
| IPTG | isopropyl-β-d-thiogalactopyranoside |
| p-NP | p-nitrophenyl |
| pNPA | p-NP acetate |
| pNPB | p-NP butyrate |
| pNPC | p-NP caprylate |
| pNPD | p-NP decanoate |
| pNPH | p-NP hexanoate |
| pNPL | p-NP laurate |
| pNPM | p-NP myristate |
| pNPP | p-NP palmitate |
| SDS-PAGE | sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| SEM | scanning electron microscopy; |
| SP | signal peptide; |
| TLC | thin-layer chromatography |
References
- Yang, G.; Zhao, F. Electrochemical sensor for chloramphenicol based on novel multiwalled carbon nanotubes@molecularly imprinted polymer. Biosens. Bioelectron. 2015, 64, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Pilehvar, S.; Mehta, J.; Dardenne, F.; Robbens, J.; Blust, R.; De, W.K. Aptasensing of chloramphenicol in the presence of its analogues: Reaching the maximum residue limit. Anal. Chem. 2012, 84, 6753–6758. [Google Scholar] [CrossRef] [PubMed]
- Abnous, K.; Danesh, N.M.; Ramezani, M.; Emrani, A.S.; Taghdisi, S.M. A novel colorimetric sandwich aptasensor based on an indirect competitive enzyme-free method for ultrasensitive detection of chloramphenicol. Biosens. Bioelectron. 2015, 78, 80–86. [Google Scholar] [CrossRef]
- Ottolina, G.; Carrea, G.; Riva, S. Synthesis of ester derivatives of chloramphenicol by lipase-catalyzed transesterification in organic solvents. J. Org. Chem. 1990, 55, 2366–2369. [Google Scholar] [CrossRef]
- Bizerra, A.M.C.; Montenegro, T.G.C.; Lemos, T.L.G.; Oliveira, M.C.F.D.; Mattos, M.C.D.; Lavandera, I.; Gotor-Fernández, V.; Gonzalo, G.D.; Gotor, V. Enzymatic regioselective production of chloramphenicol esters. Tetrahedron 2011, 67, 2858–2862. [Google Scholar] [CrossRef]
- Kumar, R.; Mahajan, S.; Kumar, A.; Singh, D. Identification of variables and value optimization for optimum lipase production by Bacillus pumilus RK31 using statistical methodology. New Biotechnol. 2011, 28, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Ghanem, E.H.; Alsayed, H.A.; Saleh, K.M. An alkalophilic thermostable lipase produced by a new isolate of Bacillus alcalophilus. World J. Microbiol. Biotechnol. 2000, 16, 459–464. [Google Scholar] [CrossRef]
- Lv, D.S.; Xue, X.T.; Wang, N.; Wu, Q.; Lin, X.F. Enzyme catalyzed synthesis of some vinyl drug esters in organic medium. Prep. Biochem. 2004, 34, 97–107. [Google Scholar] [CrossRef]
- Wang, R.; Zhang, Y.; Huang, J.; Lu, D.; Ge, J.; Zheng, L. Substrate imprinted lipase nanogel for one-step synthesis of chloramphenicol palmitate. Green Chem. 2013, 15, 1155–1158. [Google Scholar] [CrossRef]
- Esakkiraj, P.; Usha, R.; Palavesam, A.; Immanuel, G. Solid-state production of esterase using fish processing wastes by Bacillus altitudinis AP-MSU. Food Bioprod. Process. 2012, 90, 370–376. [Google Scholar] [CrossRef]
- Schmidt, M.; Henke, E.; Heinze, B.; Kourist, R.; Hidalgo, A.; Bornscheuer, U.T. A versatile esterase from Bacillus subtilis: Cloning, expression, characterization, and its application in biocatalysis. Biotechnol. J. 2010, 2, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Hung, T.C.; Giridhar, R.; Chiou, S.H.; Wu, W.T. Binary immobilization of Candida rugosa lipase on chitosan. J. Mol. Catal. B Enzym. 2003, 26, 69–78. [Google Scholar] [CrossRef]
