Comparison of Direct and Mediated Electron Transfer for Bilirubin Oxidase from Myrothecium Verrucaria. Effects of Inhibitors and Temperature on the Oxygen Reduction Reaction


Abstract
:1. Introduction
2. Results and Discussion
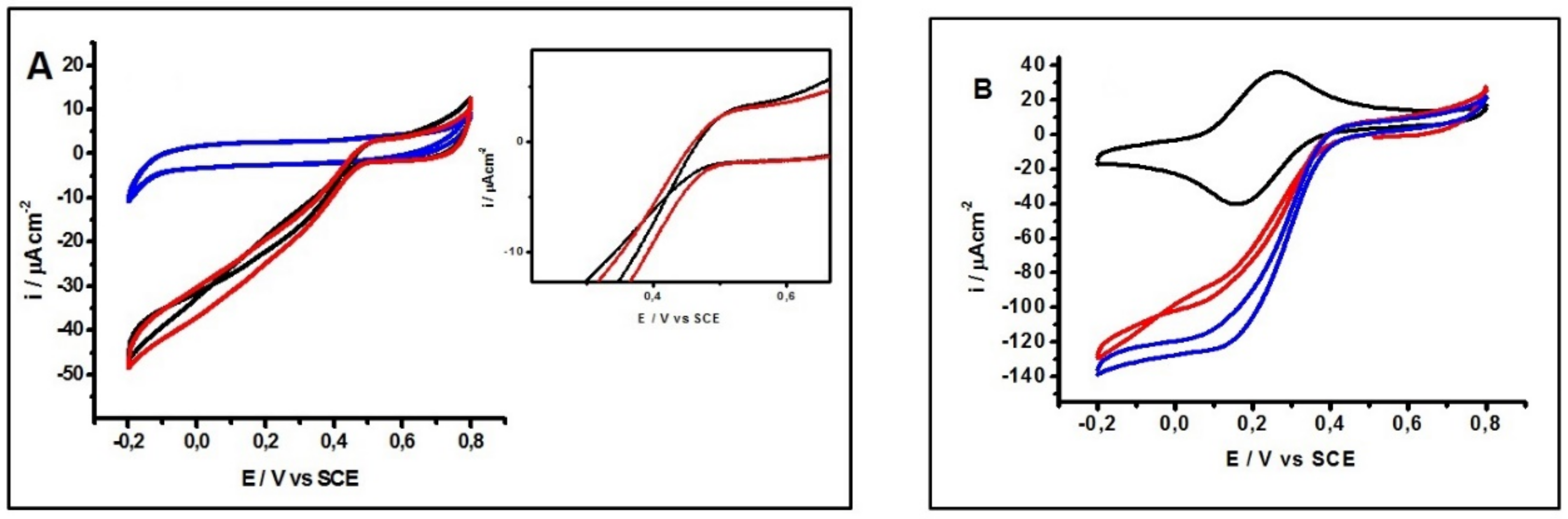
2.1. Electrochemical Characterization
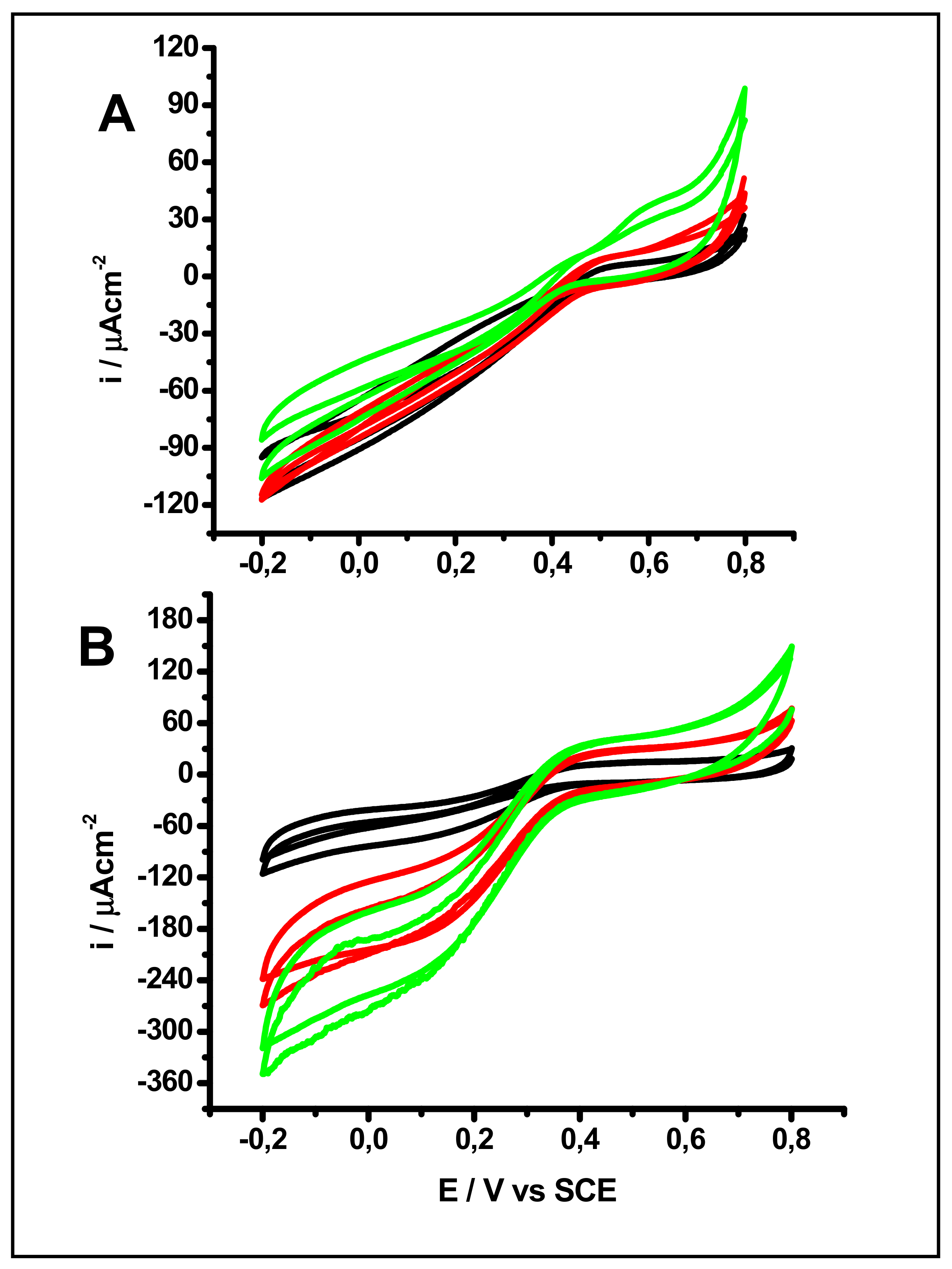
2.2. Effect of pH
2.3. Temperature Effect
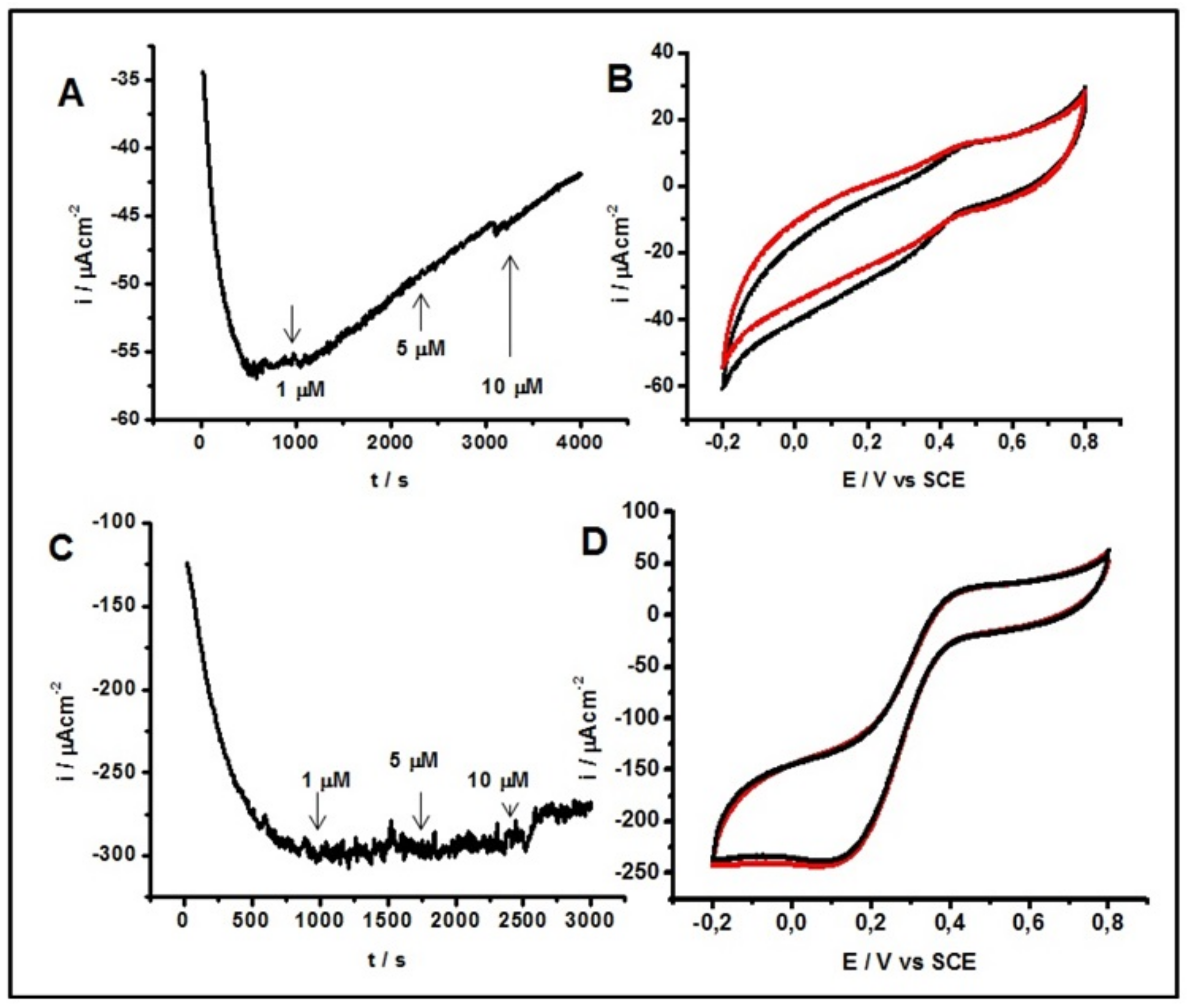
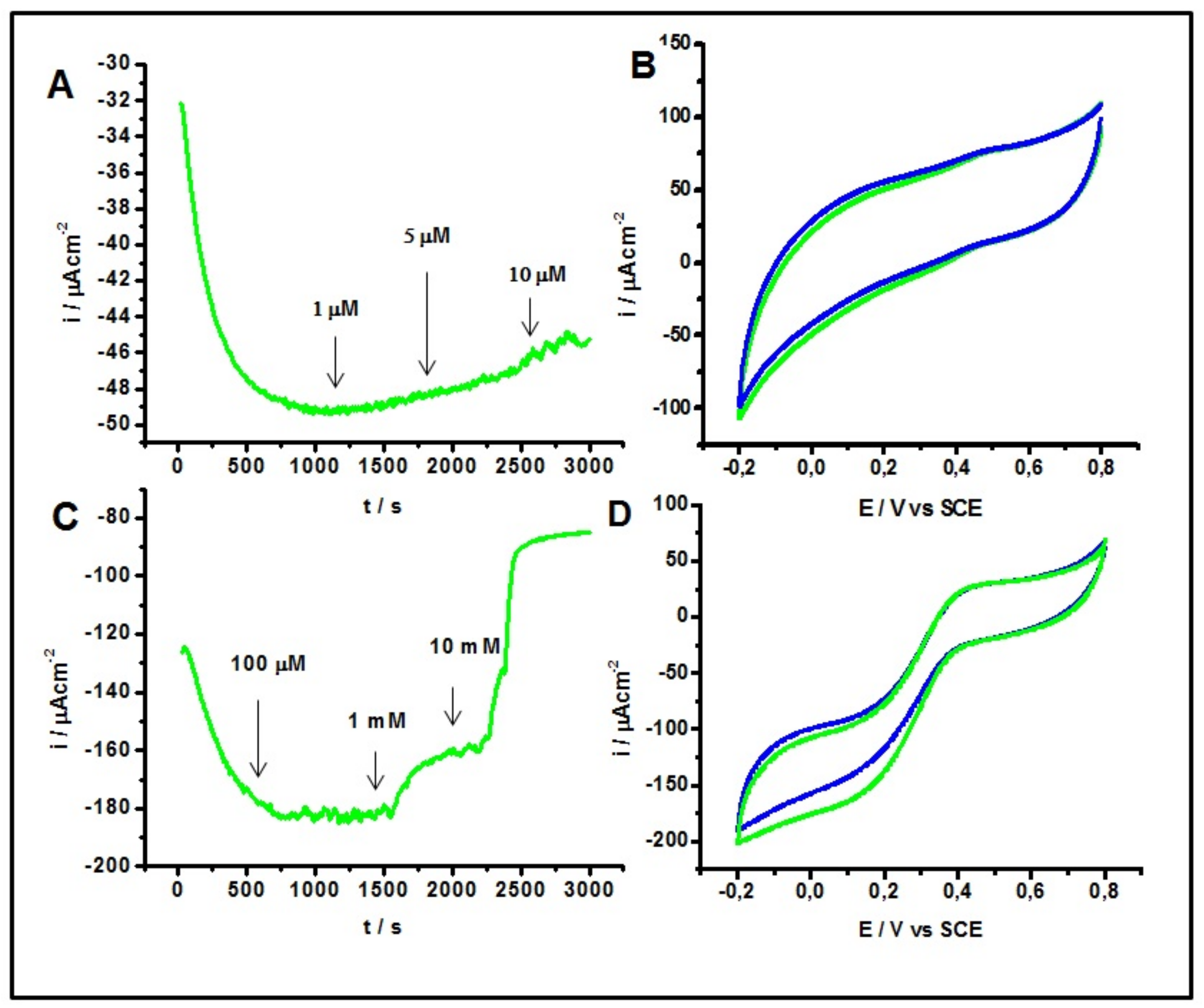
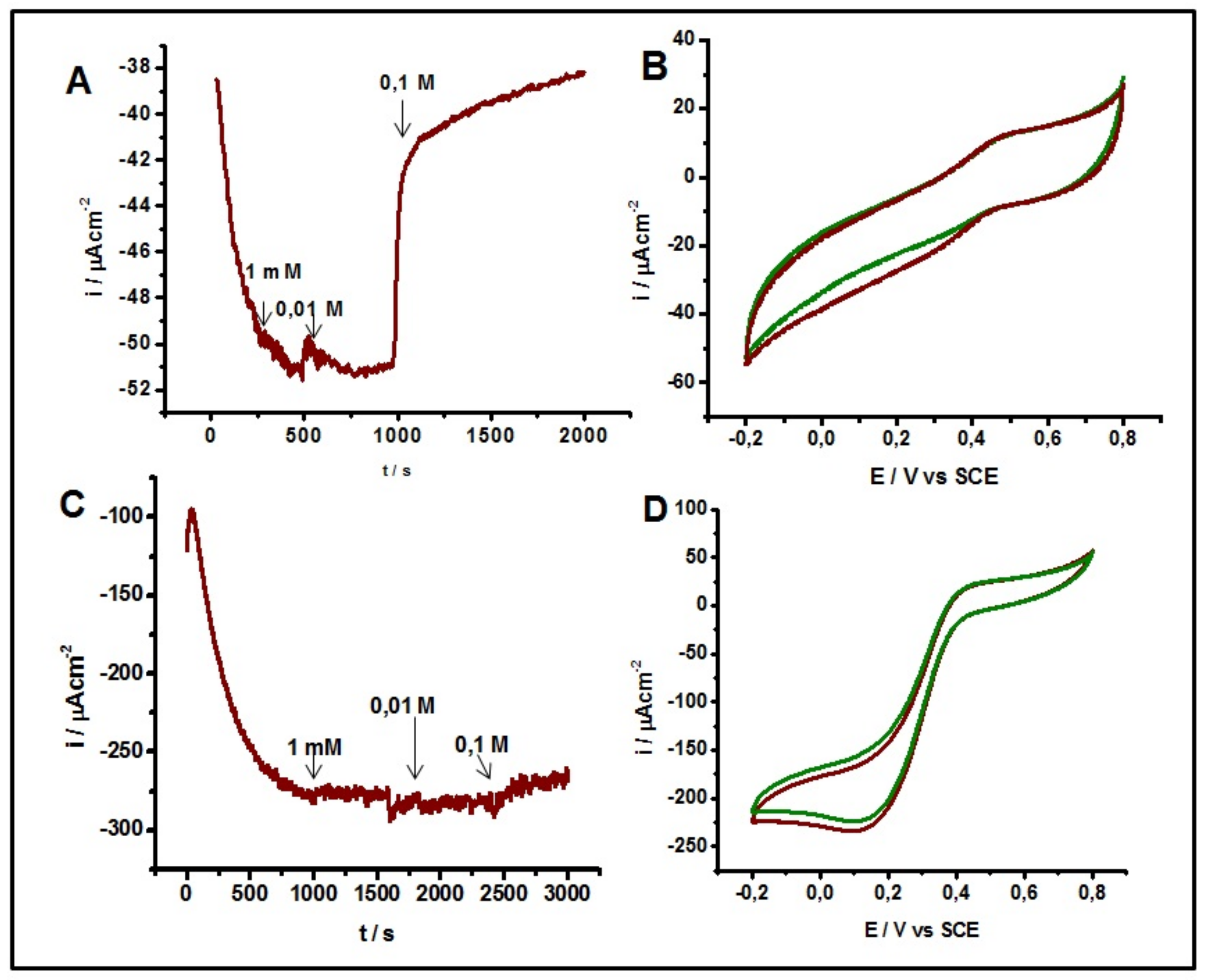
2.4. Inhibitory Effect
3. Experimental
3.1. Chemicals
3.2. Preparation of BOD and BOD-OsP Modified Electrodes
3.3. Electrochemical Measurements
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Balzani, V. (Ed.) Electron. Transfer in Chemistry; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2001. [Google Scholar]
- Gray, H.B.; Winkler, J.R. Electron. Transfer in Metalloproteins; Wiley-VCH: Weinheim, Germany, 2001. [Google Scholar]
- Gamella, M.; Koushanpour, A.; Katz, E. Biofuel cells—Activation of micro- and macro-electronic devices. Bioelectrochemistry 2018, 119, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Solomon, E.I.; Stahl, S.S. Introduction: Oxygen Reduction and Activation in Catalysis. Chem. Rev. 2018, 118, 2299–2301. [Google Scholar] [CrossRef] [PubMed]
- Gewirth, A.A.; Thorum, M.S. Electroreduction of Dioxygen for Fuel-Cell Applications: Materials and Challenges. Inorg. Chem. 2010, 49, 3557–3566. [Google Scholar] [CrossRef] [PubMed]
- Tasca, F.; Farias, D.; Castro, C.; Acuna-Rougier, C.; Antiochia, R. Bilirubin Oxidase from Myrothecium verrucaria Physically Absorbed on Graphite Electrodes. Insights into the Alternative Resting Form and the Sources of Activity Loss. PLoS ONE 2015, 10, e0132181. [Google Scholar] [CrossRef]
- Venegas, R.; Recio, F.J.; Riquelme, J.; Neira, K.; Marco, J.F.; Ponce, I.; Zagal, J.H.; Tasca, F. Biomimetic reduction of O2 in an acid medium on iron phthalocyanines axially coordinated to pyridine anchored on carbon nanotubes. J. Mater. Chem. A 2017, 5, 12054–12059. [Google Scholar] [CrossRef]
