Catalytic Decomposition of Oleic Acid to Fuels and Chemicals: Roles of Catalyst Acidity and Basicity on Product Distribution and Reaction Pathways

Abstract
:1. Introduction
2. Results and Discussion
2.1. Catalyst Characterization
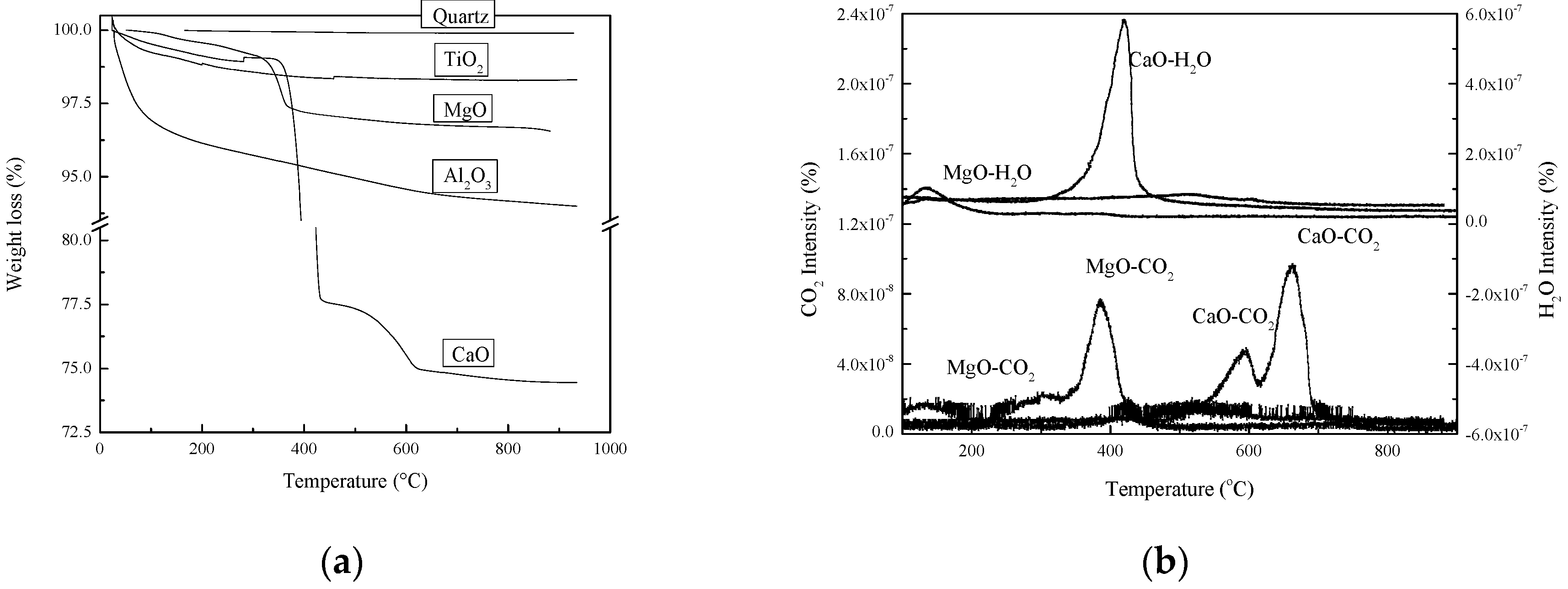
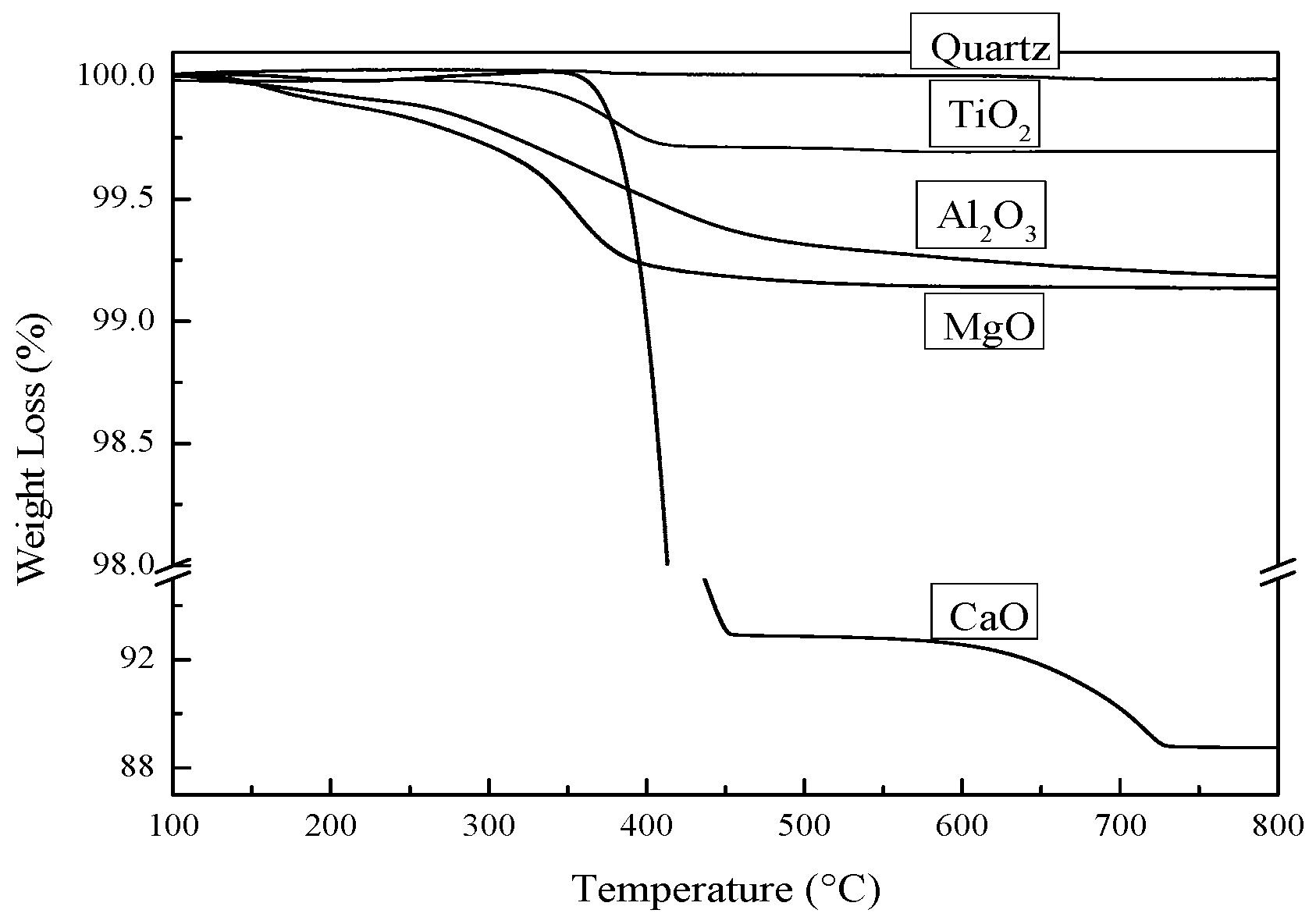
2.1.1. Thermal Treatment
2.1.2. Physical Properties
2.1.3. Amount, Types, and Strength of Active Sites
2.2. Products
2.2.1. Catalytic Upgrading by TGA
2.2.2. Product Distribution
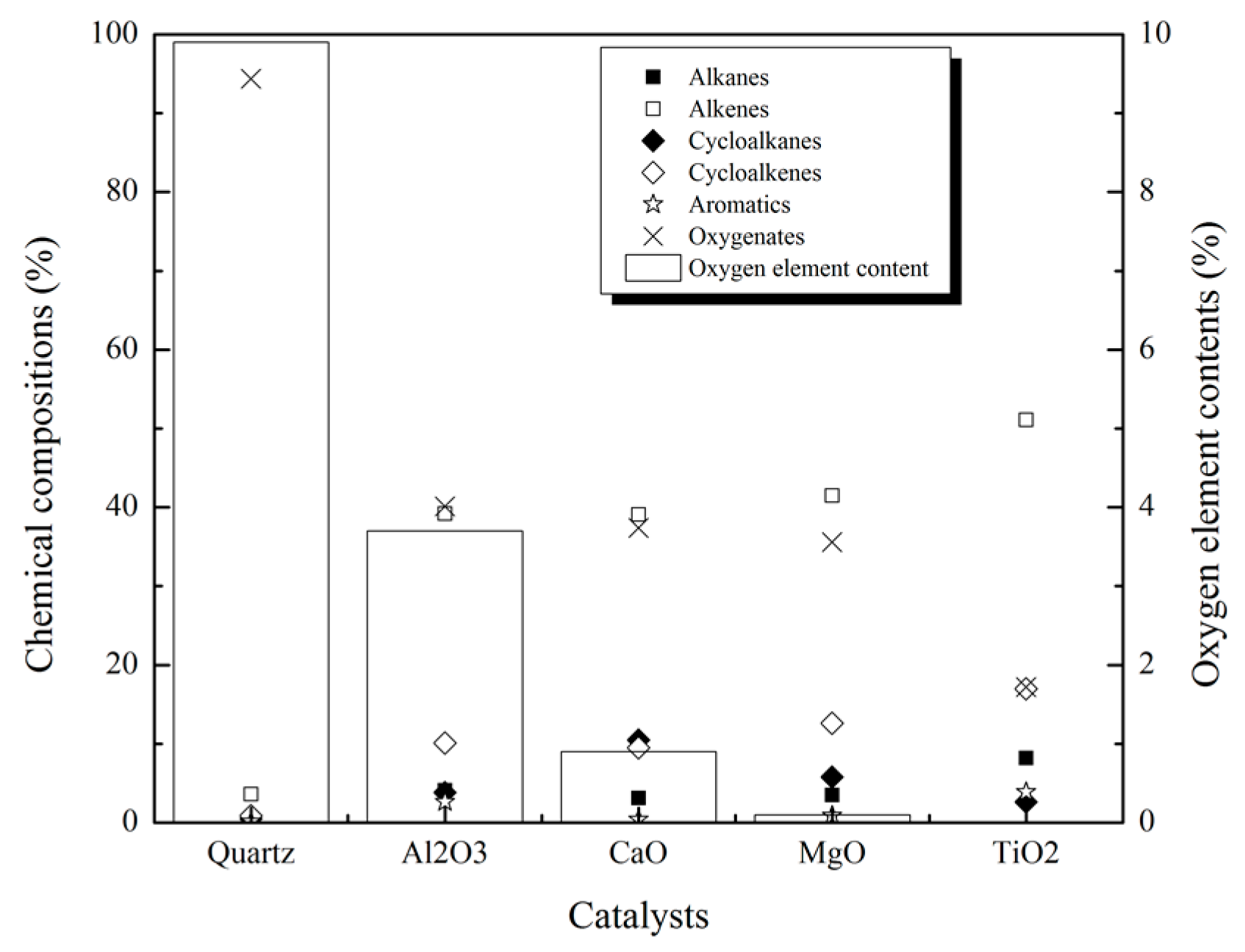
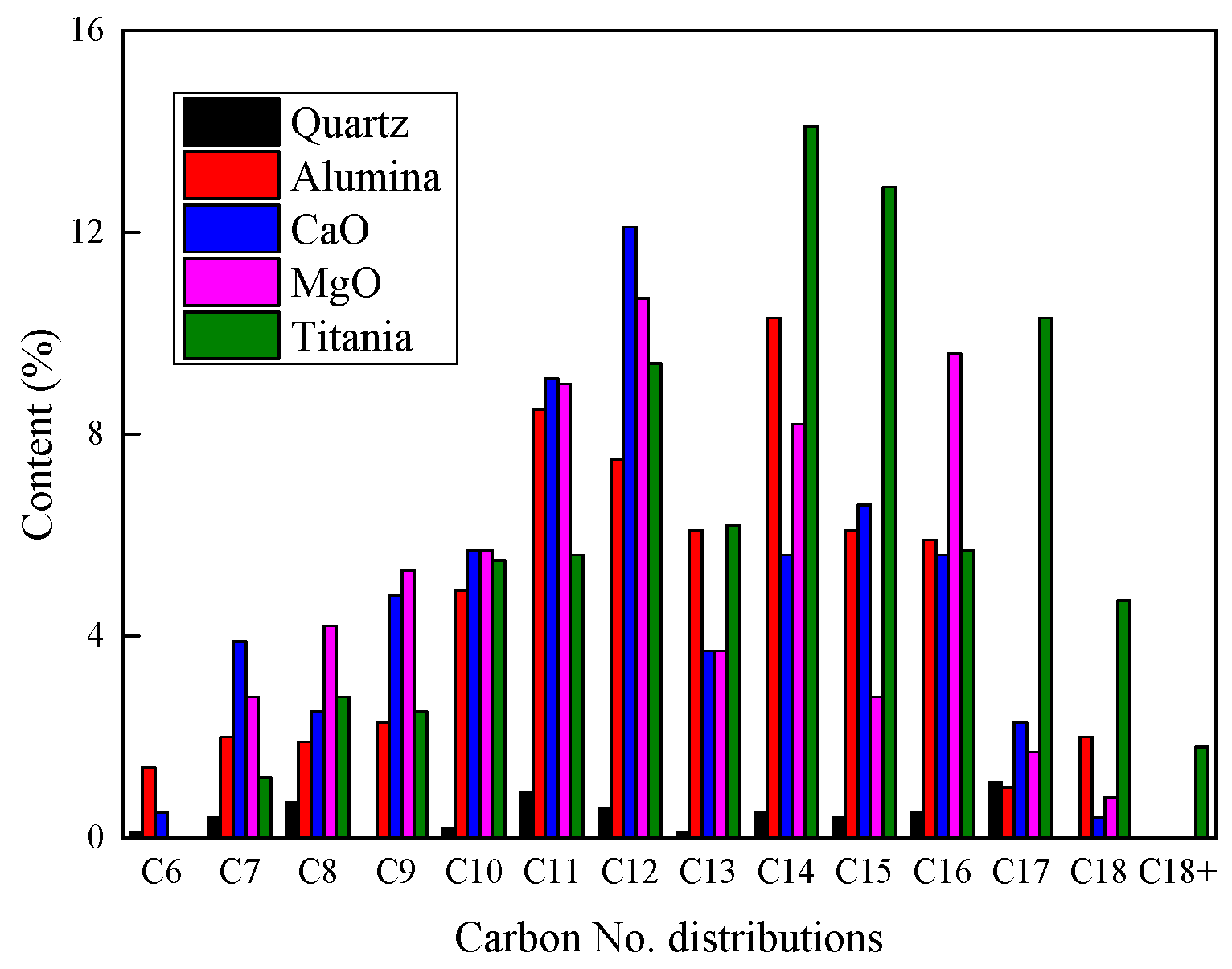
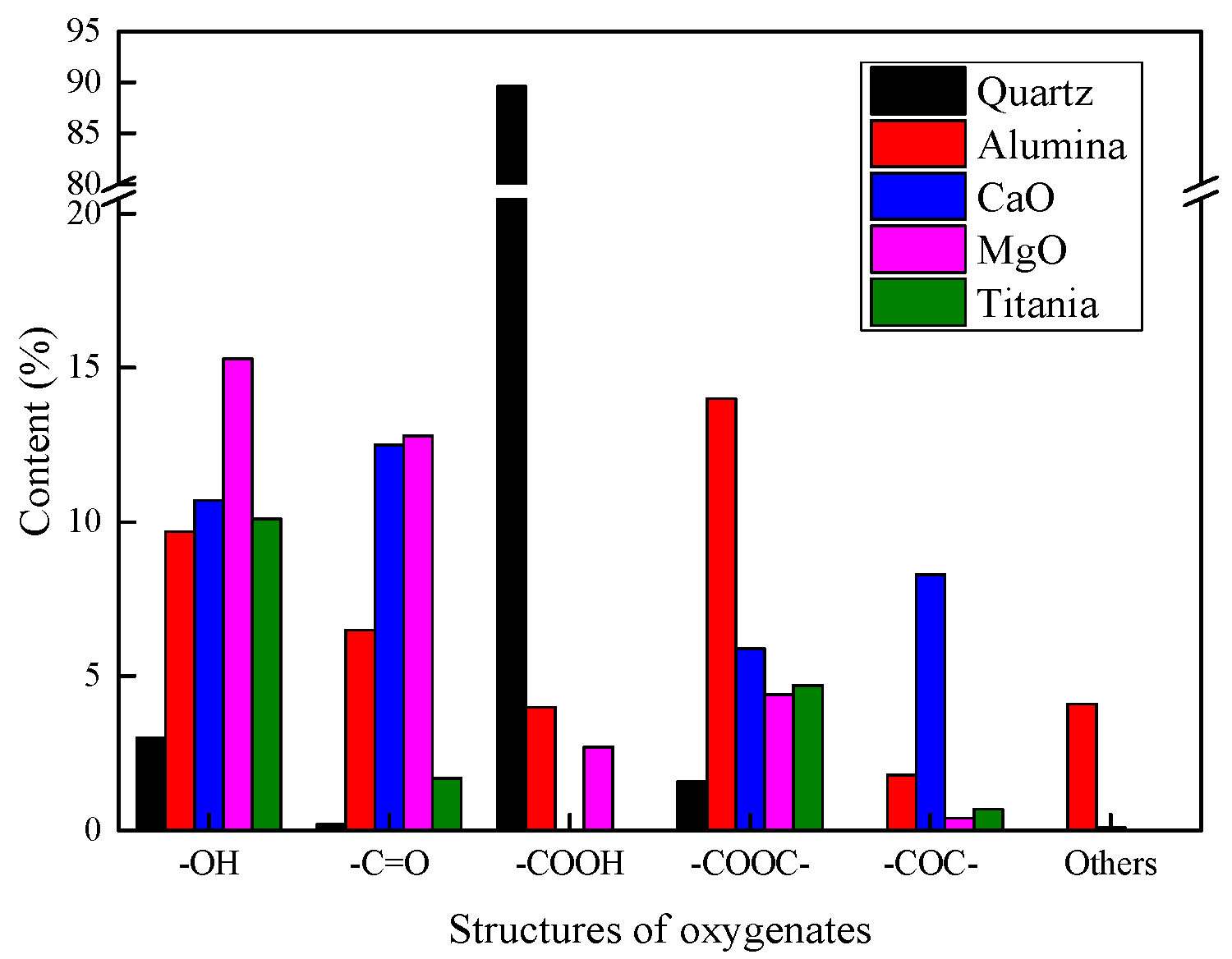
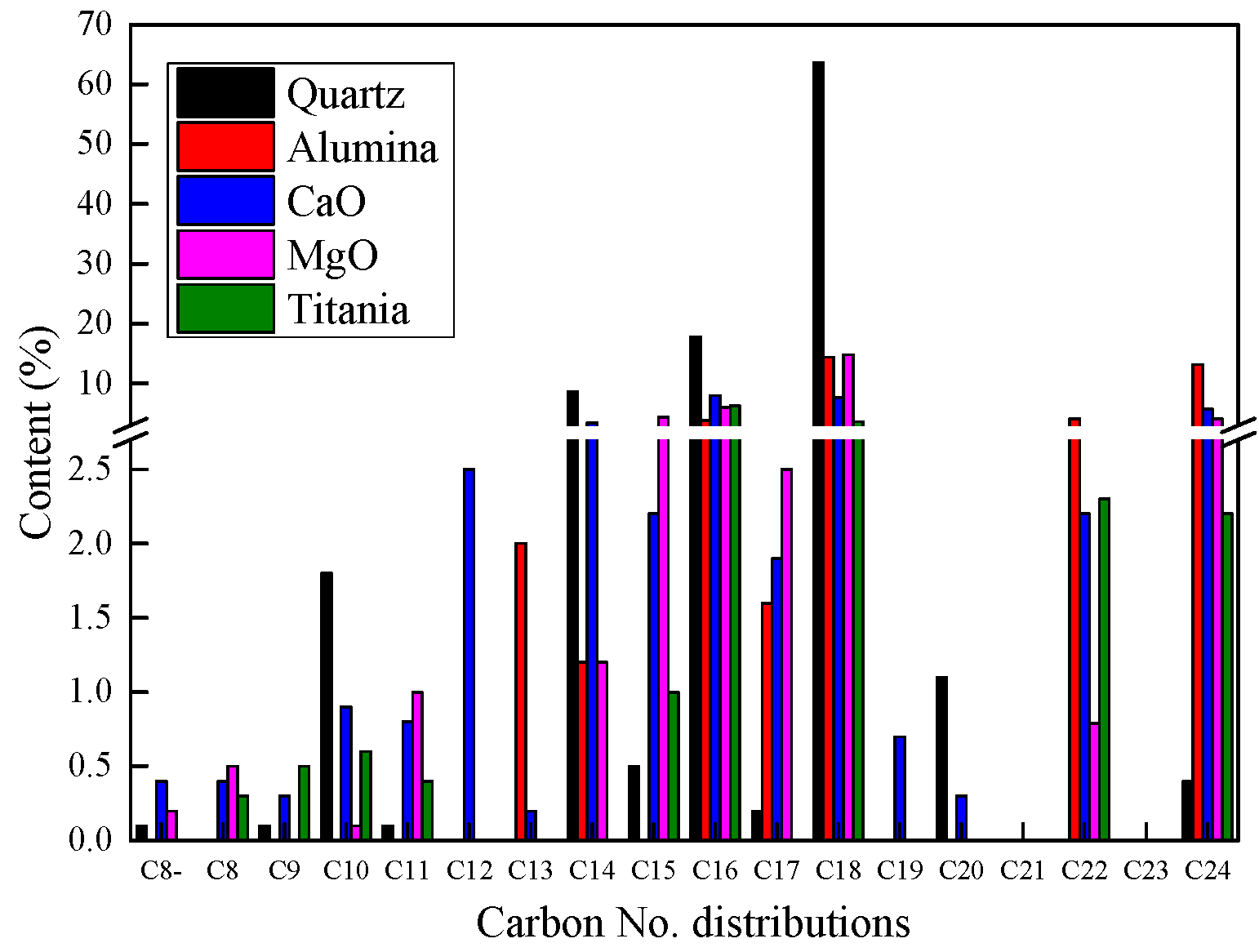
2.2.3. Product Compositions
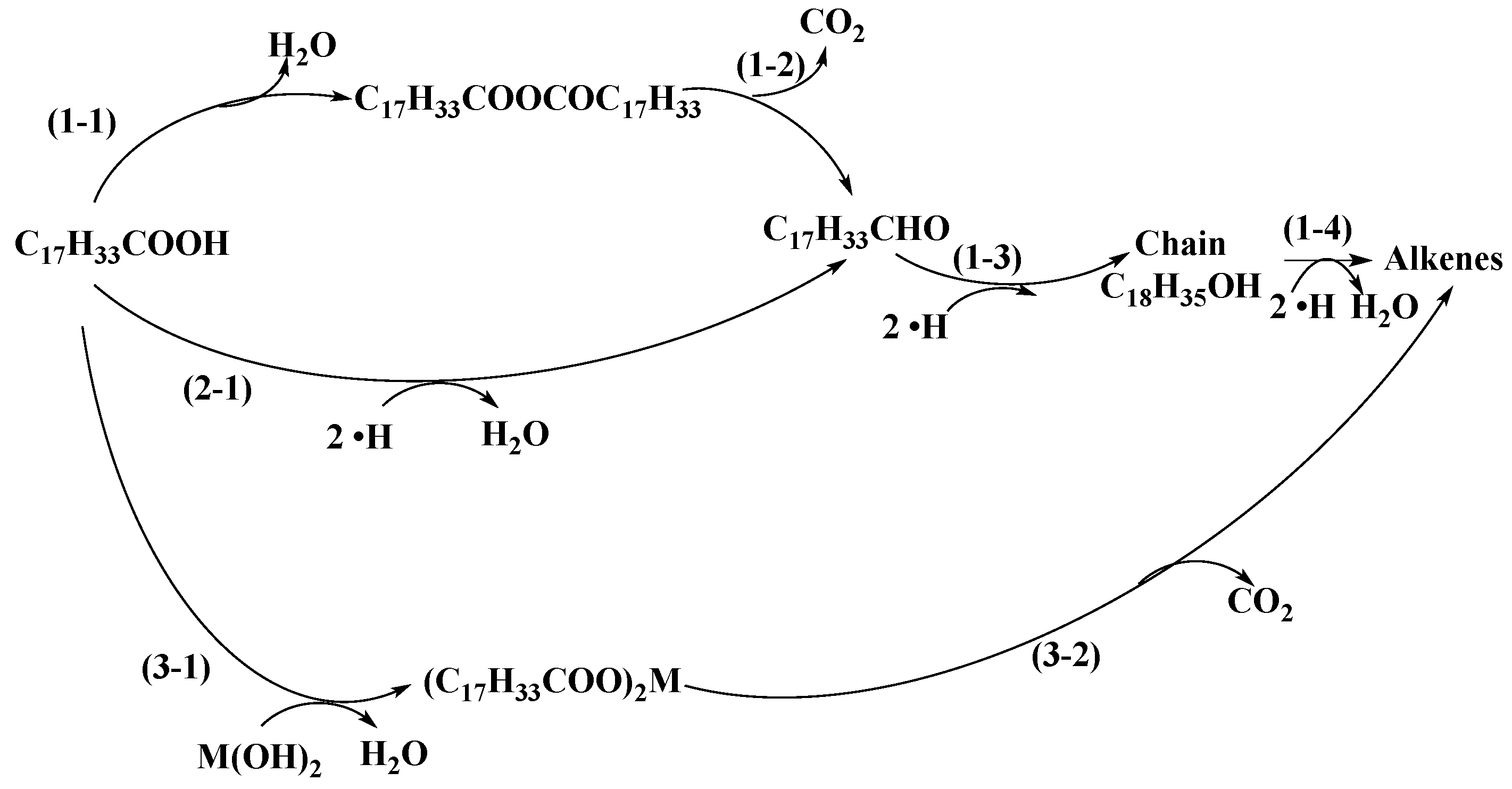
2.3. Reaction Pathways
2.3.1. Acidic Catalysts
2.3.2. Neutral Catalyst
2.3.3. Alkaline Catalysts
2.3.4. Aromatization
3. Materials and Methods
3.1. Catalyst Reaction Performance Evaluation
3.2. Catalyst Preparation
3.3. Products Analysis
3.4. Catalysts Characterization
3.5. Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wang, H.; Li, G.; Rogers, K.; Lin, H.; Zheng, Y.; Ng, S. Hydrotreating of waste cooking oil over supported CoMoS catalyst—Catalyst deactivation mechanism study. Mol. Catal. 