Kinetics of Fischer–Tropsch Synthesis in a 3-D Printed Stainless Steel Microreactor Using Different Mesoporous Silica Supported Co-Ru Catalysts

Abstract
:1. Introduction
2. Results and Discussion
2.1. Catalyst Characterization
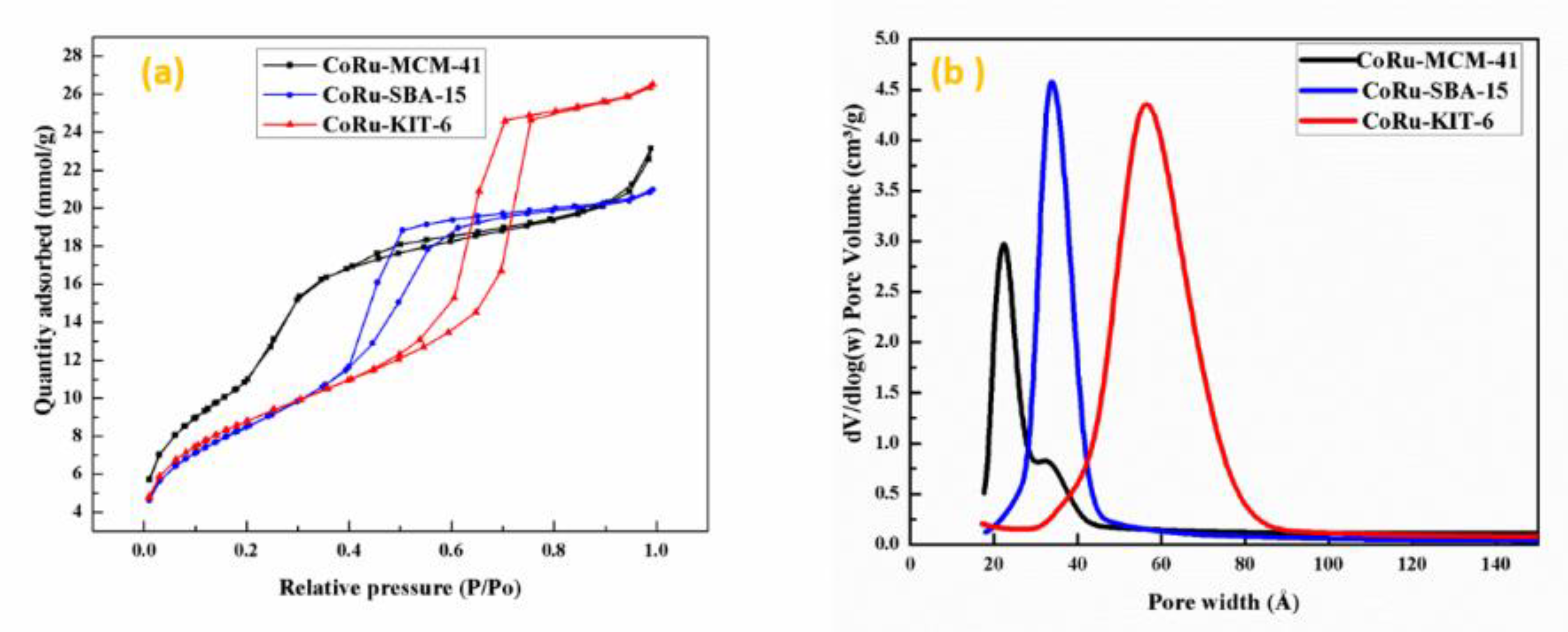
2.1.1. Textural Evaluation of Catalysts