- Santoro, C.; Babanova, S.; Erable, B.; Schuler, A.; Atanassov, P. Bilirubin oxidase based enzymatic air-breathing cathode: Operation under pristine and contaminated conditions. Bioelectrochemistry 2016, 108, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Venegas, R.; Recio, F.J.; Zuniga, C.; Viera, M.; Oyarzun, M.-P.; Silva, N.; Neira, K.; Marco, J.F.; Zagal, J.H.; Tasca, F. Comparison of the catalytic activity for O2 reduction of Fe and Co MN4 adsorbed on graphite electrodes and on carbon nanotubes. Phys. Chem. Chem. Phys. 2017, 19, 20441–20450. [Google Scholar] [CrossRef]
- Majumdar, S.; Lukk, T.; Solbiati, J.O.; Bauer, S.; Nair, S.K.; Cronan, J.E.; Gerlt, J.A. Roles of Small Laccases from Streptomyces in Lignin Degradation. Biochemistry 2014, 53, 4047–4058. [Google Scholar] [CrossRef]
- Augustine, A.J.; Kragh, M.E.; Sarangi, R.; Fujii, S.; Liboiron, B.D.; Stoj, C.S.; Kosman, D.J.; Hodgson, K.O.; Hedman, B.; Solomon, E.I. Spectroscopic Studies of Perturbed T1 Cu Sites in the Multicopper Oxidases Saccharomyces cerevisiae Fet3p and Rhus vernicifera Laccase: Allosteric Coupling between the T1 and Trinuclear Cu Sites. Biochemistry 2008, 47, 2036–2045. [Google Scholar] [CrossRef]
- Stone, J.R.; Yang, S. Hydrogen Peroxide: A Signaling Messenger. Antioxid. Redox Signal. 2006, 8, 243–270. [Google Scholar] [CrossRef]
- Johnson, D.L.; Thompson, J.L.; Brinkmann, S.M.; Schuller, K.A.; Martin, L.L. Electrochemical Characterization of Purified Rhus vernicifera Laccase: Voltammetric Evidence for a Sequential Four-Electron Transfer. Biochemistry 2003, 42, 10229–10237. [Google Scholar] [CrossRef]
- Filip, J.; Tkac, J. The pH dependence of the cathodic peak potential of the active sites in bilirubin oxidase. Bioelectrochemistry 2014, 96, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Solomon, E.I.; Sundaram, U.M.; Machonkin, T.E. Multicopper Oxidases and Oxygenases. Chem. Rev. 1996, 96, 2563–2606. [Google Scholar] [CrossRef] [PubMed]
- Mano, N.; Edembe, L. Bilirubin oxidases in bioelectrochemistry: Features and recent findings. Biosens. Bioelectron. 2013, 50, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Cracknell, J.A.; Blanford, C.F. Developing the mechanism of dioxygen reduction catalyzed by multicopper oxidases using protein film electrochemistry. Chem. Sci. 2012, 3, 1567–1581. [Google Scholar] [CrossRef]