2017, 443, 228–240. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, L.; Li, G.; Rogers, K.; Lin, H.; Seers, P.; Ledan, T.; Ng, S.; Zheng, Y. Application of uniform design experimental method in waste cooking oil (WCO) co-hydroprocessing parameter optimization and reaction route investigation. Fuel 2017, 210, 390–397. [Google Scholar] [CrossRef]
- Lu, Q.; Li, W.Z.; Zhu, X.F. Overview of fuel properties of biomass fast pyrolysis oils. Energy Convers. Manag. 2009, 50, 1376–1383. [Google Scholar] [CrossRef]
- Wang, H.; Lin, H.; Feng, P.; Han, X.; Zheng, Y. Integration of catalytic cracking and hydrotreating technology for triglyceride deoxygenation. Catal. Today 2017, 291, 172–179. [Google Scholar] [CrossRef]
- Wang, H.; Lin, H.; Zheng, Y.; Ng, S.; Brown, H.; Xia, Y. Kaolin-based catalyst as a triglyceride FCC upgrading catalyst with high deoxygenation, mild cracking, and low dehydrogenation performances. Catal. Today 2019, 319, 164–171. [Google Scholar] [CrossRef]
- Yigezu, Z.D.; Muthukumar, K. Catalytic cracking of vegetable oil with metal oxides for biofuel production. Energy Convers. Manag. 2014, 84, 326–333. [Google Scholar] [CrossRef]