- Mateo, C.; Palomo, J.M.; Fernandez-Lorente, G.; Guisan, J.M.; Fernandez-Lafuente, R. Improvement of enzyme activity, stability and selectivity via immobilization techniques. Enzym. Microb. Technol. 2007, 40, 1451–1463. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Van, P.S. Enzyme immobilisation in biocatalysis: Why, what and how. Chem. Soc. Rev. 2013, 42, 6223–6235. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhou, W.; Li, J.; Hao, D.; Su, Z.; Ma, G. Comparison of covalent and physical immobilization of lipase in gigaporous polymeric microspheres. Bioprocess Biosyst. Eng. 2015, 38, 2107–2115. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Bai, Y.; Li, Y.; Yi, L.; Yang, Y.; Xia, C. Study on immobilization of lipase onto magnetic microspheres with epoxy groups. J. Magn. Magn. Mater. 2009, 321, 252–258. [Google Scholar] [CrossRef]
- Bayramoglu, G.; Karagoz, B.; Altintas, B.; Arica, M.Y.; Bicak, N. Poly(styrene–divinylbenzene) beads surface functionalized with di-block polymer grafting and multi-modal ligand attachment: Performance of reversibly immobilized lipase in ester synthesis. Bioprocess Biosyst. Eng. 2011, 34, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Gomes, F.M.; Pereira, E.B.; de Castro, H.F. Immobilization of lipase on chitin and its use in nonconventional biocatalysis. Biomacromolecules 2004, 5, 17–23. [Google Scholar] [CrossRef]
- Matte, C.R.; Bordinhão, C.; Poppe, J.K.; Rodrigues, R.C.; Hertz, P.F.; Ayub, M.A.Z. Synthesis of butyl butyrate in batch and continuous enzymatic reactors using Thermomyces lanuginosus lipase immobilized in Immobead 150. J. Mol. Catal. B Enzym. 2016, 127, 67–75. [Google Scholar] [CrossRef]
- Zhang, D.H.; Peng, L.J.; Wang, Y.; Li, Y.Q. Lipase immobilization on epoxy-activated poly(vinyl acetate-acrylamide) microspheres. Colloids Surf. B Biointerfaces 2015, 129, 206–210. [Google Scholar] [CrossRef]
- Ma, H.Z.; Yu, X.W.; Song, C.; Xue, Q.L.; Jiang, B. Immobilization of Candida Antarctica lipase B on epoxy modified silica by sol-gel process. J. Mol. Catal. B Enzym. 2016, 127, 76–81. [Google Scholar] [CrossRef]
- Ashjari, M.; Mohammadi, M.; Badri, R. Chemical amination of Rhizopus oryzae lipase for multipoint covalent immobilization on epoxy-functionalized supports: Modulation of stability and selectivity. J. Mol. Catal. B Enzym. 2015, 115, 128–134. [Google Scholar] [CrossRef]
- Petersen, T.N.; Brunak, S.; Von, H.G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef]
- Harris, D.A. Cellular Biology of Prion Diseases. Clin. Microbiol. Rev. 1999, 12, 429–444. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Cong, Q.; Chao, C.; Fu, Y.; Ming, M.; Xie, Q. Highly sensitive phenolic biosensor based on magnetic polydopamine-laccase-Fe3O4 bionanocomposite. Sens. Actuators B 2012, 168, 46–53. [Google Scholar] [CrossRef]
- Silva, R.; Carmona-Ribeiro, A.; Petri, D. Catalytic Behavior of Lipase Immobilized onto Congo Red and PEG-Decorated Particles. Molecules 2014, 19, 8610–8628. [Google Scholar] [CrossRef] [Green Version]