- Murao, S.; Tanaka, N. A new enzyme “bilirubin oxidase” produced by Myrothecium verrucaria MT-1. Agric. Biol. Chem. 1981, 45, 2383–2384. [Google Scholar] [CrossRef] [Green Version]
- Goldfinch, M.E.; Maguire, G.A. Investigation of the use of bilirubin oxidase to measure the apparent unbound bilirubin concentration in human plasma. Ann. Clin. Biochem. 1988, 25, 73–77. [Google Scholar] [CrossRef] [Green Version]
- Tsujimura, S.; Fujita, M.; Tatsumi, H.; Kano, K.; Ikeda, T. Bioelectrocatalysis-based dihydrogen/dioxygen fuel cell operating at physiological pH. Phys. Chem. Chem. Phys. 2001, 3, 1331–1335. [Google Scholar] [CrossRef]
- Palmore, G.T.R.; Kim, H.-H. Electro-enzymatic reduction of dioxygen to water in the cathode compartment of a biofuel cell. J. Electroanal. Chem. 1999, 464, 110–117. [Google Scholar] [CrossRef]
- Mano, N.; Kim, H.-H.; Heller, A. On the Relationship between the Characteristics of Bilirubin Oxidases and O2 Cathodes Based on Their “Wiring”. J. Phys. Chem. B 2002, 106, 8842–8848. [Google Scholar] [CrossRef]
- Milton, R.D.; Giroud, F.; Thumser, A.E.; Minteer, S.D.; Slade, R.C.T. Bilirubin oxidase bioelectrocatalytic cathodes: The impact of hydrogen peroxide. Chem. Commun. (Cambridge, UK) 2014, 50, 94–96. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Kataoka, K. Basic and applied features of multicopper oxidases, CueO, bilirubin oxidase, and laccase. Chem. Rec. 2007, 7, 220–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, E.I.; Augustine, A.J.; Yoon, J. O2 Reduction to H2O by the multicopper oxidases. Dalton Trans. 2008, 3921–3932. [Google Scholar] [CrossRef] [PubMed]
- Kjaergaard, C.H.; Durand, F.; Tasca, F.; Qayyum, M.F.; Kauffmann, B.; Gounel, S.; Suraniti, E.; Hodgson, B.; Hedman, K.O.; Mano, N.; et al. Spectroscopic and Crystallographic Characterization of “Alternative Resting” and “Resting Oxidized” Enzyme Forms of Bilirubin Oxidase: Implications for Activity and Electrochemical Behavior of Multicopper Oxidases. J. Am. Chem. Soc. 2012, 134, 5548–5551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kjaergaard, C.H.; Jones, S.M.; Gounel, S.; Mano, N.; Solomon, E.I. Two-Electron Reduction versus One-Electron Oxidation of the Type 3 Pair in the Multicopper Oxidases. J. Am. Chem. Soc. 2015, 137, 8783–8794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barton, S.C.; Gallaway, J.; Atanassov, P. Enzymatic biofuel cells for implantable and microscale devices. Chem. Rev. (Washington, DC, USA) 2004, 104, 4867–4886. [Google Scholar] [CrossRef]