- Refaat, A.A. Biodiesel production using solid metal oxide catalysts. Int. J. Environ. Sci. Technol. 2011, 8, 203–221. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Bruck, T.; Lercher, J.A. Catalytic deoxygenation of microalgae oil to green hydrocarbons. Green Chem. 2013, 15, 1720–1739. [Google Scholar] [CrossRef]
- Dosanjos, J.R.S.; Gonzalez, W.D.; Lam, Y.L.; Frety, R. Catalytic decomposition of vegetable oil. Appl. Catal. 1983, 5, 299–308. [Google Scholar] [CrossRef]
- Kirszensztejn, P.; Przekop, R.; Tolinska, A.; Mackowska, E. Pyrolytic and catalytic conversion of rape oil into aromatic and aliphatic fractions in a fixed bed reactor on Al2O3 and Al2O3/B2O3 catalysts. Chem. Pap. 2009, 63, 226–232. [Google Scholar] [CrossRef]
- Vonghia, E.; Boocock, D.G.B.; Konar, S.K.; Leung, A. Pathways for the deoxygenation of triglycerides to alphatic-hydrocarbons over activated alumina. Energy Fuels 1995, 9, 1090–1096. [Google Scholar] [CrossRef]
- Idem, R.O.; Katikaneni, S.P.R.; Bakhshi, N.N. Catalytic conversion of canola oil to fuels and chemicals: Roles of catalyst acidity, basicity and shape selectivity on product distribution. Fuel Proc. Technol. 1997, 51, 101–125. [Google Scholar] [CrossRef]
