Titanium-Dioxide-Based Visible-Light-Sensitive Photocatalysis: Mechanistic Insight and Applications
Department of Applied Chemistry, Faculty of Engineering, Osaka Institute of Technology, 5-16-1 Omiya, Asahi-ku, Osaka 535-8585, Japan
Catalysts 2019, 9(2), 201; https://doi.org/10.3390/catal9020201
Submission received: 15 January 2019
/
Revised: 12 February 2019
/
Accepted: 14 February 2019
/
Published: 22 February 2019
(This article belongs to the Special Issue Emerging Trends in TiO2 Photocatalysis and Applications)
Abstract
:Titanium dioxide (TiO2) is one of the most practical and prevalent photo-functional materials. Many researchers have endeavored to design several types of visible-light-responsive photocatalysts. In particular, TiO2-based photocatalysts operating under visible light should be urgently designed and developed, in order to take advantage of the unlimited solar light available. Herein, we review recent advances of TiO2-based visible-light-sensitive photocatalysts, classified by the origins of charge separation photo-induced in (1) bulk impurity (N-doping), (2) hetero-junction of metal (Au NPs), and (3) interfacial surface complexes (ISC) and their related photocatalysts. These photocatalysts have demonstrated useful applications, such as photocatalytic mineralization of toxic agents in the polluted atmosphere and water, photocatalytic organic synthesis, and artificial photosynthesis. We wish to provide comprehension and enlightenment of modification strategies and mechanistic insight, and to inspire future work.
1. Introduction
Titanium dioxide (TiO2) is one of the most practical and prevalent photo-functional materials, since it is chemically stable, abundant (Ti: 10th highest Clarke number), nontoxic, and cost-effective. In recent years, a great deal of attention has been directed towards TiO2 photocatalysis for useful applications such as photocatalytic mineralization of toxic agents in the polluted atmosphere and water, photocatalytic organic synthesis, and artificial photosynthesis [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20].
The TiO2 involving Ti3+ sites that are oxygen-deficient at the impurity level exhibits n-type semiconductor. The photocatalytic activities of TiO2 strongly depend on crystal structures (anatase, brookite, and rutile), crystallinity, crystalline plane, morphology, particle sizes, defective sites, and surface OH groups. The valence band (V.B.) and conduction band (C.B.) of TiO2 consist of O 2p and Ti 3d orbitals, respectively, and their band gap (forbidden band) is circa ~3.0–3.2 eV (~410–380 nm). Photo-irradiation (hv > 3.2 eV) of the TiO2 photocatalyst leads to band gap excitation, resulting in charge separation of electrons into the C.B. and the holes in the V.B. These photo-formed electrons and holes simultaneously work as electron donors and acceptors, respectively, on the photocatalyst surface, thus enabling the photocatalytic reactions. Details are given in other articles and reviews [21,22,23,24,25]. UV light reaching the earth surface represents only a very small fraction (4%) of the solar energy available. Therefore, many researchers have endeavored to design several types of visible-light-responsive photocatalyst. In particular, TiO2-based photocatalysts operating under visible light should be urgently designed and developed, in order to take advantage of the unlimited solar light available.
In the late 1990s, Anpo et al. first reported that TiO2 doped with Cr, V, and Fe cations by ion implantation operates under visible light irradiation. They exhibited red shift of the band-edge of the TiO2, resulting in decomposition of NO into N2, O2, and N2O [26]. This work accelerated subsequent works for the design and development of visible-light-responsive photocatalysts. Recently, much attention has been paid to visible-light-responsive TiO2 prepared by: doping with nitrogen (N), carbon (C), and sulfur (S) ions etc.; surface plasmonic effects with Au or Ag nanoparticles (NPs); the interfacial surface complex (ISC); coupling with visible-light-sensitive hetero-semiconductors (cadmium sulfide, carbon nitride etc.); and dye-sensitized photocatalysts. In fact, some photocatalysts are considered to work under similar principles.
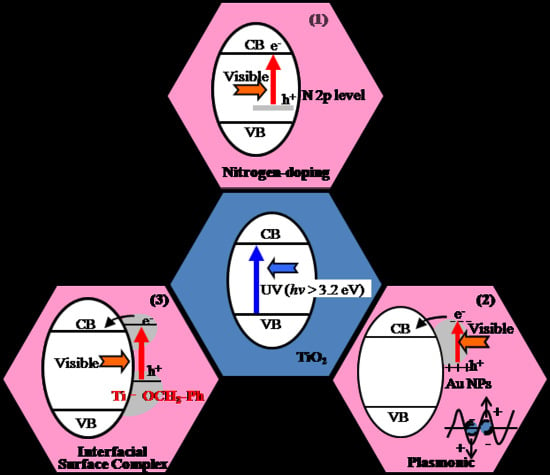
Along these backgrounds, this review focuses on the recent advances of the visible-light-sensitive TiO2 photocatalyst. These advances have been classified by the origin of charge separation photo-induced in (1) the bulk impurity (N-doping), (2) hetero-junction of metal (Au NPs), and (3) the interfacial surface complex (ISC) (See Figure 1). They have been well characterized by several spectroscopic techniques, and applied for mineralization of volatile organic compounds (VOC), water splitting to produce H2, and fine organic synthesis.
2. Nitrogen-doped TiO2 Photocatalysts
In 1986, Sato and co-workers first explored the photocatalytic activity of nitrogen-doped TiO2 (N-doped TiO2) photocatalysts for the oxidation of gaseous ethane and carbon monoxide [27]. They found that N-doped TiO2 photocatalyst exhibited a superior photocatalytic activity to pure TiO2 under visible light irradiation. Later, in 2001, Asahi et al. demonstrated visible-light-induced complete photo-oxidation of gaseous CH3CHO (one of VOCs) to CO2 with an N-doped TiO2 photocatalyst [28]. In this section, fundamental synthetic routes, characterizations, and application of photocatalytic reactions are highlighted.
2.1. Synthesis of N-doped TiO2 Photocatalyst
N-doped TiO2 was prepared by employing several procedures and materials. Details are given in Reference [13]. Preparation methods for N-doped TiO2 photocatalysts can be classified into two categories: dry processes and wet processes.
2.1.1. Dry Processes
Typically, N-doped TiO2 powder can be prepared by the nitrification of TiO2 in an ammonia (NH3) gas flow at high temperature [28,29]. The amount of N doping into the TiO2 can be controlled by annealing temperatures in the range of 550−600 °C under an NH3 flow. However, a large number of O vacancies are introduced into the N-doped TiO2 with increasing annealing temperature, since the NH3 decomposes into N2 and H2 at high temperature, and TiO2 is simultaneously reduced by H2 [30]. Figure 2 shows schematics of N-doping into TiO2, accompanied by the formation of oxygen vacancies to exhibit the n-type semiconductor.
2.1.2. Wet Processes
A sol-gel method can be employed for the preparation of N-doped TiO2 powder. Typically, NH3 aq. (NH4OH) is added to a solution of titanium (IV) isopropoxide (TTIP) [31,32,33] to form titanium hydroxide involving N-species. The precipitate was dried, followed by calcination at ~400–450 °C in air to obtain a yellowish TiO2 powder.
2.2. N-states in N-doped TiO2
One of the major concerns is to understand the physico-chemical nature of the N species in N-doped TiO2, which are responsible for the visible light sensitivity. They were characterized by density functional theory (DFT) calculations, X-rap photoelectron spectroscopy (XPS), Ultraviolet-visible (UV-vis) and electron paramagnetic resonance (EPR) spectroscopy.
2.2.1. DFT Calculations
DFT calculations demonstrated the electronic structures of the N-doped TiO2 photocatalyst (see Figure 3). The substitution of N with lattice O of the N-doped TiO2 exhibits band gap narrowing (circa 0.1 eV) caused by mixing orbitals of N 2p with O 2p, resulting in the negative shift of the valence band edge. On the other hand, the interstitial N is localized to impurity states (N 2p levels) above the V.B. (circa 0.7 eV) in the mid-band gap. Therefore, the oxidation power of photo-induced holes on the N 2p is lower than on the O 2p in the TiO2 lattice.
2.2.2. XPS Spectra
XPS analysis can confirm the oxidative states of the N species and bonding states in the N-doped TiO2 (See Figure 4I). N 1s XPS peaks at a binding energy in the range of ~396–400 eV showed different oxidative states of the N species. By the combination of the DFT calculations [31], it was identified that the N 1s XPS peaks at ~396–397 eV are due to the substitution of N with the lattice O of TiO2 [13,34], while those at ~399–400 eV are due to the interstitial N in the form of NOx or NHx [13,31,35,36].
2.2.3. Optical Properties
The UV-vis absorption spectra of the N-doped TiO2 are shown in Figure 4II. The N-doped TiO2 with the substitution of N exhibited band gap narrowing from 3.1 to 2.8 eV. On the other hand, the N-doped TiO2 prepared by the sol-gel method exhibited visible light absorption up to 540 nm (2.3 eV), due to the electronic transition from localized N doping level to the C.B. of the TiO2, while band-narrowing was not observed. These results are in good agreement with the DFT calculations.
2.2.4. Electron Paramagnetic Resonance (EPR) Spectra
N species in the N-doped TiO2 are present at either diamagnetic (N−) or paramagnetic (N•) bulk centers, which are responsible for the visible light sensitivity [31,37]. The EPR measurements can detect the paramagnetic (N•) bulk centers (see Figure 5). One type, of three lines with a hyperfine tensor (g = 2.006 and A = 32.0 G) splitting by nuclear spin of nitrogen (I = 1), was observed. The signal intensity of N• radicals increased when the light was turned on, while the signal intensity significantly decreased when the light was turned off. In general, the paramagnetic interaction between N species and O2 makes EPR signals disappear. However, they were remarkably enhanced in the presence of O2 under λ > 420 nm, while its signal intensity still remained to some extent even after the light was turned off. These results suggest that N-species are located in bulk inside the TiO2, and visible light irradiation of the N-doped TiO2 exhibits effective charge separation to form holes (N• radicals) and electrons, which participate in the oxidation and reduction of reactant molecules, respectively.
2.2.5. Photo-Electrochemical Properties
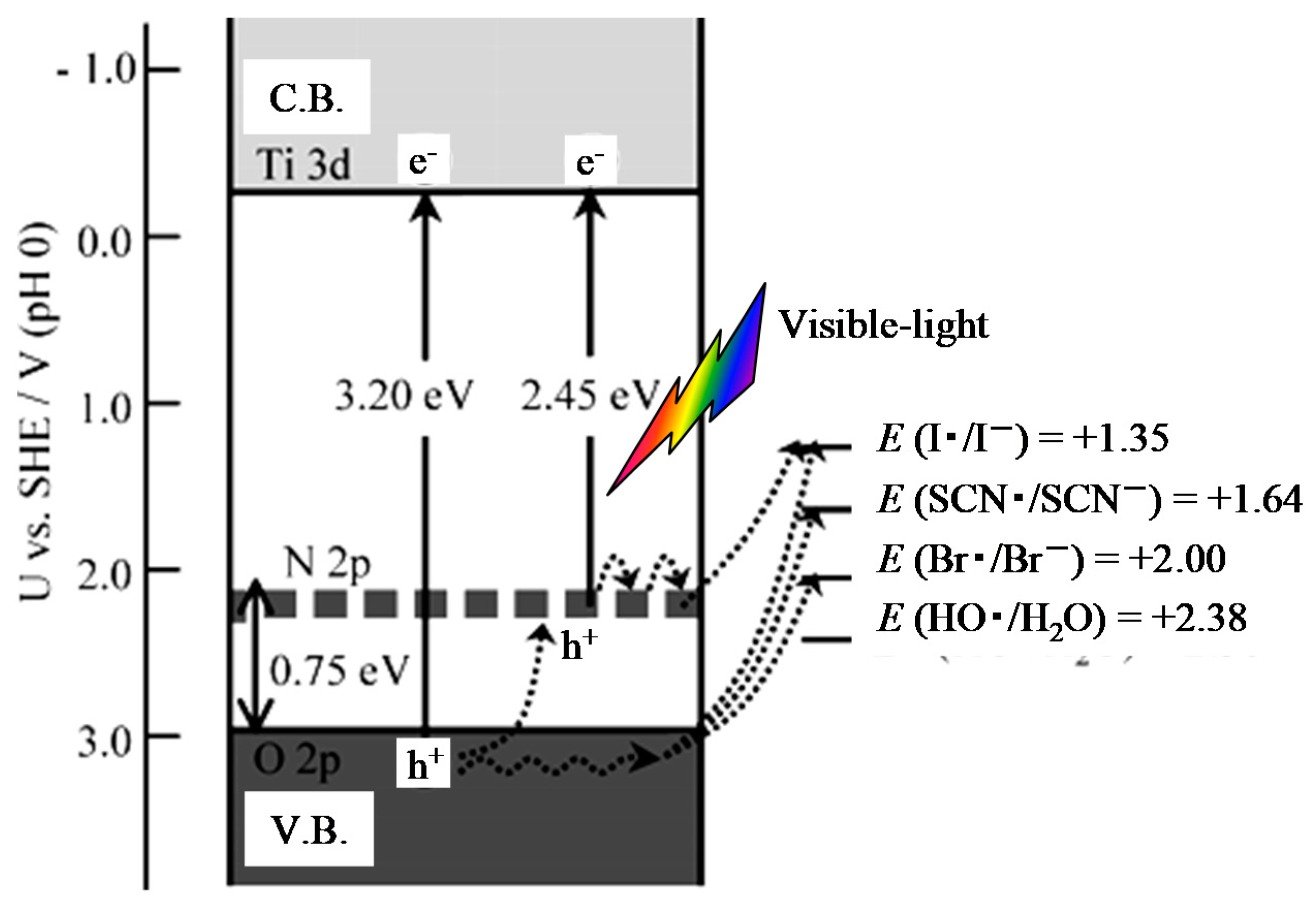
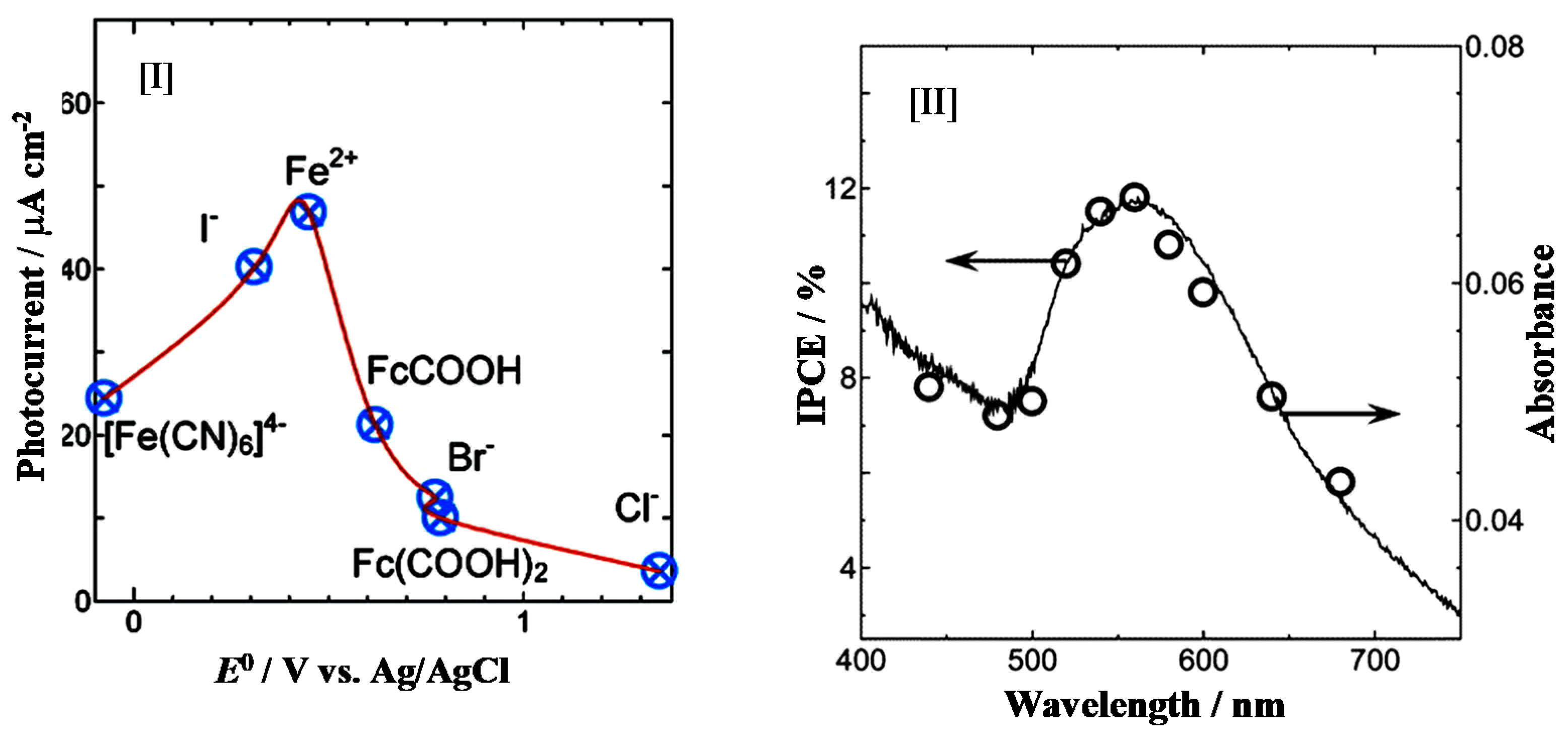
Nakamura et al. investigated the photo-electrochemical oxidation power of the N-doped TiO2 by employing several electron donors [38]. Figure 6 shows that the photo-induced hole on the N 2p level can directly oxidize only I− ions under visible light illumination, while I−, SCN−, Br−, and H2O are oxidized by the hole on the V.B. under UV light illumination. Therefore, the oxidation power of the holes induced on the N 2p level is lower than that of those on the O 2p on the V.B. Tang et al. studied the dynamics of photogenerated electrons and holes on the N-doped TiO2 using transient absorption spectroscopy [39]. They concluded that the lack of activity of nanocrystalline N-doped TiO2 film for photocatalytic water oxidation is due to rapid electron–hole recombination.