- Gallaway, J.W.; Calabrese Barton, S.A. Effect of redox polymer synthesis on the performance of a mediated laccase oxygen cathode. J. Electroanal. Chem. 2009, 626, 149–155. [Google Scholar] [CrossRef]
- Szamocki, R.; Flexer, V.; Levin, L.; Forchiasin, F.; Calvo, E.J. Oxygen cathode based on a layer-by-layer self-assembled laccase and osmium redox mediator. Electrochim. Acta 2009, 54, 1970–1977. [Google Scholar] [CrossRef] [Green Version]
- Grattieri, M.; Scodeller, P.; Adam, C.; Calvo, E.J. Non-competitive reversible inhibition of laccase by H2O2 in osmium mediated layer-by-layer multilayer O2 biocathodes. J. Electrochem. Soc. 2015, 162, G82–G86. [Google Scholar] [CrossRef]
- Scodeller, P.; Carballo, R.; Szamocki, R.; Levin, L.; Forchiassin, F.; Calvo, E.J. Layer-by-layer self-assembled osmium polymer-mediated laccase oxygen cathodes for biofuel cells: The role of hydrogen peroxide. J. Am. Chem. Soc. 2010, 132, 11132–11140. [Google Scholar] [CrossRef]
- Ackermann, Y.; Guschin, D.A.; Eckhard, K.; Shleev, S.; Schuhmann, W. Design of a bioelectrocatalytic electrode interface for oxygen reduction in biofuel cells based on a specifically adapted Os-complex containing redox polymer with entrapped Trametes hirsuta laccase. Electrochem. Commun. 2010, 12, 640–643. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, P.A.; Boland, S.; Kavanagh, P.; Leech, D. Evaluation of performance and stability of biocatalytic redox films constructed with different copper oxygenases and osmium-based redox polymers. Bioelectrochemistry 2009, 76, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Lalaoui, N.; Reuillard, B.; Philouze, C.; Holzinger, M.; Cosnier, S.; Le Goff, A. Osmium (II) Complexes Bearing Chelating N-Heterocyclic Carbene and Pyrene-Modified Ligands: Surface Electrochemistry and Electron Transfer Mediation of Oxygen Reduction by Multicopper Enzymes. Organometallics 2016, 35, 2987–2992. [Google Scholar] [CrossRef]