- Smets, K.; Roukaerts, A.; Czech, J.; Reggers, G.; Schreurs, S.; Carleer, R.; Yperman, J. Slow catalytic pyrolysis of rapeseed cake: Product yield and characterization of the pyrolysis liquid. Biomass Bioenergy 2013, 57, 180–190. [Google Scholar] [CrossRef]
- Xu, J.M.; Jiang, J.C.; Lu, Y.J.; Chen, J. Liquid hydrocarbon fuels obtained by the pyrolysis of soybean oils. Bioresour. Technol. 2009, 100, 4867–4870. [Google Scholar]
- Xu, J.; Jiang, J.; Chen, J.; Sun, Y. Biofuel production from catalytic cracking of woody oils. Bioresour. Technol. 2010, 101, 5586–5591. [Google Scholar] [CrossRef]
- Xu, J.; Jiang, J.; Sun, Y.; Chen, J. Production of hydrocarbon fuels from pyrolysis of soybean oils using a basic catalyst. Bioresour. Technol. 2010, 101, 9803–9806. [Google Scholar] [CrossRef]
- Ward, J.W. Nature of active sites on zeolite. Rare earth Y zeolite. J. Catal. 1969, 13, 321–327. [Google Scholar] [CrossRef]
- Serrano, D.P.; Dufour, J.; Iribarren, D. On the feasibility of producing hydrogen with net carbon fixation by the decomposition of vegetable and microalgal oils. Energy Environ. Sci. 2012, 5, 6126–6135. [Google Scholar] [CrossRef]