On the other hand, Higashimoto et al. investigated the photo-electrochemical reduction power of the N-doped TiO2 (see Figure 7) [33]. When the N-doped TiO2 was photo-excited under visible light irradiation, the photo-induced electrons were accumulated on the oxygen vacancies of TiO2. Subsequently, when various kinds of redox species as electron acceptors were introduced into the photo-charged N–TiO2, the accumulated electrons could reduce O2 molecules, Pt4+, Ag+, and Au3+ ions, but not MV2+, H+, and Cu2+ ions. In principle, the N-doped TiO2 has the potential to reduce H+/H2, but many oxygen vacancies involved in the bulk TiO2 could influence the drastic charge recombination. In particular, photo-induced electrons trapped at the oxygen vacancies (mainly γ region) could reduce O2 molecules to form such active oxygen species as hydrogen peroxide (H2O2), resulting in further oxidation of organic substrates.
2.3. Application to Photocatalytic Decomposition of Volatile Organic Compounds (VOC)
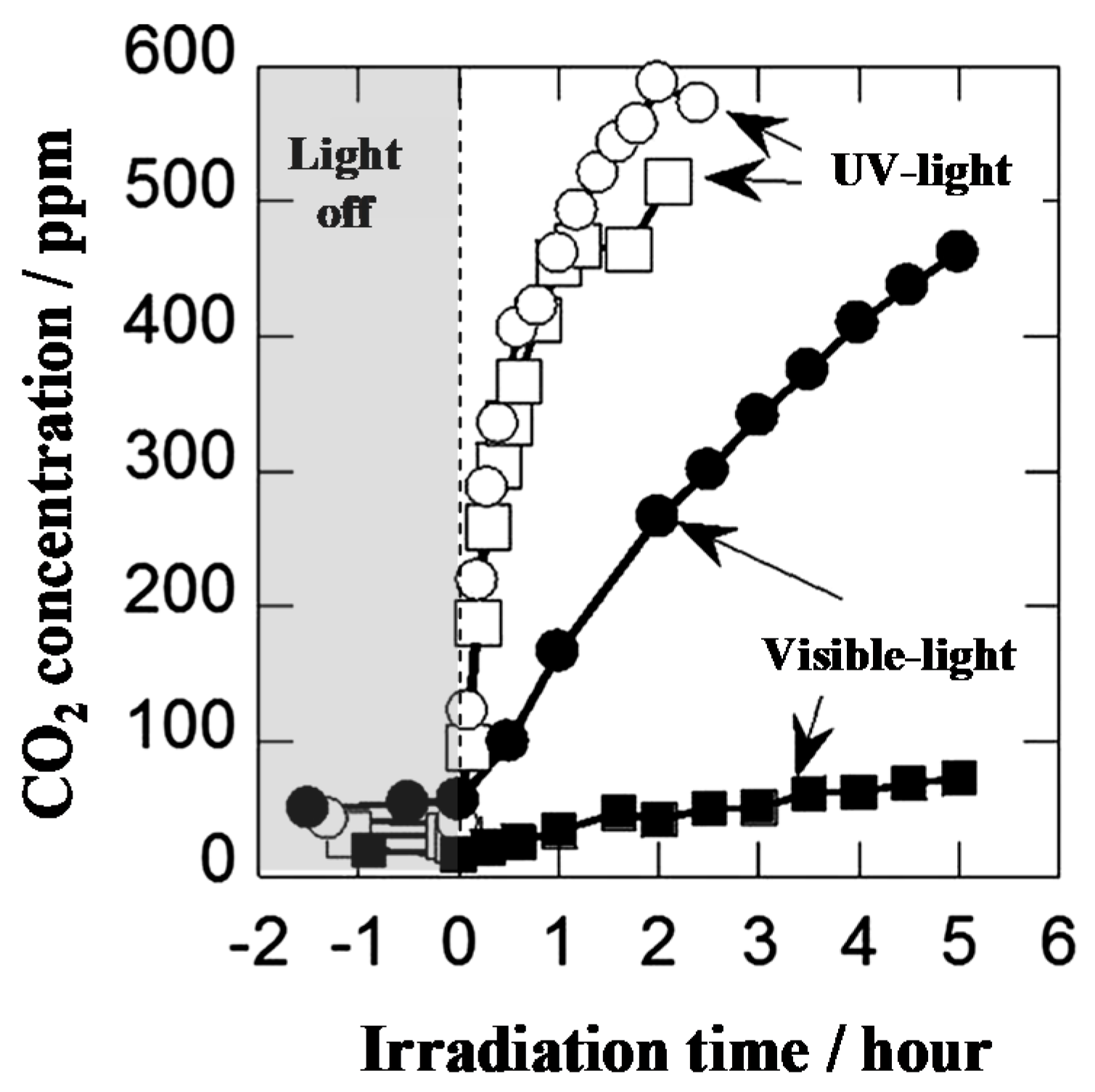
Time profile for the photocatalytic decomposition of gaseous acetaldehyde on the N-doped TiO2 is shown in Figure 8. The N-doped TiO2 exhibited photocatalytic activity 5 times greater than TiO2 under visible light irradiation, while they exhibited similar activities under UV light irradiation [28].
Table 1 shows that the N-doped TiO2 exhibited photocatalytic activity for the decomposition of several kinds of VOC into CO2 under visible light irradiation (λ > 420 nm). It was observed that the N-doped TiO2 exhibited photocatalytic activity for the decomposition of aldehydes, but little activity for alcohol, acid, ketone, and halogene compounds. The vanadium species was deposited on the N-doped TiO2 (VCl3/N-doped TiO2) by impregnation method. As shown in Table 1, VCl3/N-doped TiO2 showed higher photocatalytic activity for the decomposition of all VOC, in particular, acetic acid or acetone by ~13–16 times more than N-doped TiO2. Therefore, it was confirmed that vanadium species worked as the effective co-catalyst.
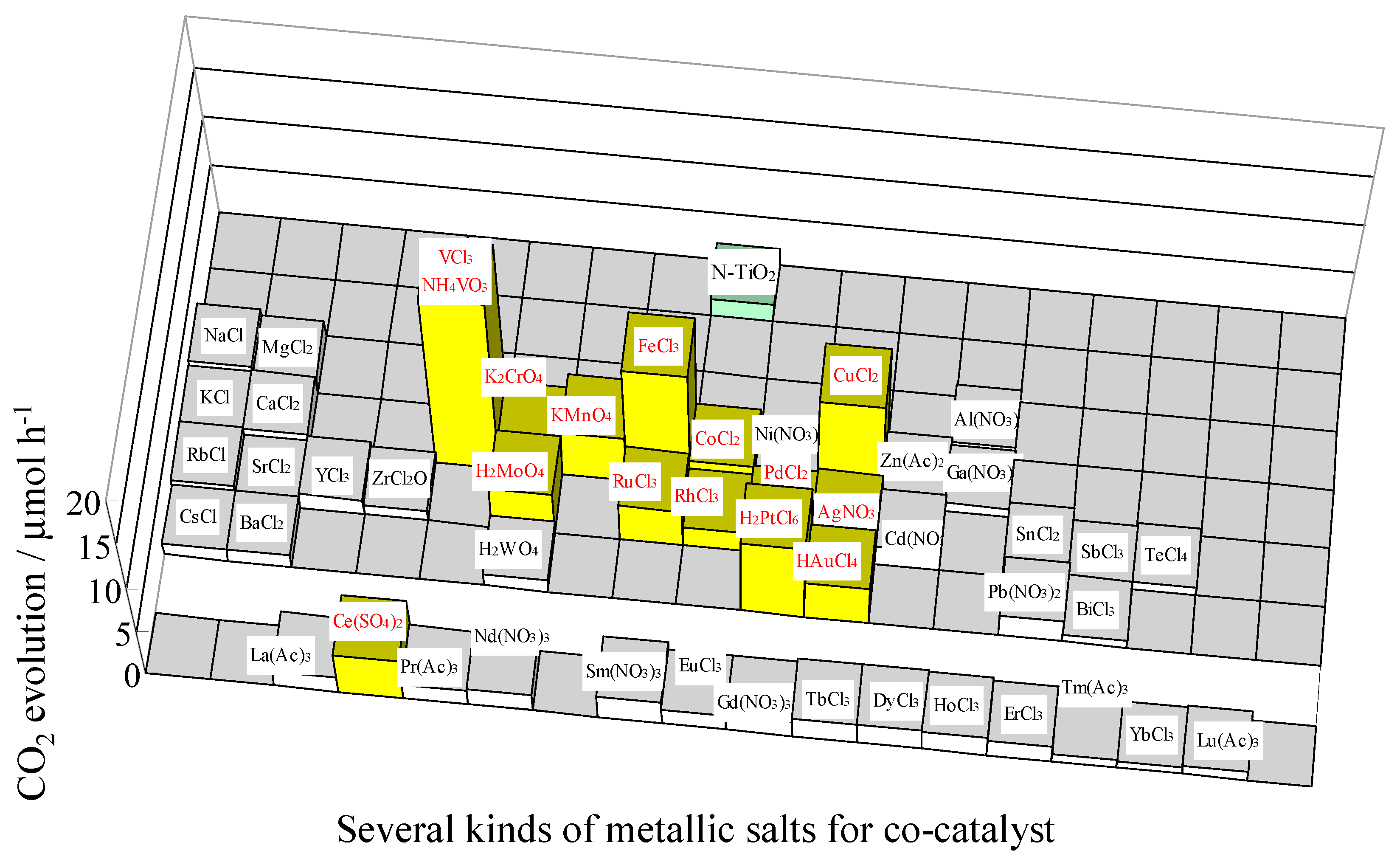
Furthermore, effects of co-catalysts (48 metal ions using nitrate, sulfate, chloride, acetate, and oxide precursors) deposited on the N-doped TiO2 for the photocatalytic activities were examined (See Figure 9) [40]. The bars marked in yellow exhibited higher photocatalytic activities than the N-doped TiO2 by itself. In particular, N-doped-TiO2-deposited Cu, Fe, V, and Pt oxides exhibited high photocatalytic activities. The local structures of the co-catalysts were characterized by XPS. It was observed that Cu loaded N-doped TiO2 involves cuprous oxide (Cu2O) or Cu hydroxides, Fe loaded N-doped TiO2 involves clusters containing Fe–O bonds or Fe2+ hydroxide [41], and Pt loaded N-doped TiO2 involves Pt4+/Pt2+ species [36]. The redox potentials of co-catalysts such as V (+IV/+V), Fe (+II/+III), Cu (+I/+II), and Pt (+III/+IV) were in the range of circa +0.6 to +1.0 V vs. SHE, while the multi-electron reduction of O2 leads to the formation of active oxygen species via O2 + 2H+ + 2e− / H2O2 (E0 = +0.687 V vs. SHE). Therefore, the co-catalysts, such as Pt, Fe, Cu, and V species, enhance the photocatalytic activity due to the effective electron transfer to O2 (O2 reduction), resulting in the formation of active oxygen species.
2.4. C3N4-Modified TiO2 Compared with N-doped TiO2
Several nitrogen sources such as urea, cyanamid, cyanuric acid, and melamine were employed for the preparation of N-containing TiO2 photocatalyst, i.e., the TiO2 surface is modified with polymerized carbon nitride (C3N4) [42,43,44,45,46,47,48,49,50,51]. The structures of the C, N-species strongly depend on their concentrations. If the C, N species are present in only a small amount, they act as a molecular photosensitizer. At higher amounts they form a C3N4 crystalline semiconductor, which chemically binds to TiO2. The C3N4–TiO2 was systematically synthesized by thermal condensation of cyanuric acid on the TiO2 surface [51]. In fact, H2 was evolved from TEA aq. on the C3N4–TiO2 photocatalyst under visible light irradiation, while the N-doped TiO2 did not exhibit H2 production. From characterization of C3N4–TiO2 by Fourier transformed-infrared (FT-IR), XPS, electrochemical measurements, and DFT calculations, the band structures and photo-induced charge separation mechanisms were demonstrated (Figure 10). The C3N4–TiO2 was found to exhibit photo-induced charge separation through the hetero-coupling of semiconductors between C3N4 and TiO2 on the surface. On the other hand, N-doped TiO2 was photo-sensitized by bulk impurity of the N-doping. It can be assumed that many oxygen vacancies promoted the charge recombination, resulting in weak reduction power in the N-doped TiO2.
3. Plasmonic Au NPs Modified TiO2
3.1. What Is Localized Surface Plasmon Resonance (LSPR)?
Localized surface plasmon resonance (LSPR) is an optical phenomenon generated by light when it interacts with conductive nanoparticles (NPs) that are smaller than the incident wavelength. The LSPR is induced by the collective oscillations of delocalized electrons in response to an external electric field. The resonance wavelength strongly depends on the size and shape of the NPs, the interparticle distance, and the dielectric property of the surrounding medium. The Au and Ag NPs exhibit unique plasmon absorption [52,53]. The plasmonic Ag NPs are considered to be unstable under illumination, and could be applicable to multi-colored rewritable devices. In this section, we focused on stable plasmonic Au NPs exploited for a visible-light-sensitive photovoltaic fuel cell or photocatalyst [54,55].
3.2. Preparation and Characterization of Au–TiO2 Photocatalyst
3.2.1. Photodeposition (PD) Methods
By using the photocatalysis of TiO2, metallic Au was deposited on the TiO2 surface, accompanied by the oxidation of methanol [56,57] or ethanol [58]. Typically, TiO2 powder was suspended in a 50 vol. % aqueous methanol in the presence of HAuCl4·6H2O, purged of air with argon. The suspension was photoirradiated with UV light under magnetic stirring. The temperature of the suspension during photoirradiation was maintained at 298K. The Au/TiO2 photocatalyst was centrifuged, washed with distilled water, dried at 393K, and ground in an agate mortar.
3.2.2. Colloid Photodeposition Operated in the Presence of a Hole Scavenger (CPH)
Colloidal Au NPs were prepared using the method reported by Frens [59]. In brief, mixtures of an aqueous tetrachloroauric acid (HAuCl4) solution and sodium citrate were heated and boiled for 1 h. The color of the solution changed from deep blue to deep red. The citrate plays a role in the reduction of Au ions, and the capping agent in suppressing the aggregation of Au NPs. The suspension of TiO2 in an aqueous solution of colloidal Au NPs and oxalic acid was then photo-irradiated at λ > 300 nm at 298 K under argon (Ar). The solids were recovered, washed, and dried to produce Au–TiO2. Details are given in Reference [60].
3.2.3. Deposition Precipitation (DP) Method
Deposition–precipitation (DP) methods were employed for the deposition of a gold (III) species on the TiO2 surface [61,62]. The [AuCl(OH)3]−, main species present at pH 8, adjusted by NaOH aq., reacts with hydroxyl groups of the TiO2 surface to form a grafted hydroxyl–gold compound. The catalyst was then recovered, filtered, washed with deionized water, and dried. Finally, the powder was calcined at ~473–673 K in air.
3.2.4. Characterization of the Au–TiO2 Photocatalyst
The Au–TiO2 photocatalysts were typically characterized by the transmittance electron microscope (TEM) for the particle sizes, and UV-vis absorption for optical properties (See Table 2).
Kowalska et al. [56,57] reported that Au–TiO2 photocatalysts with different Au particle sizes (~10–60 nm) were prepared by photo-deposition (entry 1). The particle sizes of Au strongly depend on the particle sizes of the TiO2 polycrystalline structure. The top peak of plasmonic absorption was in the range of ~530–610 nm, depending on the particle sizes of the Au NPs. Tanaka and Kominami et al. [60,63,64,65,66] reported unique CPH methods for the preparation of Au-TiO2 (entry 2). The particle sizes were uniformed to be ~12–14 nm, which exhibits plasmonic absorption at ~550–560 nm. Thus, colloidal Au NPs were successfully loaded onto TiO2 without change in the original particle size. Furthermore, the top peak of Au plasmon absorption was found to extend towards 620 nm by simple calcinations of the samples. This phenomenon is due to high contact area between TiO2 and Au NPs without change of particle size [66]. Additionally, Naya et al. [67,68] and Shiraishi et al. [69] employed precipitation deposition methods to deposit small Au NPs (~2–6 nm) on TiO2 (entry 3).
3.3. Application of LSPR of Au–TiO2 to Several Photocatalytic Reactions
Au NPs deposited on TiO2 have been used as visible-light-responsive photocatalysts for several chemical reactions: decomposition of VOCs, selective oxidation of an aromatic alcohol, direct water splitting, H2 formation from sacrificial aqueous solutions, and reduction of organic compounds (see Table 3). Several research groups concluded that photocatalytic activities are induced by LSPR of the Au NPs. Some research indicates that small Au NPs (~5 nm) effectively work for the reactions [61,69]. Tanaka and Kominami et al. suggest that two types of Au particles of different sizes loaded onto TiO2 exhibit different functionalities. That is, the larger Au particles contribute to strong light absorption, and the smaller Au particles act as a co-catalyst for H2 evolution [63].
3.4. Application to a Photovoltaic Fuel Cell Operating under Visible Light Irradiation
The Au–TiO2 films were found to exhibit the behavior of a photovoltaic fuel cell [54,55]. An anodic photocurrent was yielded on the Au−TiO2 film as the visible light was irradiated, while the current was observed neither on a TiO2 film under visible light irradiation, nor on the Au−TiO2 film when the light was turned off. The short-circuit photocurrent density (Jsc) was strongly influenced by kinds of donors, and the photocurrent efficiency was maximized in the presence of Fe2+ ions. Furthermore, the photocurrent action spectra were closely fitted with the absorption spectrum of the Au NPs deposited on the TiO2 film (See Figure 11).
3.5. Mechanisms of Charge Separation
The mechanism for the Au plasmon-induced charge separation is shown in Figure 12. Visible light irradiation generates the photo-excited state of the Au NPs by LSPR. The photo-excited electrons are injected into the C.B. of TiO2, while the holes abstracted electrons from a donor in the solution. The Au NPs behave like an intrinsic semiconductor, and the Fermi levels of Au NPs and TiO2 are leveled out, resulting in the formation of Schottky barrier at Au–TiO2 junctions. This band model seems to be similar with dye-sensitized photo-anodic electrodes.