- Shin, H.; Cho, S.; Heller, A.; Kang, C. Stabilization of a Bilirubin Oxidase-Wiring Redox Polymer by Quaternization and Characteristics of the Resulting O2 Cathode. J. Electrochem. Soc. 2009, 156, F87–F92. [Google Scholar] [CrossRef]
- Shin, H.; Kang, C. Co-electrodeposition of bilirubin oxidase with redox polymer through ligand substitution for use as an oxygen reduction cathode. Bull. Korean Chem. Soc. 2010, 31, 3118–3122. [Google Scholar] [CrossRef] [Green Version]
- Mano, N.; Kim, H.-H.; Zhang, Y.; Heller, A. An Oxygen Cathode Operating in a Physiological Solution. J. Am. Chem. Soc. 2002, 124, 6480–6486. [Google Scholar] [CrossRef] [PubMed]
- Poeller, S.; Beyl, Y.; Vivekananthan, J.; Guschin, D.A.; Schuhmann, W. A new synthesis route for Os-complex modified redox polymers for potential biofuel cell applications. Bioelectrochemistry 2002, 87, 178–184. [Google Scholar] [CrossRef]
- Wang, X.; Falk, M.; Ortiz, R.; Matsumura, H.; Bobacka, J.; Ludwig, R.; Bergelin, M.; Gorton, L.; Shleev, S. Mediatorless sugar/oxygen enzymatic fuel cells based on gold nanoparticle-modified electrodes. Biosens. Bioelectron. 2012, 31, 219–225. [Google Scholar] [CrossRef]
- Salaj-Kosla, U.; Poller, S.; Schuhmann, W.; Shleev, S.; Magner, E. Direct electron transfer of Trametes hirsuta laccase adsorbed at unmodified nanoporous gold electrodes. Bioelectrochemistry 2013, 91, 15–20. [Google Scholar] [CrossRef] [Green Version]
- Leger, C.; Jones, A.K.; Albracht, S.P.J.; Armstrong, F.A. Effect of a Dispersion of Interfacial Electron Transfer Rates on Steady State Catalytic Electron Transport in [NiFe]-hydrogenase and Other Enzymes. J. Phys. Chem. B 2002, 106, 13058–13063. [Google Scholar] [CrossRef]
- Dos Santos, L.; Climent, V.; Blanford, C.F.; Armstrong, F.A. Mechanistic studies of the ’blue’ Cu enzyme, bilirubin oxidase, as a highly efficient electrocatalyst for the oxygen reduction reaction. Phys. Chem. Chem. Phys. 2010, 12, 13962–13974. [Google Scholar] [CrossRef] [PubMed]