- Gosselink, R.W.; Hollak, S.A.W.; Chang, S.W.; van Haveren, J.; de Jong, K.P.; Bitter, J.H.; van Es, D.S. Reaction Pathways for the Deoxygenation of Vegetable Oils and Related Model Compounds. ChemSusChem 2013, 6, 1576–1594. [Google Scholar] [CrossRef]
- Rozmyslowicz, B.; Maki-Arvela, P.; Tokarev, A.; Leino, A.R.; Eranen, K.; Murzin, D.Y. Influence of Hydrogen in Catalytic Deoxygenation of Fatty Acids and Their Derivatives over Pd/C. Ind. Eng. Chem. Res. 2012, 51, 8922–8927. [Google Scholar] [CrossRef]
- Immer, J.G.; Kelly, M.J.; Lamb, H.H. Catalytic reaction pathways in liquid-phase deoxygenation of C18 free fatty acids. Appl. Catal. A Gen. 2010, 375, 134–139. [Google Scholar] [CrossRef]
- Demirbas, A.; Kara, H. New options for conversion of vegetable oils to alternative fuels. Energy Sour. Part A Recovery Util. Environ. Eff. 2006, 28, 619–626. [Google Scholar] [CrossRef]
- Benson, T.J.; Daggolu, P.R.; Hernandez, R.A.; Liu, S.T.; White, M.G. Catalytic deoxygenation chemistry: Upgrading of liquids derived from biomass processing. In Advances in Catalysis; Gates, B.C., Jentoft, F.C., Eds.; Academic Press: Cambridge, MA, USA, 2013; Volume 56, pp. 187–353. [Google Scholar]