Recently, Furube et al. studied the plasmon-induced charge transfer mechanisms between Au NPs and TiO2 by means of femtosecond visible pump/infrared probe transient absorption spectroscopy [73]. The electron transfer from the Au NPs to the C.B. of TiO2 was confirmed to occur within 50 fs, and that the electron injection yielded 20–50% upon 550 nm laser excitation.
4. Photo-Induced Interfacial Charge Transfer
4.1. Dye-Sensitized TiO2 Photocatalysis
Dye sensitized TiO2 photocatalysis was studied in the late 1990s. The Ru complex, [Ru(bipy)3]2+ grafted on the TiO2 surface exhibits visible light absorption [74,75]. In this system, the excitation of the Ru complex induces electron transfer via metal–ligand charge transfer (MLCT). The photo-induced electrons are then transferred onto TiO2, resulting in photocatalytic water splitting to produce H2. The platinum-chloride-modified TiO2 system was reported by Kisch et al. [76,77]. Photo-irradiation of Pt(IV) chloride exhibits visible-light absorption to generate the active center, (Pt4+(Cl−)4 + hν → Pt3+Cl0(Cl–)3. The photo-induced electrons are transferred from Pt3+ to C.B. of TiO2 as reductive sites, while the Cl0 work as the oxidative sites, resulting in the redox photocatalytic reactions. Important strategies to develop these types of photocatalysts are to design robust sensitizers adjusted with HOMO-LUMO levels.
4.2. Visible-Light-Responsive TiO2 Photocatalyst Modified by Phenolic Organic Compounds
Strong interaction of phenolic groups in organic compounds with Ti–OH of the TiO2 surface probably forms two types of interfacial surface complexes (ISC, Figure 13I), which exhibits visible light absorption via LMCT. The photocatalysis of the ISC is strongly influenced by the electronic structures of the ISC (Figure 13II): the ISC with EWG exhibits strong oxidizability under visible light irradiation, and it can favorably oxidize the TEA, together with H2 evolution from deaerated TEA aqueous solutions [78]. The visible light response of the ISC is attributed to electronic excitation from the donor levels (0.7 V above V.B.) to the C.B. of TiO2 (see Figure 14). Therefore, the electronic structures of sensitizers strongly influence the photocatalytic activities. Ikeda et al. [79] demonstrated that a TiO2 photocatalyst modified with 1,1′-binaphthalene-2,2′-diol (bn(OH)2) exhibited photocatalytic H2 evolution from deaerated TEA aq. under visible light irradiation. Kamegawa et al. [80] designed a 2, 3-dihydroxynaphthalene (2,3-DN)-modified TiO2 photocatalyst for the reduction of nitrobenzene to aminobenzene under visible light irradiation.
On the other hand, the phenolic compounds were degraded on the TiO2 in the presence of O2 under visible light (λ > 420 nm) illumination, producing Cl– and CO2 [81]. The ISC formed by the interaction of phenolic compounds with TiO2 exhibited self-degradation. It was proposed that an electronic transition occurs from the ISC to the C.B. of TiO2 to form active oxygen species, which also participate in the oxidative degradation of phenolic compounds.
4.3. Interfacial-Surface-Complex-Mediated Visible-Light-Sensitive TiO2 Photocatalysts
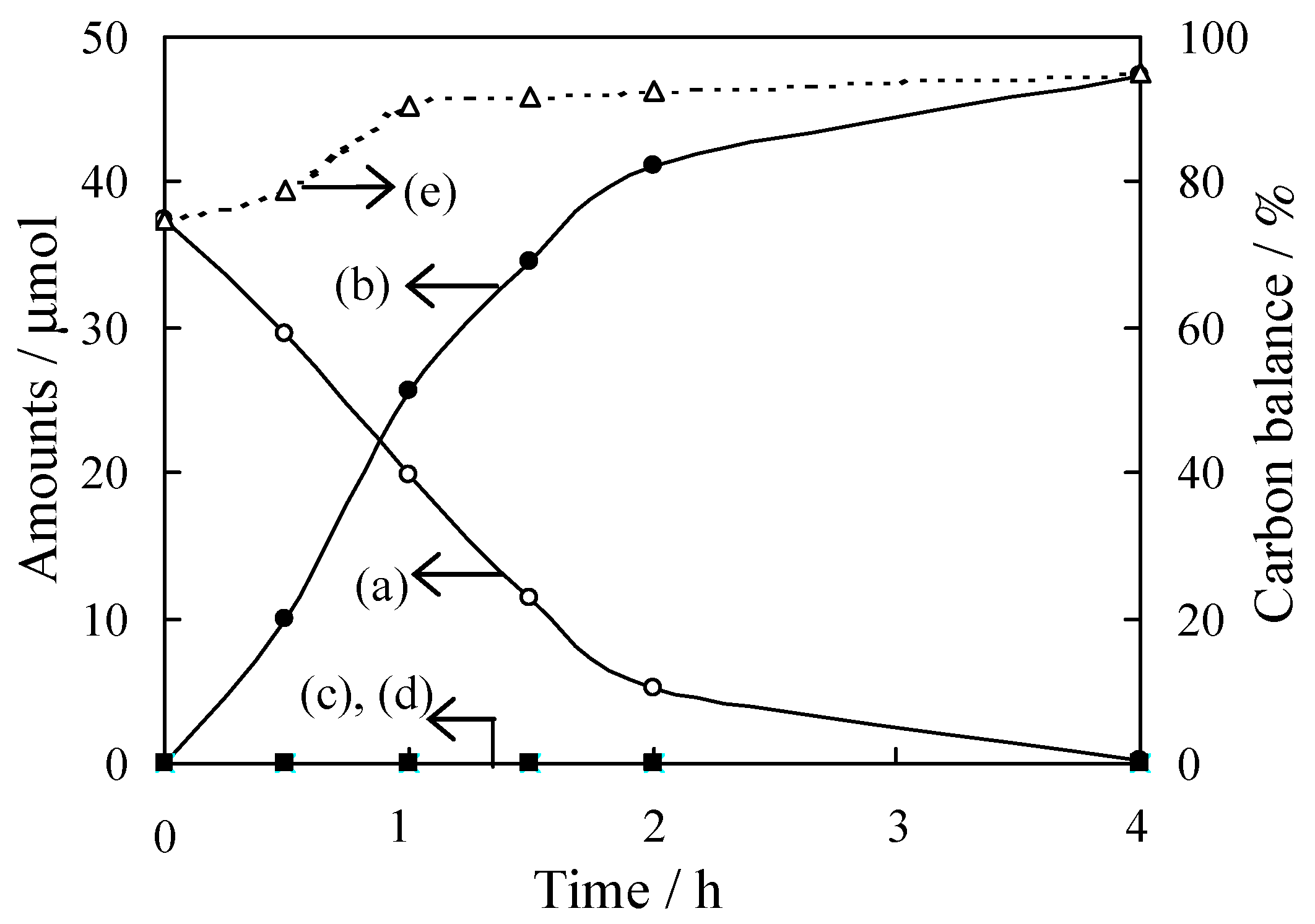
The interfacial surface complex (ISC)-mediated visible-light-sensitive TiO2 photocatalyst was applied to selective oxidation of several aromatic alcohols [82,83,84,85,86,87,88]. Unlike to the ISC in Figure 14, reactant molecules adsorbed onto the TiO2 surface (ISC) is activated under visible-light irradiation, and they are converted into products. Figure 15 shows reaction time profiles for the oxidation of benzyl alcohol in an acetonitrile solution suspended with TiO2 photocatalyst in the presence of O2 under visible light irradiation (λ > 420 nm). This reaction does not proceed without TiO2 or irradiation. It was found that the amount of benzyl alcohol decreased with an increase in the irradiation time, while the amount of benzaldehyde increased. Neither benzoic acid nor CO2 were formed as oxidative products. The yield of benzaldehyde reached circa 95%, and the carbon balance in the liquid phase was circa 95% after photo-irradiation for 4 h.
Photocatalytic oxidation of benzyl alcohol and its derivatives into corresponding aldehydes was carried out with TiO2 under visible light irradiation. Benzyl alcohol and its derivatives substituted by –OCH3, –Cl, –NO2, –CH3, –CF3, and –C(CH3)3 groups were successfully converted to corresponding aldehydes with a high conversion and high selectivity on TiO2, while no other products were observed (See Table 4). However, the phenolic compound (entry 9) was deeply oxidized, since it strongly adsorbed on the TiO2 surface [82].
4.3.1. What Is the Origin of the Visible Light Response?
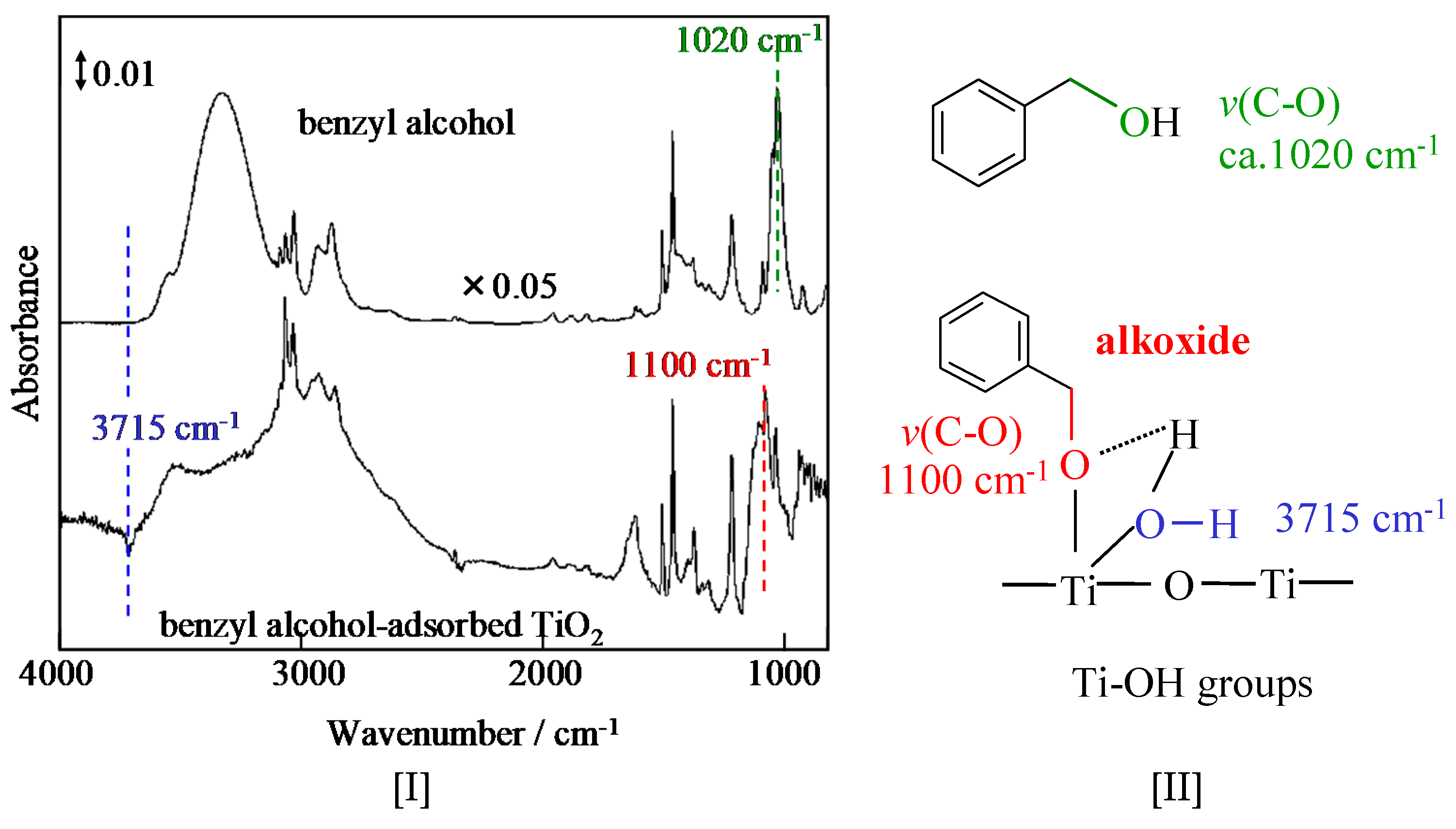
The interaction of benzyl alcohol with TiO2 was analyzed by FT-IR spectroscopy (See Figure 16). Characteristic features of the ISC are as follows: (i) a remarkable downward negative band at 3715 cm−1 attributed to the O–H stretching of the terminal OH group; (ii) a new band appeared at circa 1100 cm−1, which is attributed to the C–O stretching of the alkoxide species formed by the interaction of benzyl alcohol with TiO2, while that of benzyl alcohol by itself is 1020 cm−1.
When the TiO2 was treated by diluted HF (aq), the IR band at 3715 cm−1 on the HF–TiO2 drastically decreased, while the photocatalytic activity significantly decreased. The active sites were confirmed to be alkoxide by the interaction of benzyl alcohol with the terminal OH groups of TiO2.
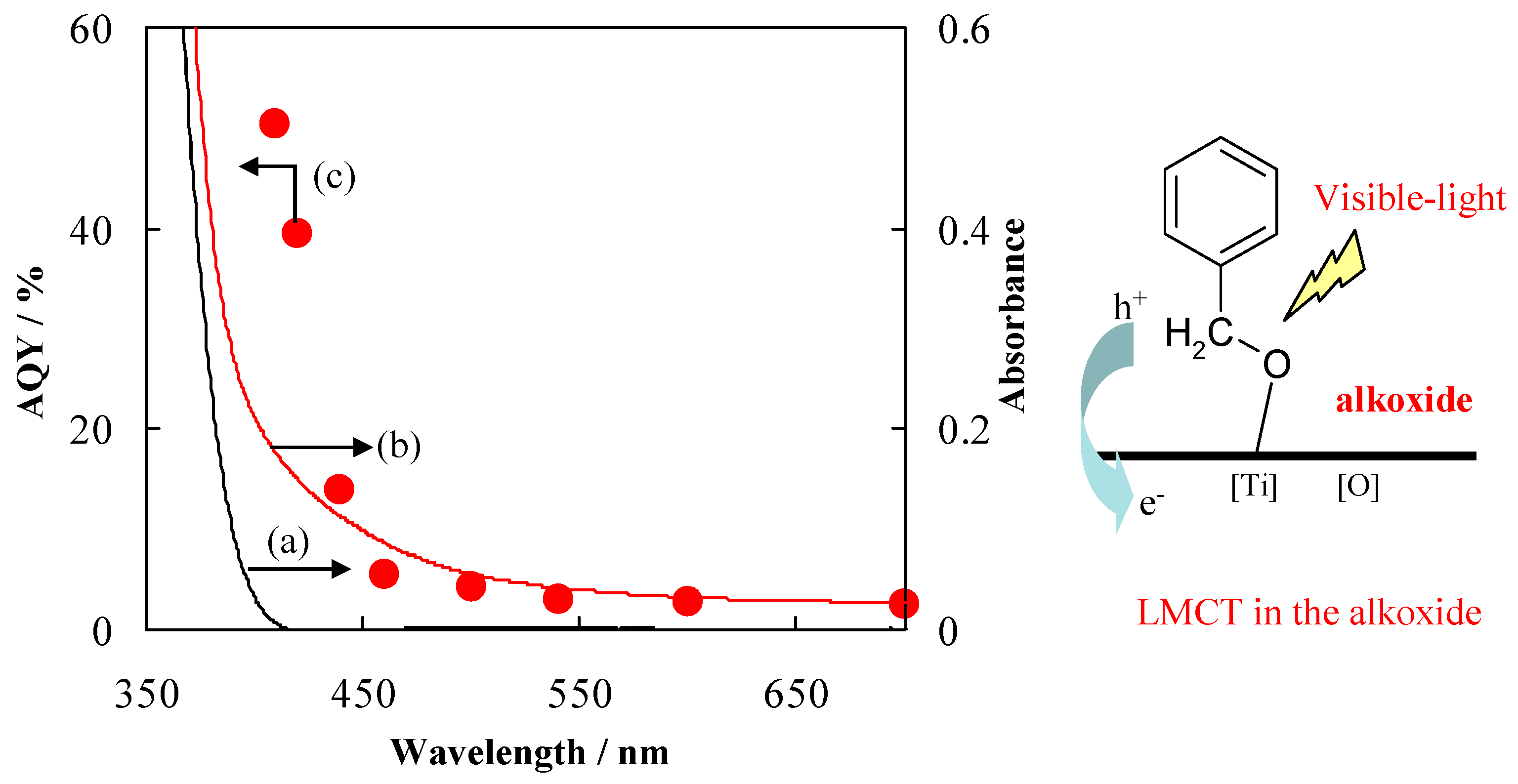
TiO2 by itself exhibited absorption only in the UV region, which is attributed to the charge transition from V.B. to C.B. When the benzyl alcohol was adsorbed on TiO2, absorption in the visible region could be observed. This absorption in the visible light region is assignable to the ISC through the LMCT (See Figure 17). The action spectra of apparent quantum yield (AQY) plots were fitted with the photo-absorption of TiO2-adsorbed benzyl alcohol, suggesting that visible light absorption directly participated in the photocatalytic reactions.
DFT calculations [87] indicated the interaction of benzyl alcohol with surface hydroxyl groups on the TiO2 surface, resulting in the formation of alkoxide species. The electron density contour maps for the alkoxide species are shown in Figure 18. The orbital #212 at −0.80 eV forms the V.B. of TiO2, while #218 at +2.25 eV forms the C.B. One type of surface state consisting of the orbital (#215) originates with the alkoxide species ([Ti]–O–CH2–ph) hybridized with the O2p AOs in the V.B. of the TiO2. The energy gap between #215 and #218 (2.8 eV) was confirmed to be the origin for the visible light response.
4.3.2. What Makes the High Selectivity for the Photocatalytic Reactions?
It was observed that benzyl alcohol is adsorbed on TiO2 more favorably than benzaldehyde in a mixture of benzyl alcohol and benzaldehyde under dark conditions. This result indicates that the interaction between benzaldehyde and TiO2 is fairly weak. According to DFT calculations [87,88], the interaction of benzyl alcohol with the TiO2 surface formed a hybridized orbital, while benzaldehyde did not form orbital mixing. Therefore, once benzaldehyde was produced by the oxidation of benzyl alcohol, benzaldehyde was immediately released into the bulk solution, and was not oxidized further to benzoic acid or CO2.
4.3.3. Reaction Mechanisms behind the Selective Photocatalytic Oxidation of Benzyl Alcohol
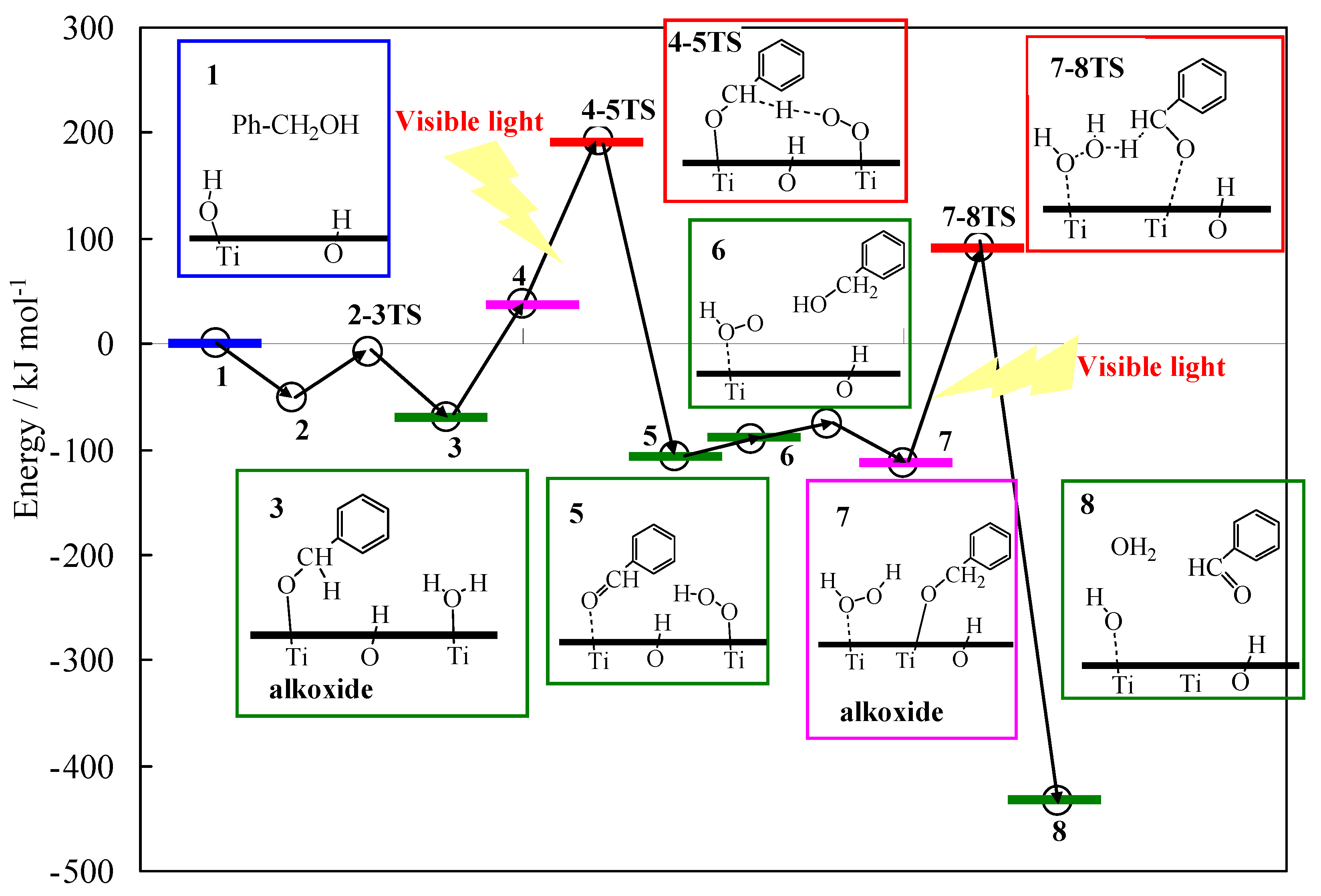
The photocatalytic activities for the oxidation of benzyl alcohol or α, α-d2 benzyl alcohol were investigated. The kinetic isotope effect (KIE) [=kC-H/kCD] was estimated to be 3.9 at 295 K. This result suggests that the process for the α-deprotonation is the rate determining step (RDS) for the overall reaction. From the experimental and theoretical studies by DFT calculations, one of the favorable reaction paths is depicted in Figure 19. When benzyl alcohol interacts with Ti–OH of the TiO2, the alkoxide species (ISC) is formed on a Ti site (3). The ISC was photo-excited under visible light irradiation via LMCT of the ISC, which induces holes (h+) and electrons (e−). Subsequently, the electrons are transferred to O2 to form superoxide anions (the bonding distance between O–O becomes longer), which induces α-deprotonation of the benzyl alcohol (4-5TS). Such hydro-peroxide species would further induce the de-protonation from another benzyl alcohol to form benzaldehyde (7-8TS), resulting in regeneration of the surface terminal OH groups. The consecutive generation of the terminal OH groups would, thus, be one of the key factors for the photocatalytic reactions.
4.4. Photocatalytic Oxidation of Benzyl Amine into Imine
Imines are important intermediates for the synthesis of pharmaceuticals and agricultural chemicals. Selective photocatalytic oxidation of benzyl amine into N-benzylidenebenzylamine takes place in the presence of O2 on the TiO2 at room temperature (Scheme 1) [89,90]. Several kinds of benzylic amines were examined, and they were converted into the corresponding imines, yielding circa 38–94% [89]. The origin of the visible light response is due to formation of amine oxide (ISC) through the interaction of benzylic amine onto the surface of TiO2, and the ISC exhibits electronic transition from the localized N 2p orbitals of the amine oxide (ISC) to the C.B. of TiO2. The photo-induced redox catalysis produces benzaldehyde in the presence of O2. Subsequently, the condensation reaction of benzaldehyde with another benzyl amine forms N-benzylidenebenzylamine under dark conditions.
5. Conclusions
This review focused on some fundamental issues behind the visible-light-sensitive TiO2 photocatalysts, highlighting the bulk and/or surface electronic structures modified by doping with nitrogen anions; plasmonic Au NPs, and interfacial surface complexes (ISC) and their related photocatalysts. Tailoring the interface and bulk properties, including surface band bending, sub-band structure, surface state distribution, and charge separation, significantly reflects on the photocatalysis. We hope that this review has provided some useful contributions for the future design and development of novel photocatalytic systems employing TiO2 as well as non-TiO2 semiconductor materials with nanoscale levels. The applications of such photocatalytic systems could not only convert unlimited solar energy into chemical energy, but also protect our environment, leading to sustainable green chemistry.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| NPs | nanoparticles |
| ISC | interfacial surface complex |
| VOCs | volatile organic compounds |
| V.B. | valence band |
| C.B. | conduction band |
| XPS | X-ray photoelectron spectroscopy |