- Xavier, A.; Tavares, A.; Daniel-Da-Silva, A.; Fortes, C. Optimisation of laccase immobilization on modified magnetic nanoparticles for biocatalytic reactions. New Biotechnol. 2016, 33, S15. [Google Scholar] [CrossRef]
- Jenjob, S.; Sunintaboon, P.; Inprakhon, P.; Anantachoke, N.; Reutrakul, V. Chitosan-functionalized poly(methyl methacrylate) particles by spinning disk processing for lipase immobilization. Carbohydr. Polym. 2012, 89, 842–848. [Google Scholar] [CrossRef]
- Li, X.; Li, D.; Wang, W.; Durrani, R.; Bo, Y.; Wang, Y. Immobilization of SMG1-F278N lipase onto a novel epoxy resin: Characterization and its application in synthesis of partial glycerides. J. Mol. Catal. B Enzym. 2016, 133, 154–160. [Google Scholar] [CrossRef]
- Dong, H.; Secundo, F.; Xue, C.; Mao, X. Whole-Cell Biocatalytic Synthesis of Cinnamyl Acetate with a Novel Esterase from the DNA Library of Acinetobacter hemolyticus. J. Agric. Food Chem. 2017, 65, 2120–2128. [Google Scholar] [CrossRef]
- Oliveranappa, A.; Andrews, B.A.; Asenjo, J.A. A mixed mechanistic-electrostatic model to explain pH dependence of glycosyl hydrolase enzyme activity. Biotechnol. Bioeng. 2010, 86, 573–586. [Google Scholar] [CrossRef]
- Lin, J.; Liu, Y.; Chen, S.; Le, X.; Zhou, X.; Zhao, Z.; Ou, Y.; Yang, J. Reversible immobilization of laccase onto metal-ion-chelated magnetic microspheres for bisphenol A removal. Int. J. Biol. Macromol. 2016, 84, 189–199. [Google Scholar] [CrossRef]
- Hou, C.; Qi, Z.; Zhu, H. Preparation of core-shell magnetic polydopamine/alginate biocomposite for Candida rugosa lipase immobilization. Colloids Surf. B Biointerfaces 2015, 128, 544–551. [Google Scholar] [CrossRef]
- Mishra, M.K.; Kumaraguru, T.; Sheelu, G. Lipase activity of Lecitase® Ultra: Characterization and applications in enantioselective reactions. Tetrahedron Asymmetry 2009, 20, 2854–2860. [Google Scholar] [CrossRef]
- Xie, W.; Ning, M. Enzymatic transesterification of soybean oil by using immobilized lipase on magnetic nano-particles. Biomass Bioenergy 2010, 34, 890–896. [Google Scholar] [CrossRef]
- Freitas, L.; Paula, A.V.; Santos, J.C.D.; Zanin, G.M.; Castro, H.F.D. Enzymatic synthesis of monoglycerides by esterification reaction using Penicillium camembertii lipase immobilized on epoxy SiO 2 -PVA composite. J. Mol. Catal. B Enzym. 2010, 65, 87–90. [Google Scholar] [CrossRef]
- Gao, W.; Wu, K.; Chen, L.; Fan, H.; Zhao, Z.; Gao, B.; Wang, H.; Wei, D. A novel esterase from a marine mud metagenomic library for biocatalytic synthesis of short-chain flavor esters. Microb. Cell Factories 2016, 15, 41. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Tan, T.; Zhang, H.; Feng, W. Analysis of the conformational stability and activity of Candida antarctica lipase B in organic solvents: Insight from molecular dynamics and quantum mechanics/simulations. J. Biol. Chem. 2010, 285, 28434–28441. [Google Scholar] [CrossRef]
- Alemdar, A.; Sain, M. Isolation and characterization of nanofibers from agricultural residues: Wheat straw and soy hulls. Bioresour. Technol. 2008, 99, 1664–1671. [Google Scholar] [CrossRef]