- Tani, H.; Hasegawa, T.; Shimouchi, M.; Asami, K.; Fujimoto, K. Selective catalytic decarboxy-cracking of triglyceride to middle-distillate hydrocarbon. Catal. Today 2011, 164, 410–414. [Google Scholar] [CrossRef]
- Asomaning, J.; Mussone, P.; Bressler, D.C. Thermal deoxygenation and pyrolysis of oleic acid. J. Anal. Appl. Pyrolysis 2014, 105, 1–7. [Google Scholar] [CrossRef]
- Billaud, F.; Minh, A.K.T.; Lozano, P.; Pioch, D. Catalytic cracking of octanoic acid, Journal of Analytical and Applied Pyrolysis. J. Anal. Appl. Pyrolysis 2001, 58, 605–616. [Google Scholar] [CrossRef]
- Omar, R.; Robinson, J.P. Conventional and microwave-assisted pyrolysis of rapeseed oil for bio-fuel production-reaction pathways analysis by GCMS results. J. Anal. Appl. Pyrolysis 2014, 105, 131–142. [Google Scholar] [CrossRef]
- Yoshioka, T.; Handa, T.; Grause, G.; Lei, Z.G.; Inomata, H.; Mizoguchi, T. Effects of metal oxides on the pyrolysis of poly(ethylene terephthalate). J. Anal. Appl. Pyrolysis 2005, 73, 139–144. [Google Scholar] [CrossRef]
- Cerny, R.; Kubu, M.; Kubicka, D. The effect of oxygenates structure on their deoxygenation over USY zeolite. Catal. Today 2013, 204, 46–53. [Google Scholar] [CrossRef]