| EPR | electron paramagnetic resonance |
| UV-vis | Ultraviolet-visible |
| LSPR | localized surface plasmon resonance |
| PD | photodeposition |
| CPH | colloid photodeposition by hole scavenger |
| DP | deposition precipitation |
| TEM | transmittance electron microscope |
| JSC | short-circuit photocurrent |
| IPCE | incident photo to current efficiency |
| DFT | density functional theory |
| MLCT | metal to ligand charge transfer |
| FT-IR | Fourier transformed-infrared |
| KIE | kinetic isotope effect |
| LMCT | ligand to metal charge transfer |
References
- Honda, K.; Fijishima, A. Electrochemical Photolysis of Water at a Semiconductor Electrode. Nature 1972, 238, 37–38. [Google Scholar]
- Inoue, T.; Fujishima, A.; Konishi, S.; Honda, K. Photoelectrocatalytic reduction of carbon dioxide in aqueous suspensions of semiconductor powders. Nature 1979, 277, 637–638. [Google Scholar] [CrossRef]
- Kamat, P.V. Photochemistry on nonreactive and reactive (semiconductor) surfaces. Chem. Rev. 1993, 93, 267–300. [Google Scholar]
- Fox, M.A.; Dulay, M.T. Heterogeneous photocatalysis. Chem. Rev. 1993, 93, 341–357. [Google Scholar]
- Hoffman, M.R.; Martin, S.T.; Choi, W.; Bahnemann, D.W. Environmental applications of semiconductor photocatalysis. Chem. Rev. 1995, 95, 69–96. [Google Scholar] [CrossRef]
- Fujishima, A.; Rao, T.N.; Tryk, A. Titanium dioxide photocatalysis. J. Photochem. Photobio. C Photochem. Rev. 2000, 1, 1–21. [Google Scholar] [CrossRef]
- Anpo, M.; Takeuchi, M. The design and development of highly reactive titanium oxide photocatalysts operating under visible light irradiation. J. Catal. 2003, 216, 505–516. [Google Scholar] [CrossRef]
- Chen, X.; Mao, S.S. Titanium Dioxide Nanomaterials: Synthesis, Properties, Modifications, and Applications. Chem. Rev. 2007, 197, 2891–2959. [Google Scholar] [CrossRef]
- Shiraishi, Y.; Hirai, T. Selective organic transformations on titanium oxide-based photocatalysts. J. Photochem. Photobiol. C Photochem. Rev. 2008, 9, 157–170. [Google Scholar] [CrossRef]
- Palmisano, G.; García-López, E.; Marcì, G.; Loddo, V.; Yurdakal, S.; Augugliaro, V.; Palmisano, L. Advances in selective conversions by heterogeneous photocatalysis. Chem. Commun. 2010, 46, 7074–7089. [Google Scholar] [CrossRef]
- Schneider, J.; Matsuoka, M.; Takeuchi, M.; Zhang, J.; Horiuchi, Y.; Anpo, M.; Bahnemann, D.W. Understanding TiO2 Photocatalysis: Mechanisms and Materials. Chem. Rev. 2014, 114, 9919–9986. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, X.; Jia, Y.; Chen, X.; Han, H.; Li, C. Titanium dioxide-based nanomaterials for: Photocatalytic fuel generations. Chem. Rev. 2014, 114, 9987–10043. [Google Scholar] [CrossRef]
- Asahi, R.; Morikawa, T.; Irie, H.; Ohwaki, T. Nitrogen-doped titanium dioxide as visiblelight-sensitive photocatalyst: Designs, developments, and prospects. Chem Rev. 2014, 114, 9824–9852. [Google Scholar] [CrossRef]
- Lang, X.; Chen, X.; Zhao, J. Heterogeneous visible light photocatalysis for selective organic transformations. Chem. Soc. Rev. 2014, 43, 473–486. [Google Scholar] [CrossRef]
- Sang, L.; Zhao, Y.; Burda, C. TiO2 Nanoparticles as Functional Building Blocks. Chem. Rev. 2014, 114, 9283–9318. [Google Scholar] [CrossRef]
- Nosaka, Y.; Nosaka, A.Y. Generation and Detection of Reactive Oxygen Species in Photocatalysis. Chem. Rev. 2017, 117, 11302–11336. [Google Scholar] [CrossRef]
- Kou, J.; Lu, C.; Wang, J.; Chen, Y.; Xu, Z.; Varma, R.S. Selectivity Enhancement in Heterogeneous Photocatalytic Transformations. Chem. Rev. 2017, 117, 1445–1514. [Google Scholar] [CrossRef]
- Prakash, J.; Sun, S.; Swart, H.C.; Gupta, R.K. Noble metals-TiO2 nanocomposites: From fundamental mechanisms to photocatalysis, surface enhanced Raman scattering and antibacterial applications. Appl. Mater. Today 2018, 11, 82–135. [Google Scholar] [CrossRef]
- Wang, W.; Tadé, M.O.; Shao, Z. Nitrogen-doped simple and complex oxides for photocatalysis: A review. Prog. Mater. Sci. 2018, 92, 33–63. [Google Scholar] [CrossRef]
- Yamashita, H.; Mori, K.; Kuwahara, Y.; Kamegawa, T.; Wen, M.; Verma, P.; Che, M. Single-site and nano-confined photocatalysts designed in porous materials for environmental uses and solar fuels. Chem. Soc. Rev. 2018, 47, 8072–8096. [Google Scholar] [CrossRef]
- Ahmed, A.Y.; Kandiel, T.A.; Oekermann, T.; Bahnemann, D. Photocatalytic Activities of Different Well-defined Single Crystal TiO2Surfaces: Anatase versus Rutile. J. Phys. Chem. Lett. 2011, 2, 2461–2465. [Google Scholar] [CrossRef]
- Tanaka, K.; Capule, M.F.V.; Hisanaga, T. Effect of Crystallinity of TiO2 on Its Photo-catalytic Action. Chem. Phys. Lett. 1991, 187, 73–76. [Google Scholar] [CrossRef]
- Luttrell, T.; Halpegamage, S.; Tao, J.; Kramer, A.; Sutter, E.; Batzill, M. Why is anatase a better photocatalyst than rutile? - Model studies on epitaxial TiO2 films. Sci. Rep. 2014, 4, 4043–4050. [Google Scholar] [CrossRef]
- Gordon, T.R.; Cargnello, M.; Paik, T.; Mangolini, F.; Weber, R.T.; Fornasiero, P.; Murray, C.B. Nonaqueous Synthesis of TiO2 Nanocrystals Using TiF4 to Engineer Morphology, Oxygen Vacancy Concentration, and Photocatalytic Activity. J. Am. Chem. Soc. 2012, 134, 6751–6761. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, C.-C.; Zakaria, R.J.; Ying, Y. Role of Particle Size in Nanocrystalline TiO2-Based Photocatalysts. J. Phys. Chem. B 1998, 102, 10871–10878. [Google Scholar] [CrossRef]
- Anpo, M.; Ichihashi, Y.; Takeuchi, M.; Yamashita, H. Design of unique titanium oxide photocatalysts by an advanced metal ion-implantation method and photocatalytic reactions under visible light irradiation. Res. Chem. Intermed. 1998, 24, 143–149. [Google Scholar] [CrossRef]
- Sato, S. Photocatalytic activity of NOx-doped TiO2 in the visible light region. Chem. Phys. Lett. 1986, 123, 126–128. [Google Scholar] [CrossRef]
- Asahi, R.; Morikawa, T.; Aoki, K.; Taga, Y. Visible-light photocatalysis in nitrogen-doped titanium oxides. Science 2001, 293, 269–271. [Google Scholar] [CrossRef]
- Irie, H.; Watanabe, Y.; Hashimoto, K. Nitrogen-Concentration Dependence on Photocatalytic Activity of TiO2-xNx Powders. J. Phys. Chem. B 2003, 107, 5483–5486. [Google Scholar] [CrossRef]
- Shin, C.; Bugli, G.; Djega-Mariadassou, G. Preparation and characterization of titanium oxynitrides with high specific surface areas. J. Solid State Chem. 1991, 95, 145–155. [Google Scholar] [CrossRef]
- Livraghi, S.; Paganini, M.C.; Giamello, E.; Selloni, A.; Di Valentin, C.; Pacchioni, G. Origin of Photoactivity of Nitrogen-Doped Titanium Dioxide under Visible Light. J. Am. Chem. Soc. 2006, 128, 15666–15671. [Google Scholar] [CrossRef]
- Wang, J.; Zhu, W.; Zhang, Y.; Liu, S. An efficient two-step technique for nitrogen-doped titanium dioxide synthesizing: Visible-light-induced photodecomposition of methylene blue. J. Phys. Chem. C 2007, 111, 1010–1014. [Google Scholar] [CrossRef]
- Higashimoto, S.; Azuma, M. Photo-induced charging effect and electron transfer to the redox species on nitrogen-doped TiO2 under visible light irradiation. Appl. Catal. B Environ. 2009, 89, 557–562. [Google Scholar] [CrossRef]
- Wang, H.; Hu, Y. The Photocatalytic Property of Nitrogen-Doped TiO2 Nanoball Film. Int. J. Photoenergy 2013. [Google Scholar] [CrossRef]
- Diwald, O.; Thompson, T.L.; Zubkov, T.; Goralski, E.G.; Walck, S.D.; Yates, J.T., Jr. Photochemical Activity of Nitrogen-Doped Rutile TiO2(110) in Visible Light. J. Phys. Chem. B 2004, 108, 6004–6008. [Google Scholar] [CrossRef]
- Higashimoto, S.; Ushiroda, Y.; Azuma, M.; Ohue, H. Synthesis, characterization and photocatalytic activity of N-doped TiO2 modified by platinum chloride. Catal. Today 2008, 132, 165–169. [Google Scholar] [CrossRef]
- Higashimoto, S.; Ushiroda, Y.; Azuma, M. Mechanism for enhancement of visible light response on nitrogen-doped TiO2 by modification with vanadium species. J. Nanosci. Nanotechnol. 2010, 10, 246–251. [Google Scholar] [CrossRef]
- Nakamura, R.; Tanaka, T.; Nakato, Y. Mechanism for Visible Light Responses in Anodic Photocurrents at N-Doped TiO2 Film Electrodes. J. Phys. Chem. B 2004, 108, 10617–10620. [Google Scholar] [CrossRef]
- Tang, J.; Cowan, A.J.; Durrant, J.R.; Klug, D.R. Mechanism of O2 production from water splitting: Nature of charge carriers in nitrogen doped nanocrystalline TiO2 films and factors limiting O2 production. J. Phys. Chem. C 2011, 115, 3143–3150. [Google Scholar] [CrossRef]
- Higashimoto, S.; Tanihata, W.; Nakagawa, Y.; Azuma, M.; Ohue, H.; Sakata, Y. Effective photocatalytic decomposition of VOC under visible-light irradiation on N-doped TiO2 modified by vanadium species. Appl. Catal. A Gen. 2008, 340, 98–104. [Google Scholar] [CrossRef]
- Morikawa, T.; Ohwaki, T.; Suzuki, K.; Moribe, S.; Tero-Kubota, S. Visible-light-induced photocatalytic oxidation of carboxylic acids and aldehydes over N-doped TiO2 loaded with Fe, Cu or Pt. Appl. Catal. B Environ. 2008, 83, 56–62. [Google Scholar] [CrossRef]
- Sreethawong, T.; Laehsalee, S.; Chavadej, S. Use of Pt/N-doped mesoporous-assembled nanocrystalline TiO2 for photocatalytic H2 production under visible light irradiation. Catal. Commun. 2009, 10, 538–543. [Google Scholar] [CrossRef]
- Dolat, D.; Quici, N.; Kusiak-Nejman, E.; Morawski, A.W.; Puma, G.L. One-step, hydrothermal synthesis of nitrogen, carbon co-doped titanium dioxide (N,C-TiO2) photocatalysts. Effect of alcohol degree and chain length as carbon dopant precursors on photocatalytic activity and catalyst deactivation. Appl. Catal. B Environ. 2012, 115, 81–89. [Google Scholar] [CrossRef]
- Virkutyte, J.; Varma, R.S. Visible light activity of Ag-loaded and guanidine nitrate-doped nano-TiO2: Degradation of dichlorophenol and antibacterial properties. RSC Adv. 2012, 2, 1533–1539. [Google Scholar] [CrossRef]
- Cong, Y.; Zhang, J.; Chen, F.; Anpo, M. Synthesis and Characterization of Nitrogen-Doped TiO2Nanophotocatalyst with High Visible Light Activity. J. Phys. Chem. C 2007, 111, 6976–6982. [Google Scholar] [CrossRef]
- Yang, X.; Cao, C.; Erickson, L.; Hohn, K.; Maghirang, R.; Klabunde, K. Synthesis of visible-light-active TiO2-based photocatalysts by carbon and nitrogen doping. J. Catal. 2008, 260, 128–133. [Google Scholar] [CrossRef]
- Mitoraj, D.; Kisch, H. On the Mechanism of Urea–Induced Titania Modification. Chem. Eur. J. 2010, 16, 261–269. [Google Scholar] [CrossRef]
- Chai, B.; Peng, T.; Mao, J.; Li, K.; Zan, L. Graphitic carbon nitride (g-C3N4)-Pt-TiO2 nanocomposite as an efficient photocatalyst for hydrogen production under visible light irradiation. Phys. Chem. Chem. Phys. 2012, 14, 16745–16752. [Google Scholar] [CrossRef]
- Han, C.; Wang, Y.; Lei, Y.; Wang, B.; Wu, N.; Shi, Q.; Li, Q. In situ synthesis of graphitic-C3N4 nanosheet hybridized N-doped TiO2 nanofibers for efficient photocatalytic H2 production and degradation. Nano Res. 2015, 8, 1199–1209. [Google Scholar] [CrossRef]
