Theoretical Study on Influence of Cobalt Oxides Valence State Change for C6H5COOH Pyrolysis


Abstract
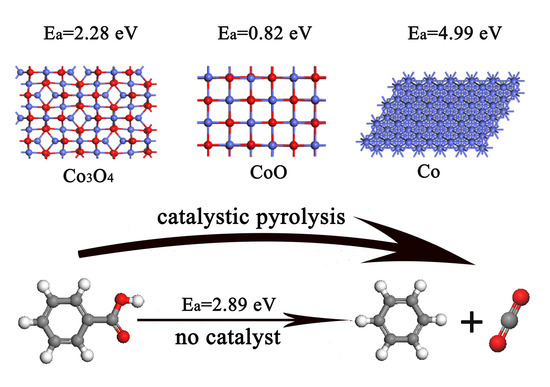
:
1. Introduction
2. Results and Discussion
2.1. C6H5COOH Pyrolysis
2.2. Catalytic Pyrolysis
2.2.1. Co3O4(110)-B Surface
2.2.2. CoO(100) Surface
2.2.3. Co(111) Surface
2.3. d-band Center Analyses
3. Computational Methods and Models
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Liu, L.L.; Kumar, S.; Wang, Z.H.; He, Y.; Liu, J.Z.; Cen, K. Catalytic Effect of Metal Chlorides on Coal Pyrolysis and Gasification Part I. Combined TG-FTIR Study for Coal Pyrolysis. Thermochim. Acta 2017, 655, 331–336. [Google Scholar] [CrossRef]
- Hu, H.Q.; Zhou, Q.; Zhu, S.W.; Meyer, B.; Krzack, S.; Chen, G.H. Product distribution and sulfur behavior in coal pyrolysis. Fuel Process. Technol. 2004, 85, 849–861. [Google Scholar] [CrossRef]
- Solomon, P.R.; Serio, M.A.; Suuberg, E.M. Coal pyrolysis: Experiments, kinetic rates and mechanisms. Prog. Energy Combust. Sci. 1992, 18, 133–220. [Google Scholar] [CrossRef]
- Zarnegar, S. A review on catalytic pyrolysis of coal and biomass for value added fuel and chemicals. Energy Sources Part A Recover. Util. Environ. Eff. 2018, 40, 1427–1433. [Google Scholar] [CrossRef]
- Wang, X.J.; Zhu, H.L.; Wang, X.M.; Liu, H.F.; Wang, F.C.; Yu, G.S. Transformation and Reactivity of a Potassium Catalyst during Coal–Steam Catalytic Pyrolysis and Gasification. Energy Technol. 2014, 2, 598–603. [Google Scholar] [CrossRef]
- Fu, Y.; Guo, Y.H.; Zhang, K.X. Effect of Three Different Catalysts (KCl, CaO, and Fe2O3) on the Reactivity and Mechanism of Low-Rank Coal Pyrolysis. Energy Fuels 2016, 30, 2428–2433. [Google Scholar] [CrossRef]
- Lei, Z.; Sha, X.L.; Lei, Z.; Wang, R.; Zhang, L.X.; Shu, X.Q. Influences of Different Preparation Conditions on Catalytic Activity of Ag2O-Co3O4/γ-Al2O3for Hydrogenation of Coal Pyrolysis. J. Spectrosc. 2014, 2014, 1–6. [Google Scholar]
- Yan, S.; Zhang, J.S.; Yan, X.Q.; Pan, D.F.; Ren, H.; Qu, X. Catalytic coal hydrogasification by cobalt-calcium catalyst in a pressurized fluidized bed: Role of hydropyrolysis and catalysis process. J. Anal. Appl. Pyrolysis 2018, 135, 251–259. [Google Scholar] [CrossRef]
- Liang, L.T.; Huai, J.T.; Zhang, Q.; Liu, J.W.; Huang, W.; Zhang, Z.L.; Hao, X.G.; Guan, G.Q. Catalytic depolymerization of a typical lignite for improving tar yield by Co and Zn catalyst. Sci. Rep. 2017, 7, 14433. [Google Scholar] [CrossRef] [Green Version]
- Takarada, T.; Onoyama, Y.; Takayama, K.; Sakashita, T. Hydropyrolysis of coal in a pressurized powder-particle fluidized bed using several catalysts. Catal. Today 1997, 39, 127–136. [Google Scholar] [CrossRef]
- Li, G.; Li, L.; Shi, L.; Jin, L.J.; Tang, Z.C.; Fan, H.J.; Hu, H.Q. Experimental and Theoretical Study on the Pyrolysis Mechanism of Three Coal-Based Model Compounds. Energy Fuels 2014, 28, 980–986. [Google Scholar] [CrossRef]