- Frety, R.; da Rocha, M.D.G.C.; Brandao, S.T.; Pontes, L.A.M.; Padilha, J.F.; Borges, L.E.P.; Gonzalez, W.A. Cracking and hydrocracking of triglycerides for renewable liquid fuels: Alternative processes to transesterification. J. Braz. Chem. Soc. 2011, 22, 1206–1220. [Google Scholar] [CrossRef]
- Hu, Y. Study on Direct Catalytic Cracking of High TAN Crude Oil. Ph.D. Thesis, China University of Petroleum (East China), Qingdao, China, 2011. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Properties | Surface Area (m2/g) | Pore Volume (cm3/g) | Amount of Active Sites (× 10−2 mmol/g) * | ||
|---|---|---|---|---|---|
| Weak | Medium | Strong | |||
| Quartz 1 | 0.1 | 4.8 × 10−4 | 0.0 | 0.0 | 0.0 |
| MgO 1 | 11.6 | 0.2 | 20.7 | 0.0 | 0.0 |
| CaO 1 | 13.7 | 0.1 | 0.0 | 0.0 | 153.8 |
| TiO2 2 | 56.9 | 0.9 | 39.8 | 10.8 | 8.0 |
| Al2O3 2 | 152.9 | 0.5 | 14.0 | 3.9 | 7.5 |
| Yields (%) | Oil | Coke | Gas Hydrocarbons | Water | CO/CO2 | H2 | Light Oil * Yields | OR |
|---|---|---|---|---|---|---|---|---|
| Quartz | 93.4 | 0.1 | 0.9 | 0.5 | 2.6 | 0.1 | 20.8 | 18.5 |
| Al2O3 | 82.5 | 5.1 | 3.0 | 1.6 | 10.6 | 0.4 | 40.8 | 73.0 |
| TiO2 | 75.3 | 2.0 | 3.6 | 0.9 | 16.3 | 1.4 | 29.1 | 100.0 |
| MgO | 67.5 | 5.3 | 5.5 | 2.3 | 14.9 | 0.1 | 22.0 | 99.4 |