- Yan, H.; Yang, H. TiO2-g-C3N4 composite materials for photocatalytic H2 evolution under visible light irradiation. J. Alloy. Comp. 2010, 509, L26–L29. [Google Scholar] [CrossRef]
- Higashimoto, S.; Hikita, K.; Azuma, M.; Yamamoto, M.; Takahashi, M.; Sakata, Y.; Matsuoka, M.; Kobayashi, H. Visible Light-Induced Photocatalysis on Carbon Nitride Deposited Titanium Dioxide: Hydrogen Production from Sacrificial Aqueous Solutions. Chin. J. Chem. 2017, 35, 165–172. [Google Scholar] [CrossRef]
- Zhang, Q.; Gangadharan, D.T.; Liu, Y.; Xu, Z.; Chaker, M.; Ma, D. Recent advancements in plasmon-enhanced visible light-driven water splitting. J. Materiomics 2017, 3, 33–50. [Google Scholar] [CrossRef]
- Ohko, Y.; Tatsuma, T.; Fujii, T.; Naoi, K.; Niwa, C.; Kubota, Y.; Fujishima, A. Multicolour photochromism of TiO2 films loaded with silver nanoparticles. Nat. Mater. 2003, 2, 29–31. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Tatsuma, T. Plasmon-induced photoelectrochemistry at metal nanoparticles supported on nanoporous TiO2. Chem. Commun. 2004, 0, 1810–1811. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Tatsuma, T. Mechanisms and Applications of Plasmon-Induced Charge Separation at TiO2 Films Loaded with Gold Nanoparticles. J. Am. Chem. Soc. 2005, 127, 7632–7637. [Google Scholar] [CrossRef] [PubMed]
- Kowalska, E.; Abe, R.; Ohtani, B. Visible light-induced photocatalytic reaction of gold-modified titanium(IV) oxide particles: Action spectrum analysis. Chem. Commun. 2009, 0, 241–243. [Google Scholar] [CrossRef] [PubMed]
- Kowalska, E.; Mahaney, O.O.P.; Abe, R.; Ohtani, B. Visible-light-induced photocatalysis through surface plasmon excitation of gold on titania surfaces. Phys. Chem. Chem. Phys. 2010, 12, 2344–2355. [Google Scholar] [CrossRef] [PubMed]
- Kolinko, P.A.; Selishchev, D.S.; Kozlov, D.V. Visible Light Photocatalytic Oxidation of Ethanol Vapor on Titanium Dioxide Modified with Noble Metals. Theor. Exp. Chem. 2015, 51, 96–103. [Google Scholar] [CrossRef]
- Frens, G. Controlled Nucleation for the Regulation of the Particle Size in Monodisperse Gold Suspensions. Nat. Phys. Sci. 1973, 241, 20–22. [Google Scholar] [CrossRef]
- Tanaka, A.; Ogino, A.; Iwaki, M.; Hashimoto, K.; Ohnuma, A.; Amano, F.; Ohtani, B.; Kominami, H. Gold–Titanium(IV) Oxide Plasmonic Photocatalysts Prepared by a Colloid-Photodeposition Method: Correlation Between Physical Properties and Photocatalytic Activities. Langmuir 2012, 28, 13105–13111. [Google Scholar] [CrossRef]
- Silva, C.G.; Juarez, R.; Marino, T.; Molinari, R.; Garcia, H. Influence of Excitation Wavelength (UV or Visible Light) on the Photocatalytic Activity of Titania Containing Gold Nanoparticles for the Generation of Hydrogen or Oxygen from Water. J. Am. Chem. Soc. 2011, 133, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Zanella, R.; Delannoy, L.; Louis, C. Mechanism of deposition of gold precursors onto TiO2 during the preparation by cation adsorption and deposition–precipitation with NaOH and urea. Appl. Catal. A Gen. 2005, 291, 62–72. [Google Scholar] [CrossRef]
- Tanaka, A.; Sakaguchi, S.; Hashimoto, K.; Kominami, H. Preparation of Au/TiO2 with Metal Cocatalysts Exhibiting Strong Surface Plasmon Resonance Effective for Photoinduced Hydrogen Formation under Irradiation of Visible Light. ACS Catal. 2013, 3, 79–85. [Google Scholar] [CrossRef]
- Tanaka, A.; Nakanishi, K.; Hamada, R.; Hashimoto, K.; Kominami, H. Simultaneous and Stoichiometric Water Oxidation and Cr(VI) Reduction in Aqueous Suspensions of Functionalized Plasmonic Photocatalyst Au/TiO2–Pt under Irradiation of Green Light. ACS Catal. 2013, 3, 1886–1891. [Google Scholar] [CrossRef]
- Tanaka, A.; Nishino, Y.; Sakaguchi, S.; Yoshikawa, T.; Imamura, K.; Hashimoto, K.; Kominami, H. Functionalization of a plasmonic Au/TiO2 photocatalyst with an Ag co-catalyst for quantitative reduction of nitrobenzene to aniline in 2-propanol suspensions under irradiation of visible light. Chem. Commun. 2013, 49, 2551–2553. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Hashimoto, K.; Kominami, H. A very simple method for the preparation of Au/TiO2 plasmonic photocatalysts working under irradiation of visible light in the range of 600–700 nm. Chem. Commun. 2017, 53, 4759–4762. [Google Scholar] [CrossRef]
- Naya, S.; Teranishi, M.; Isobe, T.; Tada, H. Light wavelength-switchable photocatalytic reaction by gold nanoparticle-loaded titanium(IV) dioxide. Chem. Commun. 2010, 46, 815–817. [Google Scholar] [CrossRef]
- Naya, S.; Kimura, K.; Tada, H. One-Step Selective Aerobic Oxidation of Amines to Imines by Gold Nanoparticle-Loaded Rutile Titanium(IV) Oxide Plasmon Photocatalyst. ACS Catal. 2013, 3, 10–13. [Google Scholar] [CrossRef]
- Tsukamoto, D.; Shiraishi, Y.; Sugano, Y.; Ichikawa, S.; Tanaka, S.; Hirai, T. Gold Nanoparticles Located at the Interface of Anatase/Rutile TiO2 Particles as Active Plasmonic Photocatalysts for Aerobic Oxidation. J. Am. Chem. Soc. 2012, 134, 6309–6315. [Google Scholar] [CrossRef]
- Zheng, Z.; Huang, B.; Qin, X.; Zhang, X.; Dai, Y.; Whangbo, M.-H. Facile in situ synthesis of visible-light plasmonic photocatalysts M@TiO2 (M = Au, Pt, Ag) and evaluation of their photocatalytic oxidation of benzene to phenol. J. Mater. Chem. 2011, 21, 9079–9087. [Google Scholar] [CrossRef]
- Tanaka, A.; Sakaguchi, S.; Hashimoto, K.; Kominami, H. Preparation of Au/TiO2 exhibiting strong surface plasmon resonance effective for photoinduced hydrogen formation from organic and inorganic compounds under irradiation of visible light. Catal. Sci. Technol. 2012, 2, 907–909. [Google Scholar] [CrossRef]
- Tanaka, A.; Teramura, K.; Hosokawa, S.; Kominami, H.; Tanaka, T. Visible light-induced water splitting in an aqueous suspension of a plasmonic Au/TiO2 photocatalyst with metal co-catalysts. Chem. Sci. 2017, 8, 2574–2580. [Google Scholar] [CrossRef] [PubMed]
- Furube, A.; Du, L.; Hara, K.; Katoh, R.; Tachiya, M. Ultrafast Plasmon-Induced Electron Transfer from Gold Nanodots into TiO2 Nanoparticles. J. Am. Chem. Soc. 2007, 129, 14852–14853. [Google Scholar] [CrossRef] [PubMed]
- Borgarello, E.; Kiwi, J.; Pelizzetti, E.; Visca, M.; Grätzel, M. Photochemical cleavage of water by photocatalysis. Nature 1981, 289, 158–160. [Google Scholar] [CrossRef]
- Vinodgopal, K.; Hua, X.; Dahlgren, R.L. Photochemistry of Ru(bpy)2(dcbpy)2+ on A12O3 and TiO2 surfaces. an insight into the mechanism of photosensitization. J. Phys. Chem. 1995, 99, 10883–10889. [Google Scholar] [CrossRef]
- Sakthivel, S.; Kisch, H. Daylight photocatalysis by carbon-modified titanium dioxide. Angew. Chem. Int. Ed. 2003, 42, 4908–4911. [Google Scholar] [CrossRef] [PubMed]
- Macyk, W.; Burgeth, G.; Kisch, H. Photoelectrochemical properties of platinum(IV) chloride surface modified TiO2. Photochem. Photobiol. Sci. 2003, 2, 322–328. [Google Scholar] [CrossRef]
- Higashimoto, S.; Nishi, T.; Yasukawa, M.; Azuma, M.; Sakata, Y.; Kobayashi, H. Photocatalysis of titanium dioxide modified by interfacial surface complexes (ISC) with different substituted groups. J. Catal. 2015, 329, 286–290. [Google Scholar] [CrossRef]
- Ikeda, S.; Abe, C.; Torimoto, T.; Ohtani, B. Photochemical hydrogen evolution from aqueous triethanolamine solutions sensitized by binaphthol-modified titanium(IV) oxide under visible-light irradiation. J. Photochem. Photobiol. A Chem. 2003, 160, 61–67. [Google Scholar] [CrossRef] [Green Version]
- Kamegawa, T.; Seto, H.; Matsuura, S.; Yamashita, H. Preparation of hydroxynaphthalene modified TiO2 via formation of surface complexes and their applications in the photocatalytic reduction of nitrobenzene under visible-light irradiation. ACS Appl. Mater. Interfaces 2012, 4, 6635–6639. [Google Scholar] [CrossRef]
- Kim, S.; Choi, W. Visible-light-induced photocatalytic degradation of 4-chlorophenol and phenolic compounds in aqueous suspension of pure titania: Demonstrating the existence of a surface-complex-mediated path. J. Phys. Chem. B 2005, 109, 5143–5149. [Google Scholar] [CrossRef] [PubMed]
- Higashimoto, S.; Kitao, N.; Yoshida, N.; Sakura, T.; Azuma, M.; Ohue, H.; Sakata, Y. Selective photocatalytic oxidation of benzyl alcohol and its derivatives into corresponding aldehydes by molecular oxygen on titanium dioxide under visible light irradiation. J. Catal. 2009, 266, 279–285. [Google Scholar] [CrossRef]
- Higashimoto, S.; Okada, K.; Morisugi, T.; Azuma, M.; Ohue, H.; Kim, T.-H.; Matsuoka, M.; Anpo, M. Effect of surface treatment on the selective photocatalytic oxidation of benzyl alcohol infrared study of hydroxy groups on coordinative defect sites. Top. Catal. 2010, 53, 578–583. [Google Scholar] [CrossRef]
- Higashimoto, S.; Okada, K.; Azuma, M.; Ohue, H.; Terai, T.; Sakata, Y. Characteristics of the charge transfer surface complex on titanium(IV) dioxide for the visible light induced chemoselective oxidation of benzyl alcohol. RSC Adv. 2012, 2, 669–676. [Google Scholar] [CrossRef]
- Higashimoto, S.; Suetsugu, N.; Azuma, M.; Ohue, H.; Sakata, Y. Efficient and selective oxidation of benzylic alcohol by O2 into corresponding aldehydes on a TiO2 photocatalyst under visible light irradiation: Effect of phenyl-ring substitution on the photocatalytic activity. J. Catal. 2010, 274, 76–83. [Google Scholar] [CrossRef]
- Higashimoto, S.; Shirai, R.; Osano, Y.; Azuma, M.; Ohue, H.; Sakata, Y.; Kobayashi, H. Influence of metal ions on the photocatalytic activity: Selective oxidation of benzyl alcohol on iron (III) ion-modified TiO2, using visible light. J. Catal. 2014, 311, 137–143. [Google Scholar] [CrossRef]
- Kobayashi, H.; Higashimoto, S. DFT study on the reaction mechanisms behind the catalytic oxidation of benzyl alcohol into benzaldehyde by O2 over anatase TiO2 surfaces with hydroxyl groups: Role of visible-light irradiation. Appl. Catal. B Environ. 2015, 170, 135–143. [Google Scholar] [CrossRef]
- Li, R.; Kobayashi, H.; Guo, J.; Fan, J. Visible-light induced high-yielding benzyl alcohol-to benzaldehyde transformation over mesoporous crystalline TiO2: A self-adjustable photooxidation system with controllable hole-generation. J. Phys. Chem. C 2011, 115, 23408–23416. [Google Scholar] [CrossRef]
- Lang, X.; Ma, W.; Zhao, Y.; Chen, C.; Ji, H.; Zhao, J. Visible-light-induced selective catalytic aerobic oxidation of amines into imines on TiO2. Chem. Eur. J. 2012, 18, 2624–2631. [Google Scholar] [CrossRef]
- Higashimoto, S.; Hatada, Y.; Ishikawa, R.; Azuma, M.; Sakata, Y.; Kobayashi, H. Selective Photocatalytic Oxidation of Benzyl Amine by O2 into N-Benzylidenebenzylamine on TiO2 Using Visible Light. Curr. Org. Chem. 2013, 17, 2374–2381. [Google Scholar] [CrossRef]
Figure 1.
Visible-light-sensitive TiO2 photocatalyst modified by (1) nitrogen-doping, (2) plasmonic Au nanoparticles (NPs), and (3) interfacial surface complex (ISC).
Figure 1.
Visible-light-sensitive TiO2 photocatalyst modified by (1) nitrogen-doping, (2) plasmonic Au nanoparticles (NPs), and (3) interfacial surface complex (ISC).