- Reiss, R.; Ihssen, J.; Thony-Meyer, L. Bacillus pumilus laccase: A heat stable enzyme with a wide substrate spectrum. BMC Biotechnol. 2011, 11, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mano, N.; de Poulpiquet, A. O2 Reduction in Enzymatic Biofuel Cells. Chem. Rev. (Washington, DC, USA) 2018, 118, 2392–2468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentil, S.; Carriere, M.; Cosnier, S.; Gounel, S.; Mano, N.; Le Goff, A. Direct electrochemistry of bilirubin oxidase from Magnaporthe orizae on covalently-functionalized MWCNT for the design of high-performance oxygen-reducing biocathodes. Chem. Eur. J. 2018, 24, 8404–8408. [Google Scholar] [CrossRef] [PubMed]
- De Poulpiquet, A.; Kjaergaard, C.H.; Rouhana, J.; Mazurenko, I.; Infossi, P.; Gounel, S.; Gadiou, R.; Giudici-Orticoni, M.T.; Solomon, E.I.; Mano, N.; et al. Mechanism of chloride inhibition of bilirubin oxidases and its dependence on potential and pH. ACS Catal. 2017, 7, 3916–3923. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Zhao, M.; Lu, L.; Liu, Y. Purification, characterization and decolorization of bilirubin oxidase from Myrothecium verrucaria 3.2190. Fungal Biol. 2012, 116, 863–871. [Google Scholar] [CrossRef]
- Zafar, M.N.; Tasca, F.; Boland, S.; Kujawa, M.; Patel, I.; Peterbauer, C.K.; Leech, D.; Gorton, L. Wiring of pyranose dehydrogenase with osmium polymers of different redox potentials. Bioelectrochemistry 2010, 80, 38–42. [Google Scholar] [CrossRef]
- Mano, N. Features and applications of bilirubin oxidases. Appl. Microbiol. Biotechnol. 2012, 96, 301–307. [Google Scholar] [CrossRef]
- De Poulpiquet, A.; Ciaccafava, A.; Gadiou, R.; Gounel, S.; Giudici-Orticoni, M.T.; Mano, N.; Lojou, E. Design of a H2/O2 biofuel cell based on thermostable enzymes. Electrochem. Commun. 2014, 42, 72–74. [Google Scholar] [CrossRef]
- Chen, X.; Yin, L.; Lv, J.; Gross, A.J.; Le, M.; Gutierrez, N.G.; Li, Y.; Jeerapan, I.; Giroud, F.; Berezovska, A.; et al. Stretchable and Flexible Buckypaper-Based Lactate Biofuel Cell for Wearable Electronics. Adv. Funct. Mater. 2019, 29, 1905785. [Google Scholar] [CrossRef]
- Kang, C.; Shin, H.; Heller, A. On the stability of the “wired” bilirubin oxidase oxygen cathode in serum. Bioelectrochemistry 2006, 68, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Milton, R.D.; Minteer, S.D. Investigating the reversible inhibition model of laccase by hydrogen peroxide for bioelectrocatalytic applications. J. Electrochem. Soc. 2014, 161, H3011–H3014. [Google Scholar] [CrossRef] [Green Version]