2.1.2. SEM-EDX Analysis
2.1.3. Transmission Electron Microscopic (TEM) Imaging
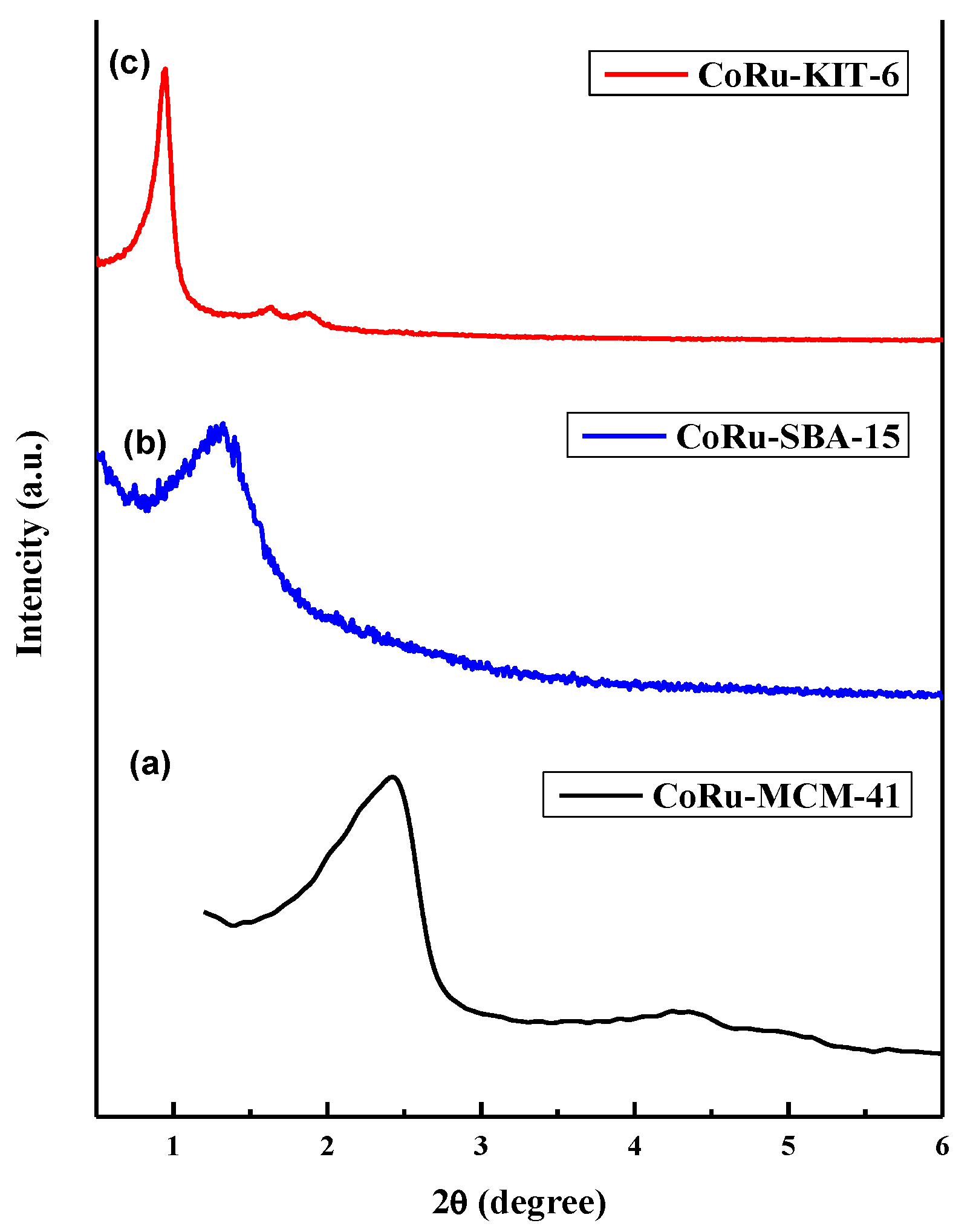
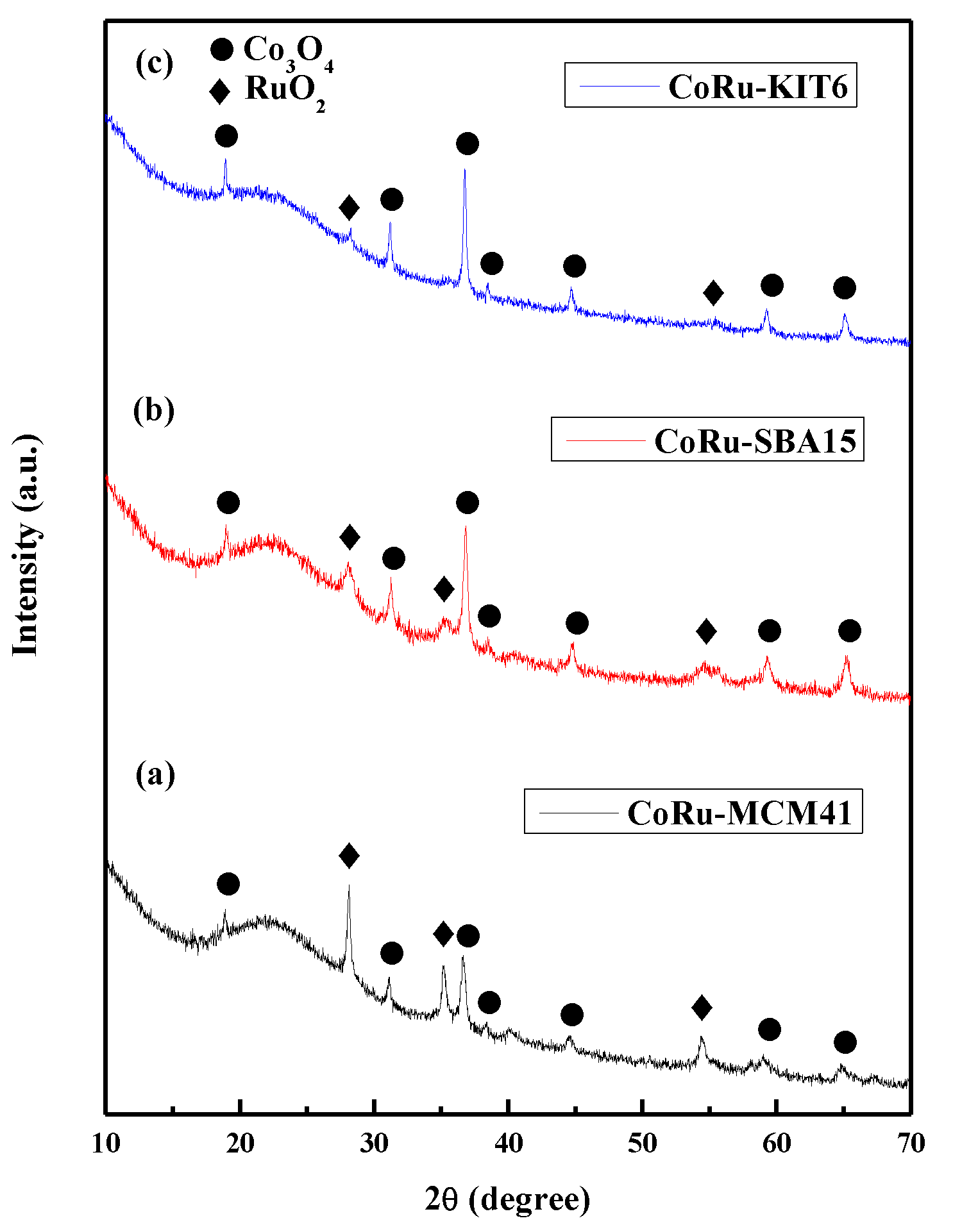