| CaO | 29.0 | 51.2 | 3.8 | 0.0 | 19.8 | 1.0 | 16.6 | 97.7 |
| Molarity (mol/mol Oleic Acid) | CO | CO2 | H2O |
|---|---|---|---|
| Quartz | 0.13 | 0.05 | 0.06 |
| Al2O3 | 0.52 | 0.21 | 0.19 |
| TiO2 | 1.11 | 0.39 | 0.14 |
| MgO | 0.82 | 0.27 | 0.30 |
| CaO | 0.02 | 0.89 | 0.00 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, W.; Wang, H.; Lin, H.; Zheng, Y.; Ng, S.; Shi, M.; Zhao, Y.; Xu, R. Catalytic Decomposition of Oleic Acid to Fuels and Chemicals: Roles of Catalyst Acidity and Basicity on Product Distribution and Reaction Pathways. Catalysts 2019, 9, 1063. https://doi.org/10.3390/catal9121063
Hu W, Wang H, Lin H, Zheng Y, Ng S, Shi M, Zhao Y, Xu R. Catalytic Decomposition of Oleic Acid to Fuels and Chemicals: Roles of Catalyst Acidity and Basicity on Product Distribution and Reaction Pathways. Catalysts. 2019; 9(12):1063. https://doi.org/10.3390/catal9121063
Chicago/Turabian StyleHu, Wanpeng, Hui Wang, Hongfei Lin, Ying Zheng, Siauw Ng, Manlin Shi, Ying Zhao, and Ruoqian Xu. 2019. "Catalytic Decomposition of Oleic Acid to Fuels and Chemicals: Roles of Catalyst Acidity and Basicity on Product Distribution and Reaction Pathways" Catalysts 9, no. 12: 1063. https://doi.org/10.3390/catal9121063