- Di Bari, C.; Mano, N.; Shleev, S.; Pita, M.; De Lacey, A.L. Halides inhibition of multicopper oxidases studied by FTIR spectroelectrochemistry using azide as an active infrared probe. JBIC J. Biol. Inorg. Chem. 2017, 22, 1179–1186. [Google Scholar] [CrossRef] [PubMed]
- Vaz-Dominguez, C.; Campuzano, S.; Ruediger, O.; Pita, M.; Gorbacheva, M.; Shleev, S.; Fernandez, V.M.; De Lacey, A.L. Laccase electrode for direct electrocatalytic reduction of O2 to H2O with high-operational stability and resistance to chloride inhibition. Biosens. Bioelectron. 2008, 24, 531–537. [Google Scholar] [CrossRef] [Green Version]
- Tominaga, M.; Sasaki, A.; Togami, M. Bioelectrocatalytic oxygen reaction and chloride inhibition resistance of laccase immobilized on single-walled carbon nanotube and carbon paper electrodes. Electrochemistry (Tokyo, Jpn.) 2016, 84, 315–318. [Google Scholar] [CrossRef] [Green Version]
- Enaud, E.; Trovaslet, M.; Naveau, F.; Decristoforo, A.; Bizet, S.; Vanhulle, S.; Jolivalt, C. Laccase chloride inhibition reduction by an anthraquinonic substrate. Enzym. Microb. Technol. 2011, 49, 517–525. [Google Scholar] [CrossRef]
- Jensen, U.B.; Vagin, M.; Koroleva, O.; Sutherland, D.S.; Besenbacher, F.; Ferapontova, E.E. Activation of laccase bioelectrocatalysis of O2 reduction to H2O by carbon nanoparticles. J. Electroanal. Chem. 2012, 667, 11–18. [Google Scholar] [CrossRef]
- Boland, S. Integrating Enzymes, Electron Transfer Mediators and Mesostructured Surfaces to Provide Bio-Electroresponsive Systems. Ph.D. Thesis, National University of Ireland, Galway, Ireland, 2008. [Google Scholar]
- Zafar, M.N.; Tasca, F.; Gorton, L.; Patridge, E.V.; Ferry, J.G.; Noll, G. Tryptophan Repressor-Binding Proteins from Escherichia coli and Archaeoglobus fulgidus as New Catalysts for 1,4-Dihydronicotinamide Adenine Dinucleotide-Dependent Amperometric Biosensors and Biofuel Cells. Anal. Chem. 2009, 81, 4082–4088. [Google Scholar] [CrossRef]
- Antiochia, R.; Tasca, F.; Mannina, L. Osmium-Polymer modified carbon nanotube paste electrode for detection of sucrose and fructose. Mater. Sci. Appl. 2013, 4, 15–22. [Google Scholar] [CrossRef]