2.1.4. Powder X-Ray Diffraction (XRD) Studies of Calcined Catalysts
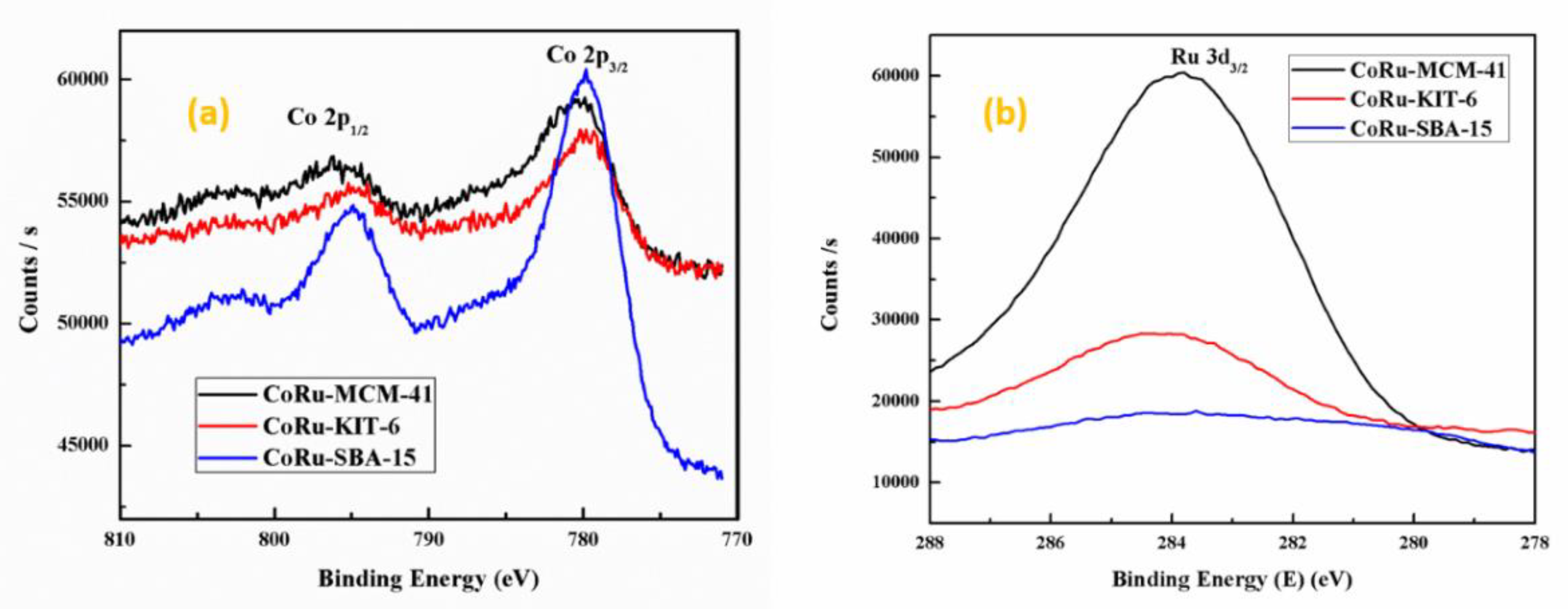
2.1.5. X-Ray Photoelectron Spectroscopy (XPS)