- Liu, S.Y.; Zhang, Z.Q.; Wang, H.F. Quantum chemical investigation of the thermal pyrolysis reactions of the carboxylic group in a brown coal model. J. Mol. Model. 2012, 18, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Murata, S.; Hosokawa, M.; Kidena, K.; Nomura, M. Analysis ofoxygen-functional groups in brown coals. Fuel Process. Technol. 2000, 67, 231–243. [Google Scholar] [CrossRef]
- Li, L.; Fan, H.J.; Hu, H.Q. A theoretical study on bond dissociation enthalpies of coal based model compounds. Fuel 2015, 153, 70–77. [Google Scholar] [CrossRef]
- Li, J.; Zhang, F.; Fang, W.H. Probing Photophysical and Photochemical Processes of Benzoic Acid from ab Initio Calculations. J. Phys. Chem. A 2005, 109, 7718–7724. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.H.; Wang, J.; Liu, J.Z.; Yang, Y.M.; Cheng, J.; Wang, Z.H.; Zhou, J.H.; Cen, K.F. Moisture removal mechanism of low-rank coal by hydrothermal dewatering: Physicochemical property analysis and DFT calculation. Fuel 2017, 187, 242–249. [Google Scholar] [CrossRef]
- Li, B.; Liu, S.Y.; Guo, J.Y.; Zhang, L. Interaction between low rank coal and kaolinite particles: A DFT simulation. Appl. Surf. Sci. 2018, 456, 215–220. [Google Scholar] [CrossRef]
- Kong, L.H.; Li, G.; Jin, L.J.; Hu, H.Q. Pyrolysis behaviors of two coal-related model compounds on a fixed-bed reactor. Fuel Process. Technol. 2015, 129, 113–119. [Google Scholar] [CrossRef]
- Gao, M.J.; Li, X.X.; Guo, L. Pyrolysis simulations of Fugu coal by large-scale ReaxFF molecular dynamics. Fuel Process. Technol. 2018, 178, 197–205. [Google Scholar] [CrossRef]
- Wang, M.F.; Zuo, Z.J.; Ren, R.P.; Gao, Z.H.; Huang, W. Theoretical Study on Catalytic Pyrolysis of Benzoic Acid as a Coal-Based Model Compound. Energy Fuels 2016, 30, 2833–2840. [Google Scholar] [CrossRef]
- Cui, L.P.; Liu, J.T.; Liu, S.Z.; Wang, M.F.; Gao, Z.H.; Zuo, Z.J.; Huang, W. A DFT study of the catalytic pyrolysis of benzaldehyde on ZnO, γ-Al2O3. J. Mol. Model. 2018, 24, 65. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.T.; Wang, M.F.; Gao, Z.H.; Zuo, Z.J.; Huang, W. The role of catalysts in the decomposition of phenoxy compounds in coal: A density functional theory study. Appl. Surf. Sci. 2018, 428, 541–548. [Google Scholar] [CrossRef]
- Wang, B.W.; Liu, S.H.; Hu, Z.Y.; Li, Z.H.; Ma, X.B. Active phase of highly active Co3O4 catalyst for synthetic natural gas production. RSC Adv. 2014, 4, 57185–57191. [Google Scholar] [CrossRef]
- Garces, L.J.; Hincapie, B.; Zerger, R.; Suib, S.L. The Effect of Temperature and Support on the Reduction of Cobalt Oxide: An in Situ X-ray Diffraction Study. J. Phys. Chem. C 2015, 119, 5484–5490. [Google Scholar] [CrossRef]
- Manion, J.A.; McMillen, D.F.; Malhotra, R. Decarboxylation and coupling reactions of aromatic acids under coal-liquefaction conditions. Energy Fuels 1996, 10, 776–788. [Google Scholar] [CrossRef]
- Eskay, T.P.; Britt, P.F.; Buchanan, A.C., III. Does Decarboxylation Lead to Cross-Linking in Low-Rank Coals? Energy Fuels 1996, 10, 1257–1261. [Google Scholar] [CrossRef]
- Eskay, T.P.; Britt, P.F.; Buchanan, A.C., III. Pyrolysis of Coal Model Compounds Containing Aromatic Carboxylic Acids: The Role of Carboxylic Acids in Cross-Linking Reactions in Low-Rank Coal; Oak Ridge National Lab.: Oak Ridge, TN, USA, 1997.