- Kurbanoglu, S.; Zafar, M.N.; Tasca, F.; Aslam, I.; Spadiut, O.; Leech, D.; Haltrich, D.; Gorton, L. Amperometric Flow Injection Analysis of Glucose and Galactose Based on Engineered Pyranose 2-Oxidases and Osmium Polymers for Biosensor Applications. Electroanalysis 2018, 30, 1496–1504. [Google Scholar] [CrossRef] [Green Version]
- Heller, A. Electrical connection of enzyme redox centers to electrodes. J. Phys. Chem. 1992, 96, 3579–3587. [Google Scholar] [CrossRef]
- Recio, F.J.; Canete, P.; Tasca, F.; Linares-Flores, C.; Zagal, J.H. Tuning the Fe (II)/(I) formal potential of the FeN4 catalysts adsorbed on graphite electrodes to the reversible potential of the reaction for maximum activity: Hydrazine oxidation. Electrochem. Commun. 2013, 30, 34–37. [Google Scholar] [CrossRef]
- Tasca, F.; Recio, F.J.; Venegas, R.; Geraldo, D.A.; Sancy, M.; Zagal, J.H. Linear versus volcano correlations for the electrocatalytic oxidation of hydrazine on graphite electrodes modified with MN4 macrocyclic complexes. Electrochim. Acta 2014, 140, 314–319. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DET | MET | |||
|---|---|---|---|---|
| pH | High Potential Hysteresis, V | Low Potential Hysteresis, V | High Potential Hysteresis, V | Low Potential Hysteresis, V |
| 7 | 0.440 ± 5 | 0.100 ± 5 | 0.300 ± 5 | 0.000 ± 5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antiochia, R.; Oyarzun, D.; Sánchez, J.; Tasca, F. Comparison of Direct and Mediated Electron Transfer for Bilirubin Oxidase from Myrothecium Verrucaria. Effects of Inhibitors and Temperature on the Oxygen Reduction Reaction. Catalysts 2019, 9, 1056. https://doi.org/10.3390/catal9121056
Antiochia R, Oyarzun D, Sánchez J, Tasca F. Comparison of Direct and Mediated Electron Transfer for Bilirubin Oxidase from Myrothecium Verrucaria. Effects of Inhibitors and Temperature on the Oxygen Reduction Reaction. Catalysts. 2019; 9(12):1056. https://doi.org/10.3390/catal9121056
Chicago/Turabian StyleAntiochia, Riccarda, Diego Oyarzun, Julio Sánchez, and Federico Tasca. 2019. "Comparison of Direct and Mediated Electron Transfer for Bilirubin Oxidase from Myrothecium Verrucaria. Effects of Inhibitors and Temperature on the Oxygen Reduction Reaction" Catalysts 9, no. 12: 1056. https://doi.org/10.3390/catal9121056