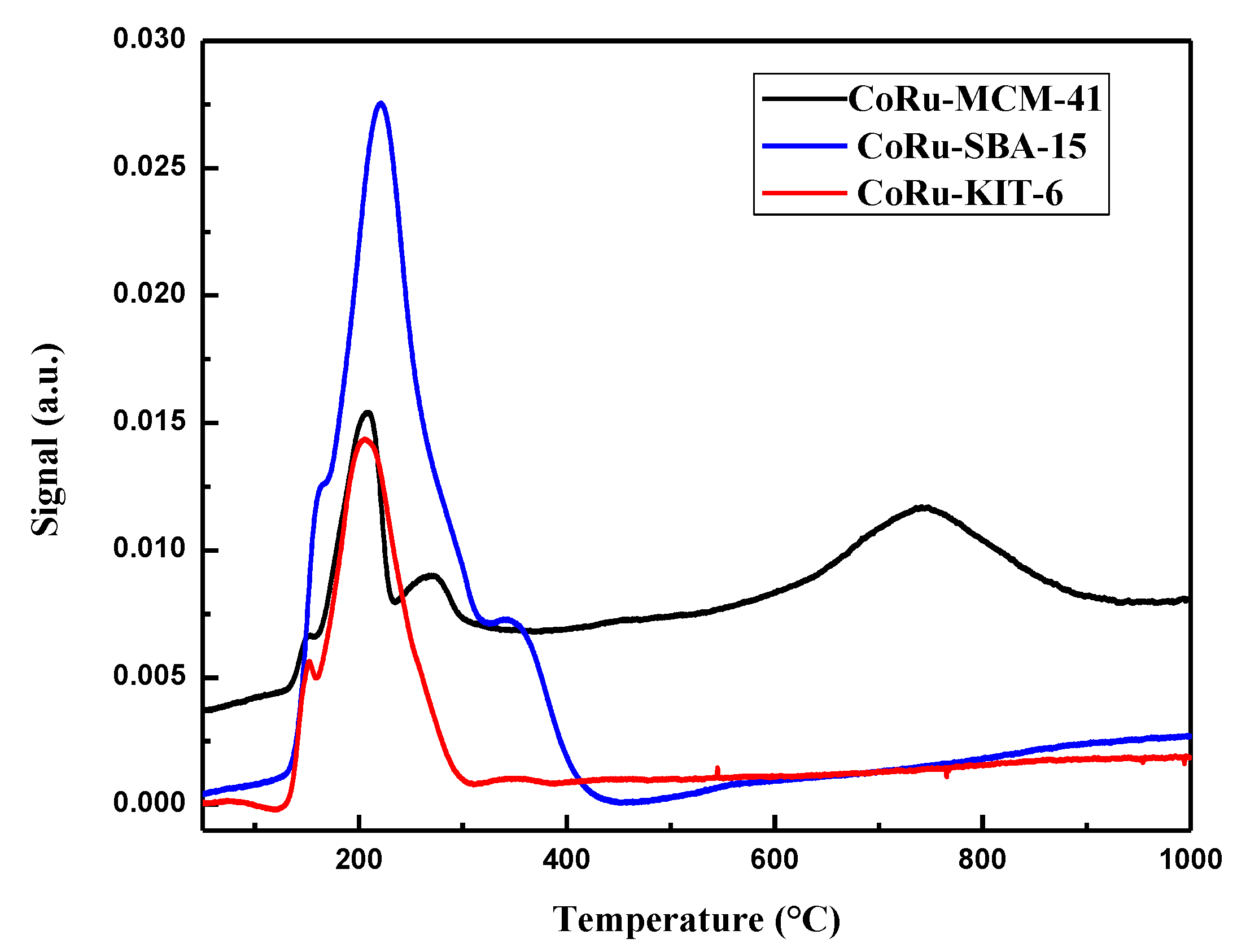
2.1.6. H2 Temperature Programmed Reduction (H2 TPR)
2.2. FT Reaction Mechanism
2.2.1. Reaction Mechanism
- = Wt. of the catalyst