Figure 2.
When the N3− is replaced with lattice O2− ions in the TiO2 lattice, the hole (h+) is formed in order to compensate for the charge balance (p-type semiconductor) (a). However, an oxygen vacancy is produced by the reduction with H2, which is formed by the decomposition of NH3 to produce an oxygen vacancy and excess electrons (b). As a consequence, N3− doped into TiO2 (N-doped TiO2) involves electrons located at N 2p and Ti 3d sites at impurity levels (n-type semiconductor) (c).
Figure 2.
When the N3− is replaced with lattice O2− ions in the TiO2 lattice, the hole (h+) is formed in order to compensate for the charge balance (p-type semiconductor) (a). However, an oxygen vacancy is produced by the reduction with H2, which is formed by the decomposition of NH3 to produce an oxygen vacancy and excess electrons (b). As a consequence, N3− doped into TiO2 (N-doped TiO2) involves electrons located at N 2p and Ti 3d sites at impurity levels (n-type semiconductor) (c).

Figure 3.
Schematic illustration of structures and their corresponding energy bands for substitutional and interstitial N species in the N-doped TiO2, together with photo-induced electronic processes.
Figure 3.
Schematic illustration of structures and their corresponding energy bands for substitutional and interstitial N species in the N-doped TiO2, together with photo-induced electronic processes.