- Wang, W.F.; Li, T.; Qin, X.L.; Ning, Z.X.; Yang, B.; Wang, Y.H. Production of lipase SMG1 and its application in synthesizing diacylglyecrol. J. Mol. Catal. B Enzym. 2012, 77, 87–91. [Google Scholar] [CrossRef]
- Cai, X.H.; Jing, M.; Wei, D.Z.; Lin, J.P.; Wei, W. Functional expression of a novel alkaline-adapted lipase of Bacillus amyloliquefaciens from stinky tofu brine and development of immobilized enzyme for biodiesel production. Antonie Van Leeuwenhoek 2014, 106, 1049–1060. [Google Scholar] [CrossRef]
- Dong, F.; Li, L.; Lin, L.; He, D.; Chen, J.; Wei, W.; Wei, D. Transesterification Synthesis of Chloramphenicol Esters with the Lipase from Bacillus amyloliquefaciens. Molecules 2017, 22, 1523. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ion | Residual | Activity (%) a | ||||
|---|---|---|---|---|---|---|
| (1 mM) | (5 mM) | (10 mM) | ||||
| F b | L c | F b | L c | F b | L c | |
| None | 100 ± 0.0 | 100 ± 0.0 | 100 ± 0.0 | 100 ± 0.0 | 100 ± 0.0 | 100 ± 0.0 |
| Mg2+ | 105 ± 2.1 | 108 ± 1.9 | 103 ± 1.9 | 104 ± 2.1 | 88.2 ± 1.9 | 99.8 ± 1.7 |
| Zn2+ | 96.1 ± 3.7 | 97 ± 2.0 | 85.1 ± 2.7 | 87 ± 2.0 | 68.5 ± 3.5 | 70 ± 1.2 |
| Co2+ | 92.7 ± 2.9 | 100 ± 1.7 | 81.8 ± 3.5 | 96 ± 2.5 | 71.4 ± 2.4 | 74 ± 1.5 |
| Ni2+ | 91.5 ± 2.3 | 92 ± 2.5 | 90.6 ± 2.4 | 91 ± 2.3 | 83.2 ± 2.0 | 87 ± 2.2 |
| Fe2+ | 92.8 ± 3.4 | 93 ± 1.8 | 83.4 ± 2.6 | 84 ± 1.7 | 68.0 ± 1.7 | 76 ± 1.6 |
| Detergent | Residual | Activity (%) a | ||
|---|---|---|---|---|
| 1% | 10% | |||
| F b | L c | F b | L c | |
| None | 100 ± 0.0 | 100 ± 0.0 | 100 ± 0.0 | 100 ± 0.0 |
| Tween20 | 94.0 ± 2.1 | 95 ± 1.9 | 40.5 ± 1.9 | 41 ± 1.1 |
| Tween60 | 94.5 ± 1.6 | 96 ± 2.1 | 45.6 ± 1.8 | 47 ± 1.9 |
| Tween80 | 94.8 ± 1.8 | 95 ± 2.3 | 53.3 ± 2.1 | 55 ± 2.1 |
| SDS | 18.9 ± 1.2 | 20 ± 1.7 | 0 | 0 |
| TritonX-100 | 36.1 ± 3.7 | 38 ± 2.9 | ND d | 8 ± 0.9 |
| Organic Solvents | LogP a | Residual | Activity (%) b |
|---|---|---|---|
| F b | L c | ||
| Control | 100 ± 1.2 | 100 ± 0.9 | |
| 1, 4-dioxane | −1.0 | 12.6 ± 0.7 | 80.2 ± 2.9 |
| Ethanol | −0.24 | 52.5 ± 1.8 | 92.5 ± 1.6 |
| Acetone | −0.23 | 25.0 ± 1.1 | 93.8 ± 2.6 |
| Acetonitrile | −0.15 | ND d | 92.0 ± 1.5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, F.; Tang, X.; Yang, X.; Lin, L.; He, D.; Wei, W.; Wei, D. Immobilization of a Novel ESTBAS Esterase from Bacillus altitudinis onto an Epoxy Resin: Characterization and Regioselective Synthesis of Chloramphenicol Palmitate. Catalysts 2019, 9, 620. https://doi.org/10.3390/catal9070620
Dong F, Tang X, Yang X, Lin L, He D, Wei W, Wei D. Immobilization of a Novel ESTBAS Esterase from Bacillus altitudinis onto an Epoxy Resin: Characterization and Regioselective Synthesis of Chloramphenicol Palmitate. Catalysts. 2019; 9(7):620. https://doi.org/10.3390/catal9070620
Chicago/Turabian StyleDong, Fengying, Xudong Tang, Xiaohui Yang, Lin Lin, Dannong He, Wei Wei, and Dongzhi Wei. 2019. "Immobilization of a Novel ESTBAS Esterase from Bacillus altitudinis onto an Epoxy Resin: Characterization and Regioselective Synthesis of Chloramphenicol Palmitate" Catalysts 9, no. 7: 620. https://doi.org/10.3390/catal9070620