- = Molar feed rate of CO
- = Conversion of CO
- = Disappearance rate of CO
- W = 0; Fi =Fi(inlet)
- W = Wcat; Fi = Fi(exit)
2.2.2. Mechanism and Kinetics
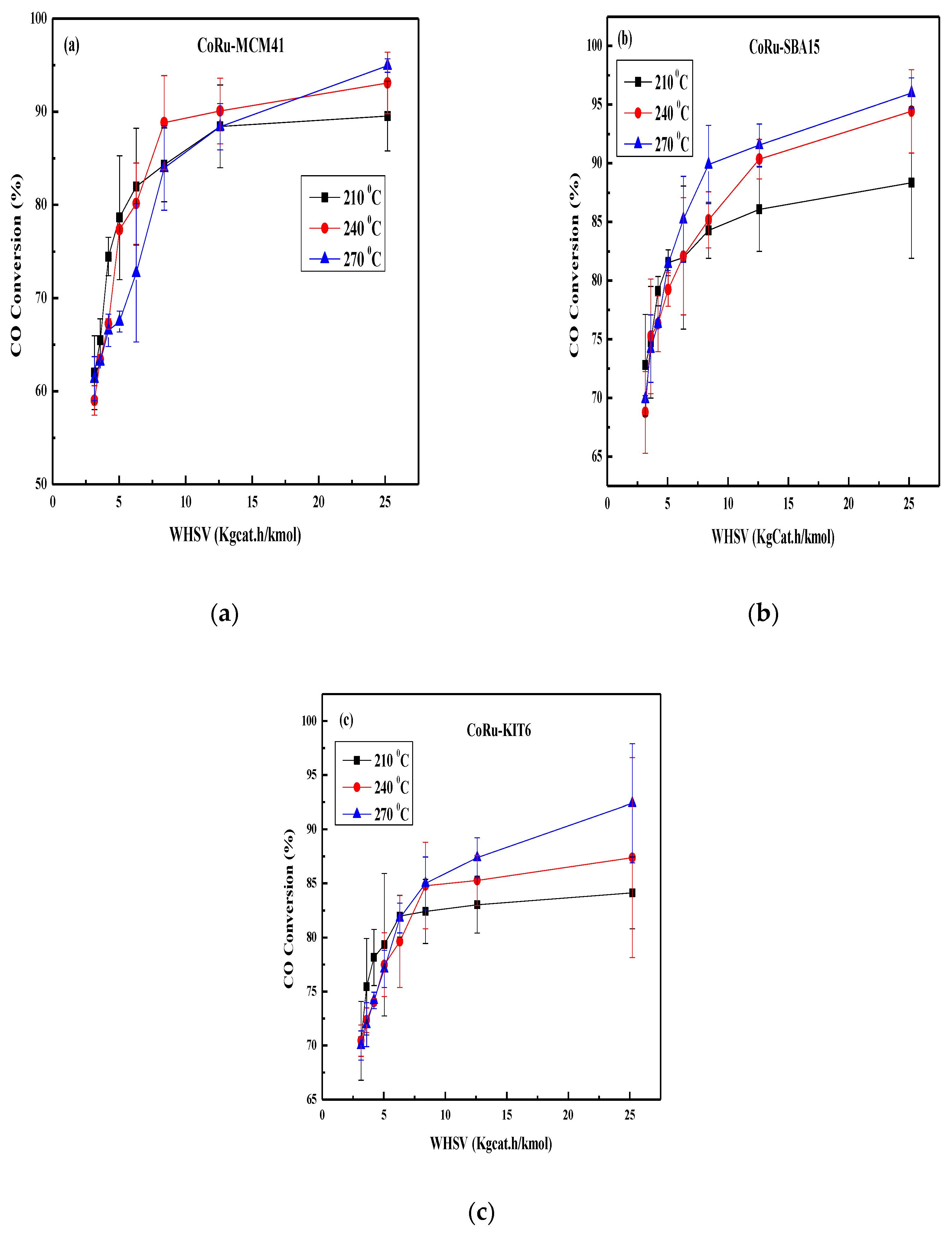
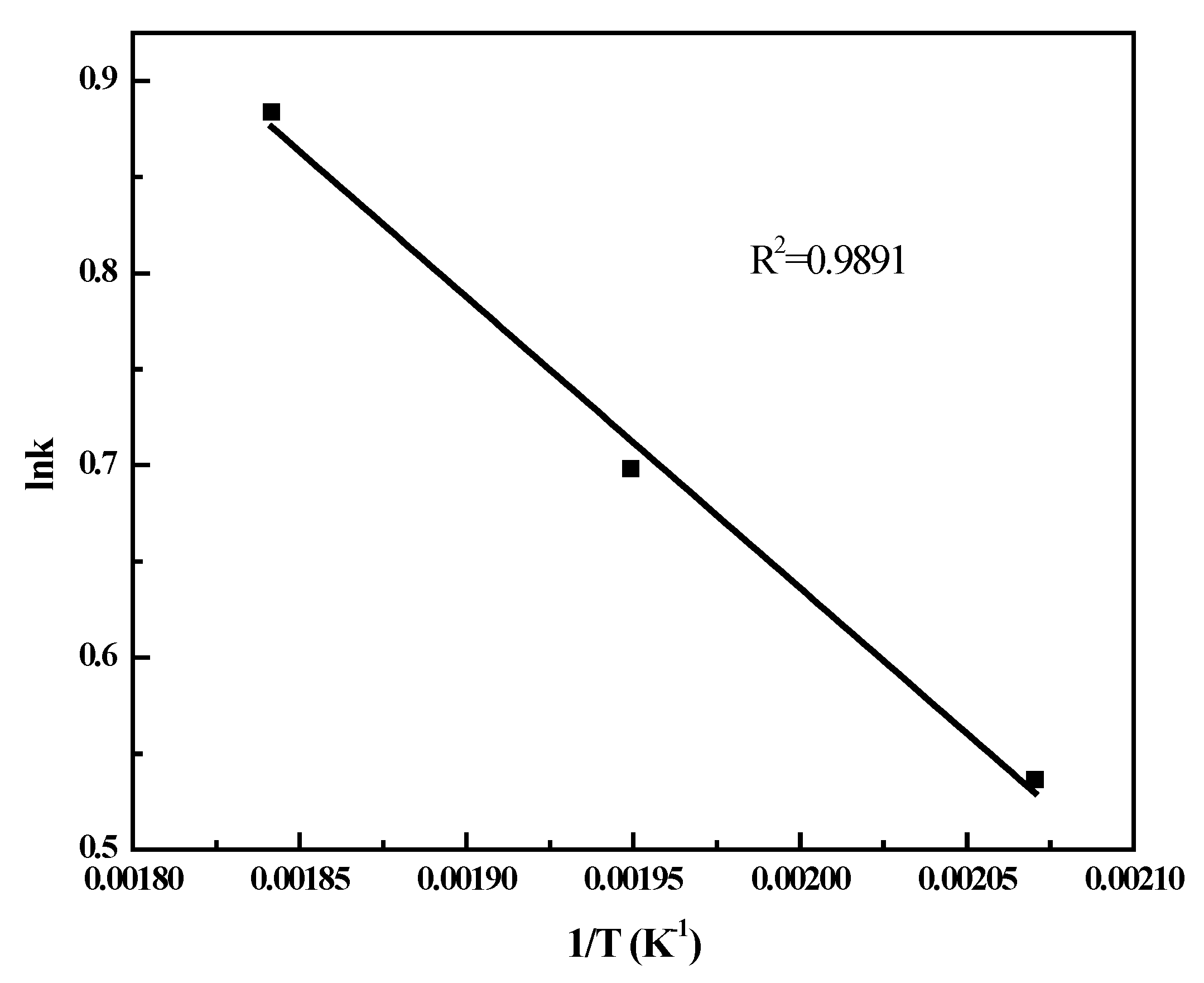
2.3. Effect of Space Velocity on CO Conversion
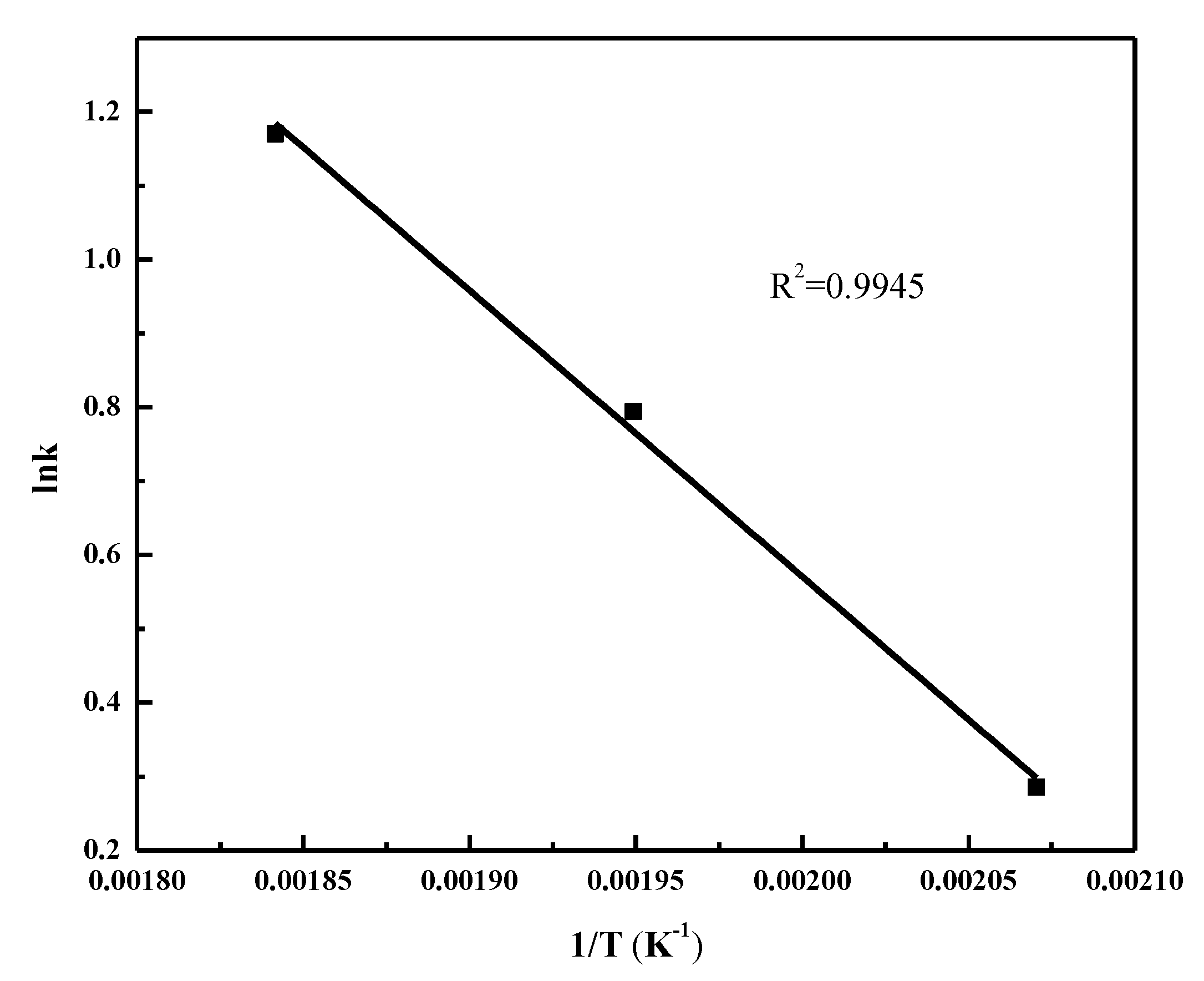