Figure 4.
XPS [I] and UV-vis absorption spectra [II] of (a) N-doped TiO2 nanoball film [34], and (b) N-doped TiO2 prepared by the sol-gel method [36].

Figure 5.
Schematic illustration of [I] formation of paramagnetic ·N by the excitation of diamagnetic N− species. Electron paramagnetic resonance (EPR) signal [II] of ·N radicals on N–TiO2, and the relative signal intensity of IN/IN0 [III] under vacuum, in the presence of argon (Ar) or O2 (400 Pa) [37]. IN0 and IN show the intensity due to ·N radicals at the initial and measured time, respectively.
Figure 5.
Schematic illustration of [I] formation of paramagnetic ·N by the excitation of diamagnetic N− species. Electron paramagnetic resonance (EPR) signal [II] of ·N radicals on N–TiO2, and the relative signal intensity of IN/IN0 [III] under vacuum, in the presence of argon (Ar) or O2 (400 Pa) [37]. IN0 and IN show the intensity due to ·N radicals at the initial and measured time, respectively.

Figure 6.
Schematic illustration of proposed energy bands for the N-doped TiO2, together with some photo-induced electronic processes. E: equilibrium redox potentials for one electron transfer [38].
Figure 6.
Schematic illustration of proposed energy bands for the N-doped TiO2, together with some photo-induced electronic processes. E: equilibrium redox potentials for one electron transfer [38].