- Labat, F.; Ciofini, I.; Adamo, C. Modeling ZnO phases using a periodic approach: From bulk to surface and beyond. J. Chem. Phys. 2009, 131, 044708. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, M.; Li, Q.; Noei, H.; Nefedov, A.; Wang, Y.M.; Muhler, M.; Fink, K.; Wöll, C. The Interaction of Formic Acid with Zinc Oxide: A Combined Experimental and Theoretical Study on Single Crystal and Powder Samples. Top. Catal. 2015, 58, 174–183. [Google Scholar] [CrossRef]
- Yildirim, H.; Greber, T.; Kara, A. Trends in Adsorption Characteristics of Benzene on Transition Metal Surfaces: Role of Surface Chemistry and van der Waals Interactions. J. Phys. Chem. C 2013, 117, 20572–20583. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, M.; Hohner, C.; Mohr, S.; Libuda, J. Dissociative Adsorption of Benzoic Acid on Well-Ordered Cobalt Oxide Surfaces: Role of the Protons. J. Phys. Chem. C 2017, 121, 28317–28327. [Google Scholar] [CrossRef]
- Chen, C.B.; Wang, Q.; Wang, G.R.; Hou, B.; Jia, L.T.; Li, D.B. Mechanistic Insight into the C2 Hydrocarbons Formation from Syngas on fcc-Co(111) Surface: A DFT Study. J. Phys. Chem. C 2016, 120, 9132–9147. [Google Scholar] [CrossRef]
- Kuttiyiel, K.A.; Choi, Y.; Hwang, S.M.; Park, G.G.; Yang, T.H.; Su, D.; Sasaki, K.; Liu, P.; Adzic, R.R. Enhancement of the oxygen reduction on nitride stabilized pt-M (M=Fe, Co, and Ni) core–shell nanoparticle electrocatalysts. Nano Energy 2015, 13, 442–449. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phy. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, D.; Xiao, P.H.; Chemelewski, W.; Johnson, D.D.; Henkelman, G. A generalized solid-state nudged elastic band method. J. Chem. Phys. 2012, 136, 074103. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Lan, J.; Guo, Y.; Cao, X.M.; Hu, P. Origin of Efficient Catalytic Combustion of Methane over Co3O4(110) Active Low-Coordination Lattice Oxygen and Cooperation of Multiple Active Sites. ACS Catal. 2016, 6, 5508–5519. [Google Scholar] [CrossRef]
- Shojaee, K.; Haynes, B.S.; Montoya, A. Molecular modelling of the decomposition of NH3 over CoO(100). Mater. Chem. Phys. 2015, 156, 141–149. [Google Scholar] [CrossRef]
- Selcuk, S.; Selloni, A. DFT+U Study of the Surface Structure and Stability of Co3O4(110): Dependence on U. J. Phys. Chem. C 2015, 119, 9973–9979. [Google Scholar] [CrossRef]
- Wang, L.; Maxisch, T.; Ceder, G. Oxidation energies of transition metal oxides within the GGA+U framework. Phys. Rev. B 2006, 73, 195107. [Google Scholar] [CrossRef]
- Dudarev, S.L.; Botton, G.A.; Savrasov, S.Y.; Humphreys, C.J.; Sutton, A.P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 1998, 57, 1505. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | Species | Site | Eads(eV) | Bond Length d (Å) |
|---|---|---|---|---|
| Co3O4(110)-B | C6H5COO | Cooct | −1.91 | dO–Co = 1.835 |
| C6H5 | O2f | −3.37 | dC–O = 1.362 | |
| H | O2f | −3.59 | dH–O = 0.971 | |
| C6H6 | no bond | −0.01 | ||
| CO2 | O2f, Cooct | −0.64 | dC–O = 1.392, dO–Co = 1.954 | |
| CoO(100) | C6H5COO | Cobri | −3.06 | dO–Co = 1.934 |
| C6H5 | Otop | −2.03 | dC–O = 1.387 | |
| H | Otop | −2.59 | dH–O = 0.976 | |
| C6H6 | no bond | −0.26 | ||
| CO2 | Cotop, Otop | −0.40 | dC–O = 1.425, dO–Co = 2.107 | |
| Co(111) | C6H5COOH | top | −0.18 | dO–Co = 2.033 |
| C6H6COO | bri | −2.18 | dO–Co = 1.956 | |
| C6H5COO | bri | −3.08 | dO–Co = 1.960 | |
| C6H5 | top | −2.29 | dC–Co = 1.922 | |
| H | fcc | −2.76 | dH–Co = 1.622 | |
| H | hcp | −2.73 | dH–Co = 1.745 | |
| C6H6 | no bond | −0.01 | ||
| CO2 | no bond | −0.28 |
| Catalysts | Number of Layers | Surface Energies(J/m2) |
|---|---|---|
| Co3O4(110)-B | 4 | 1.24 |
| 6 | 1.22 | |
| CoO(100) | 5 | 0.54 |
| 6 | 0.52 | |
| Co(111) | 3 | 1.84 |
| 4 | 1.78 | |
| 5 | 1.76 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, S.-M.; Zhao, Y.; Liu, J.-T.; Liang, W.-S.; Li, G.-S.; Huang, W.; Zuo, Z.-J. Theoretical Study on Influence of Cobalt Oxides Valence State Change for C6H5COOH Pyrolysis. Catalysts 2019, 9, 197. https://doi.org/10.3390/catal9020197
Fu S-M, Zhao Y, Liu J-T, Liang W-S, Li G-S, Huang W, Zuo Z-J. Theoretical Study on Influence of Cobalt Oxides Valence State Change for C6H5COOH Pyrolysis. Catalysts. 2019; 9(2):197. https://doi.org/10.3390/catal9020197
Chicago/Turabian StyleFu, Si-Mei, Yue Zhao, Jiang-Tao Liu, Wen-Sheng Liang, Gang-Sen Li, Wei Huang, and Zhi-Jun Zuo. 2019. "Theoretical Study on Influence of Cobalt Oxides Valence State Change for C6H5COOH Pyrolysis" Catalysts 9, no. 2: 197. https://doi.org/10.3390/catal9020197