Figure 7.
Energy levels for sub-band structures of N-doped TiO2 and photo-induced charge transfer into various kinds of redox species under visible light irradiation. The energy levels of sub-bands at the α, β, and γ potential regions (oxygen vacancies) and N-doping levels are also shown. Oxygen vacancies were estimated from the photo-electrochemical measurements. Signs of circle and cross stand for energetically favorable and unfavorable electron transfers, respectively [33].
Figure 7.
Energy levels for sub-band structures of N-doped TiO2 and photo-induced charge transfer into various kinds of redox species under visible light irradiation. The energy levels of sub-bands at the α, β, and γ potential regions (oxygen vacancies) and N-doping levels are also shown. Oxygen vacancies were estimated from the photo-electrochemical measurements. Signs of circle and cross stand for energetically favorable and unfavorable electron transfers, respectively [33].

Figure 8.
Photocatalytic decomposition of gaseous acetaldehyde on the N-doped TiO2 photocatalyst. Evolved CO2 concentration (○, ●, N-doped TiO2; □, ■, TiO2) [28].
Figure 8.
Photocatalytic decomposition of gaseous acetaldehyde on the N-doped TiO2 photocatalyst. Evolved CO2 concentration (○, ●, N-doped TiO2; □, ■, TiO2) [28].

Figure 9.
Photocatalytic activities for the decomposition of acetic acid under visible light irradiation (λ > 420 nm) on N-doped TiO2, modified by various kinds of metal species as co-catalysts. Each metal salt used in this study is shown [40].
Figure 9.
Photocatalytic activities for the decomposition of acetic acid under visible light irradiation (λ > 420 nm) on N-doped TiO2, modified by various kinds of metal species as co-catalysts. Each metal salt used in this study is shown [40].

Figure 10.
Photo-induced charge separation on the C3N4 deposited TiO2 surface [51].
Figure 10.
Photo-induced charge separation on the C3N4 deposited TiO2 surface [51].

Figure 11.
Short-circuit photocurrent densities [I] vs. apparent formal potential of different donors on the Au−TiO2 photoanode in acetonitrile/ethylene glycol (v/v 60/40) containing 0.1 M LiNO3 and 0.1 M donors; IPCE [II] of the Au−TiO2 film in a N2-saturated acetonitrile and ethylene glycol (v/v: 60/40) solution containing 0.1 M FeCl2 and 0.05 M FeCl3 [55].
Figure 11.
Short-circuit photocurrent densities [I] vs. apparent formal potential of different donors on the Au−TiO2 photoanode in acetonitrile/ethylene glycol (v/v 60/40) containing 0.1 M LiNO3 and 0.1 M donors; IPCE [II] of the Au−TiO2 film in a N2-saturated acetonitrile and ethylene glycol (v/v: 60/40) solution containing 0.1 M FeCl2 and 0.05 M FeCl3 [55].

Figure 12.
Schematic illustration [I] and its energy band levels [II] for the photo-induced charge separation on the Au–TiO2 in the presence of donors [55].
Figure 12.
Schematic illustration [I] and its energy band levels [II] for the photo-induced charge separation on the Au–TiO2 in the presence of donors [55].

Figure 13.
Schematic illustration for the formation of two types of ISCs [I], and photocatalytic H2 evolution [II] from aq. TEA (10 vol. %) on (a) BC/TiO2, (b) MC/TiO2, (c) CA/TiO2, (d) BA/TiO2, (e) BN/TiO2, and (f) TN/TiO2 [78]. BC: 4-t-butyl catechol, MC: 3-methoxy catechol, CA: catecol, BA: 2,3-dihydroxy benzoic acid; BN: 3,4-dihydroxy benzonitrile; TN: tiron.
Figure 13.
Schematic illustration for the formation of two types of ISCs [I], and photocatalytic H2 evolution [II] from aq. TEA (10 vol. %) on (a) BC/TiO2, (b) MC/TiO2, (c) CA/TiO2, (d) BA/TiO2, (e) BN/TiO2, and (f) TN/TiO2 [78]. BC: 4-t-butyl catechol, MC: 3-methoxy catechol, CA: catecol, BA: 2,3-dihydroxy benzoic acid; BN: 3,4-dihydroxy benzonitrile; TN: tiron.

Figure 14.
Schematic illustration of photo-induced charge separation on the BN/TiO2 for H2 evolution from TEA aq. in the presence of Pt as co-catalyst under visible light irradiation [78].
Figure 14.
Schematic illustration of photo-induced charge separation on the BN/TiO2 for H2 evolution from TEA aq. in the presence of Pt as co-catalyst under visible light irradiation [78].

Figure 15.
Selective oxidation of benzyl alcohol on TiO2 (50 mg) under visible light irradiation [82]. The initial amount of benzyl alcohol was 50 μmol. Amounts of: benzyl alcohol (a); benzaldehyde (b); benzoic acid (c); CO2 (d); and percentage of total organic compounds in solution (e).
Figure 15.
Selective oxidation of benzyl alcohol on TiO2 (50 mg) under visible light irradiation [82]. The initial amount of benzyl alcohol was 50 μmol. Amounts of: benzyl alcohol (a); benzaldehyde (b); benzoic acid (c); CO2 (d); and percentage of total organic compounds in solution (e).

Figure 16.
FT-IR spectra [I] of benzyl alcohol by itself and benzyl alcohol adsorbed on TiO2; and [II] their peak identification [82].
Figure 16.
FT-IR spectra [I] of benzyl alcohol by itself and benzyl alcohol adsorbed on TiO2; and [II] their peak identification [82].

Figure 17.
UV-vis absorption spectra of TiO2 (a), TiO2 adsorbed with benzyl alcohol (b), and apparent quantum yield (AQY) for the formation of benzaldehyde (c); and schematic illustration of photo-induced charge transfer through LMCT in the alkoxide [82].
Figure 17.
UV-vis absorption spectra of TiO2 (a), TiO2 adsorbed with benzyl alcohol (b), and apparent quantum yield (AQY) for the formation of benzaldehyde (c); and schematic illustration of photo-induced charge transfer through LMCT in the alkoxide [82].

Figure 18.
Photo-induced electron transfer from the hybridized orbital to the C.B. of TiO2 under visible light irradiation [87]. Density maps of V.B., C.B., and hybridized orbital are shown here.
Figure 18.
Photo-induced electron transfer from the hybridized orbital to the C.B. of TiO2 under visible light irradiation [87]. Density maps of V.B., C.B., and hybridized orbital are shown here.

Figure 19.
Possible reaction path for the selective oxidation of benzyl alcohol in the presence of O2 on the TiO2 under visible light irradiation [87].
Figure 19.
Possible reaction path for the selective oxidation of benzyl alcohol in the presence of O2 on the TiO2 under visible light irradiation [87].

Scheme 1.
Selective oxidation of benzyl amine into N-benzylidenebenzylamine on the TiO2 photocatalyst under visible light irradiation.
Scheme 1.
Selective oxidation of benzyl amine into N-benzylidenebenzylamine on the TiO2 photocatalyst under visible light irradiation.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Yields of CO2 for the photocatalytic decomposition of various kinds of volatile organic compounds (VOC) in aqueous solutions with N–TiO2 and VCl3/N-doped TiO2 under visible light irradiation (λ > 420 nm) for 3 h [40].
Table 1.
Yields of CO2 for the photocatalytic decomposition of various kinds of volatile organic compounds (VOC) in aqueous solutions with N–TiO2 and VCl3/N-doped TiO2 under visible light irradiation (λ > 420 nm) for 3 h [40].
| Entry | Reactant Molecules | Yields of CO2/μmol | |
|---|---|---|---|
| N-doped TiO2 | VCl3/N-doped TiO2 | ||
| 1 | methanol a | 0.2 | 1.1 |
| 2 | ethanol a | 0.3 | 0.5 |
| 3 | formaldehyde a | 4.6 | 21.6 |
| 4 | acetaldehyde a | 4.1 | 35.0 |
| 5 | formic acid a | 0.7 | 4.8 |
| 6 | acetic acid a | 1.2 | 17.0 |
| 7 | acetone a | 0.7 | 11.4 |
| 8 | ethyl acetate a | 1.3 | 10.6 |
| 9 | dichloromethane b | 2.4 | 4.1 |
| 10 | trichloromethane b | 1.5 | 4.1 |
| 11 | 1, 1-dichloroethane b | 0.7 | 4.8 |
| 12 | trans-1, 2-dichloroethylene b | 1.0 | 5.7 |
Concentrations of VOC are (a) 0.5 M and (b) 50 mM.
Table 2.
Particle sizes of Au nanoparticles (NPs) and optical properties of the Au–TiO2 prepared by several techniques.
Table 2.
Particle sizes of Au nanoparticles (NPs) and optical properties of the Au–TiO2 prepared by several techniques.
| Entry | Au Deposition Methods | Particle Sizes/nm | Top Peak/nm | Ref. |
|---|---|---|---|---|
| 1 | PD | ~10–60 | ~530–610 | [56,57,58] |
| 2 | CPH | ~12–14 | ~550–560 | [60,63,64,65,66] |
| 13 | ~550–620 | [67] | ||
| 3 | DP | ~2–6 | ~550–560 | [61] |
| < 5 | 550 | [68,69,70] |
Table 3.
Applications to several photocatalytic reactions on the Au–TiO2 photocatalyst.
| Entry | Photocatalytic Reactions | Au Deposition Methods | References |
|---|---|---|---|
| 1 | oxidations of 2-propanol and ethanol oxidation of formic acid | PD CPH | [56,57,58] [60] |
| 2 | oxidation of thiol to disulfide | DP | [67] |
| oxidation of amine to imine | DP | [68] | |
| oxidation of aromatic alcohol to aldehyde | CPH | [66] | |
| DP | [69] | ||
| oxidation of benzene to phenol | PD | [70] | |
| 3 | H2 formation from alcohols | CPH | [63,71] |
| water splitting into H2 and O2 | DP CPH | [61] [64,72] | |
| 4 | reduction of nitrobenzene to aniline | CPH | [65] |
Table 4.
Chemoselective photocatalytic oxidation of different kinds of benzylic alcohols on TiO2 [82].
Table 4.
Chemoselective photocatalytic oxidation of different kinds of benzylic alcohols on TiO2 [82].

| Entry | R1 | R2 | Conversion (%) | Selectivity (%) |
|---|---|---|---|---|
| 1 | H | H | > 99 | > 99 |
| 2 | H | C(CH3)3 | > 99 | > 99 |
| 3 | H | OCH3 | > 99 | > 99 |
| 4 | H | CH3 | > 99 | > 99 |
| 5 | H | Cl | > 99 | > 99 |
| 6 | H | NO2 | > 99 | > 99 |
| 7 | H | CF3 | > 99 | > 99 |
| 8 | CH3 | H | > 99 | > 99 |
| 9 | H | OH | > 85 | 23 |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Higashimoto, S. Titanium-Dioxide-Based Visible-Light-Sensitive Photocatalysis: Mechanistic Insight and Applications. Catalysts 2019, 9, 201. https://doi.org/10.3390/catal9020201
AMA Style
Higashimoto S. Titanium-Dioxide-Based Visible-Light-Sensitive Photocatalysis: Mechanistic Insight and Applications. Catalysts. 2019; 9(2):201. https://doi.org/10.3390/catal9020201
Chicago/Turabian StyleHigashimoto, Shinya. 2019. "Titanium-Dioxide-Based Visible-Light-Sensitive Photocatalysis: Mechanistic Insight and Applications" Catalysts 9, no. 2: 201. https://doi.org/10.3390/catal9020201
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.