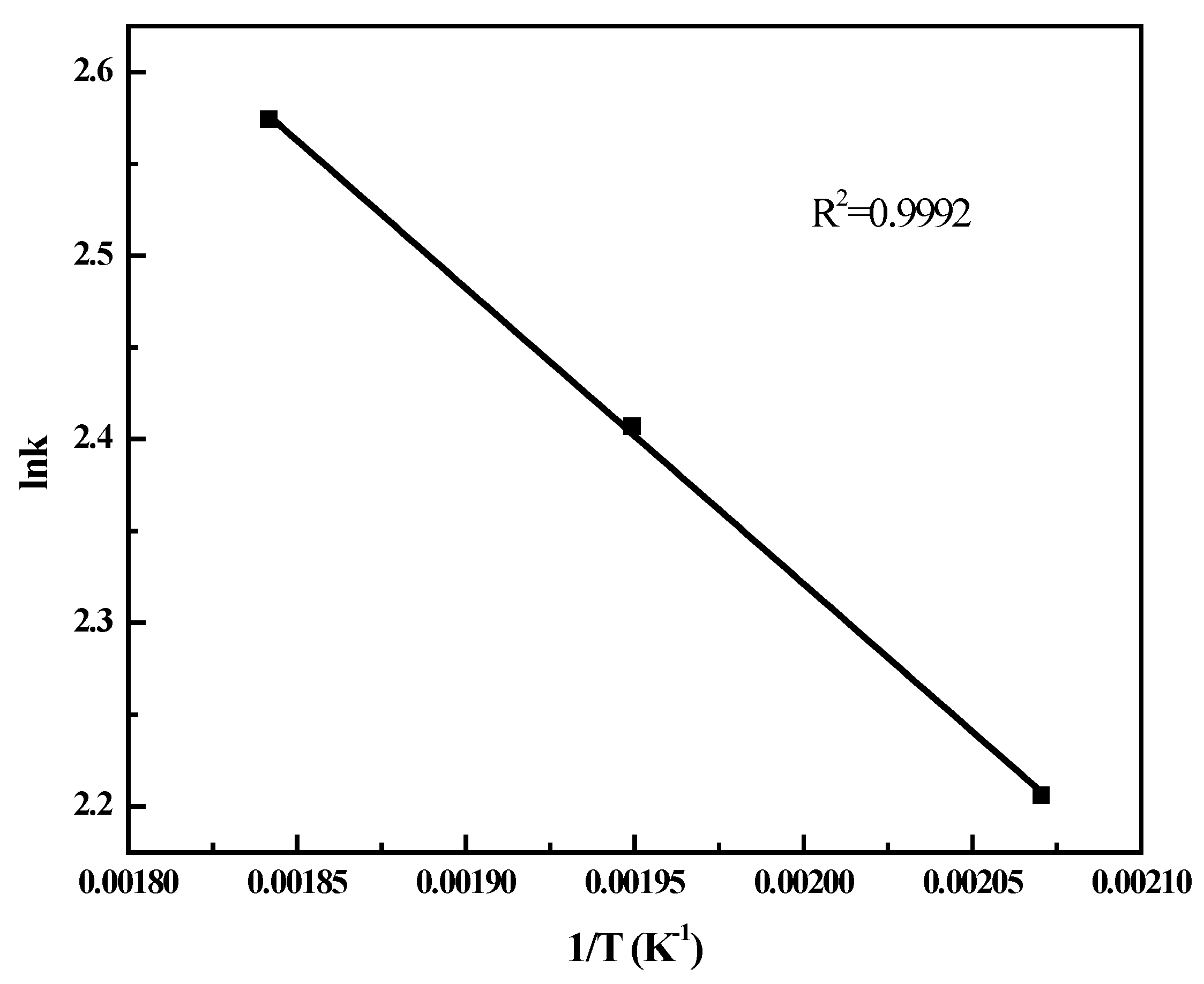
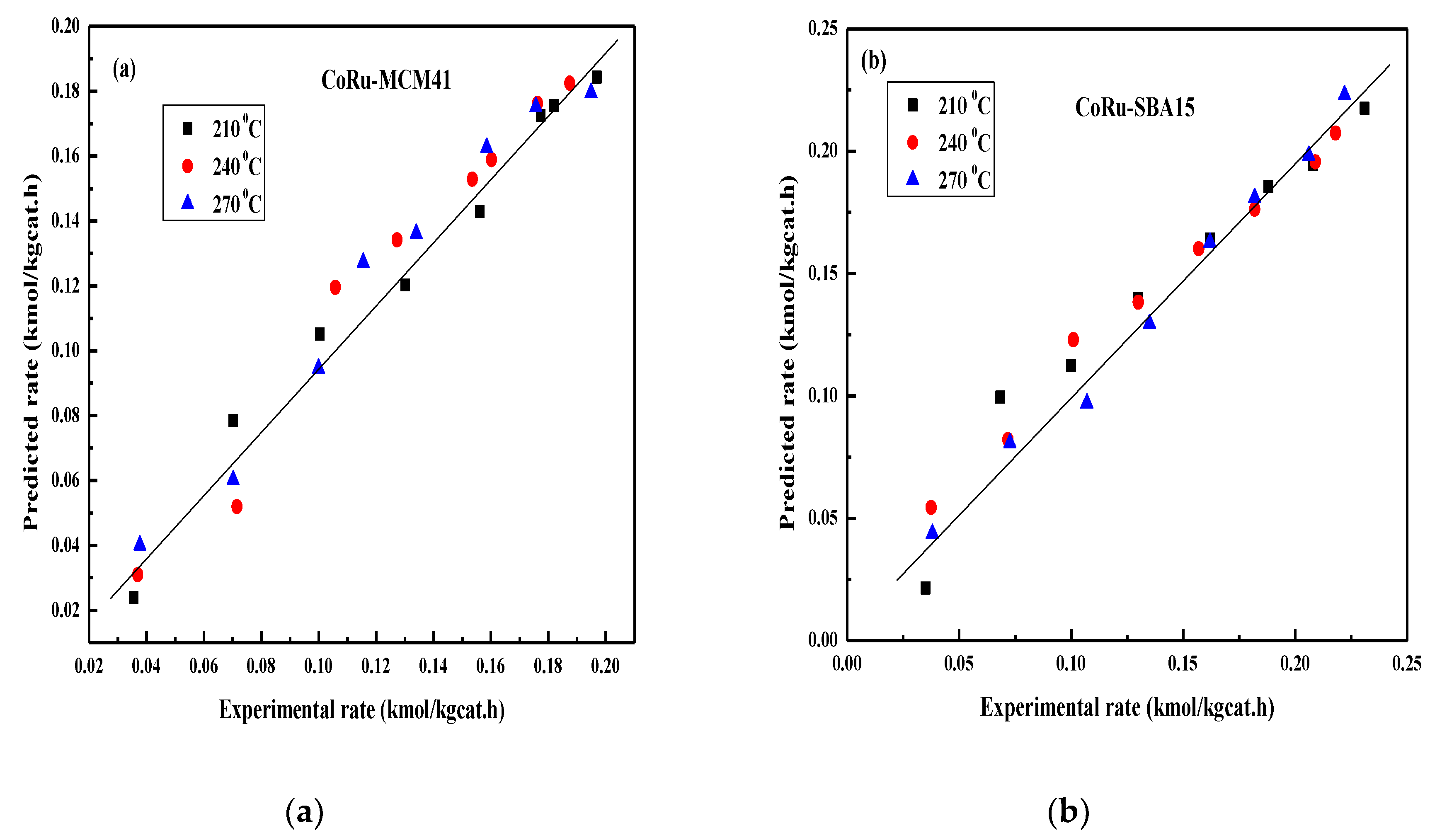
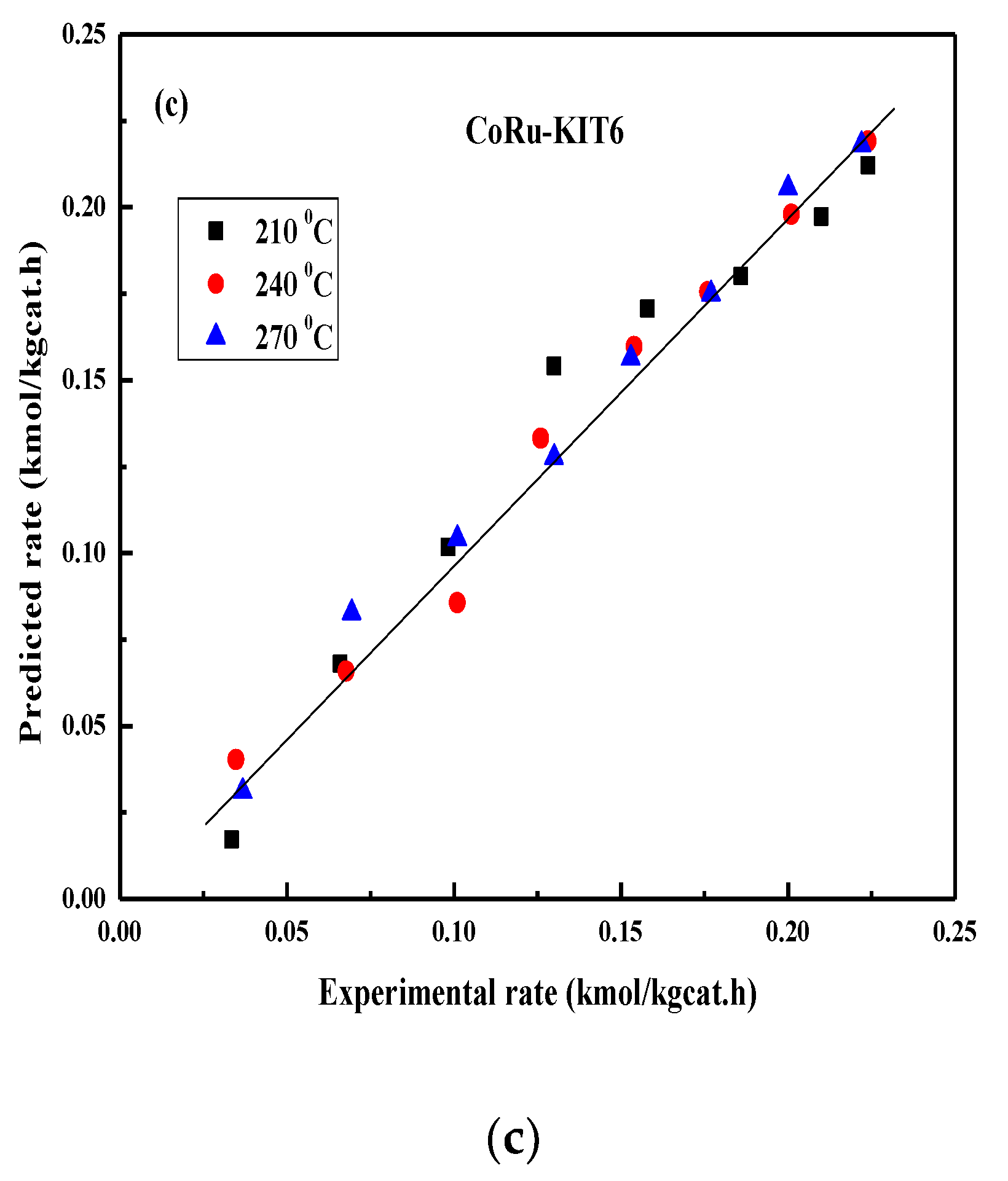
2.4. FT Kinetic Model
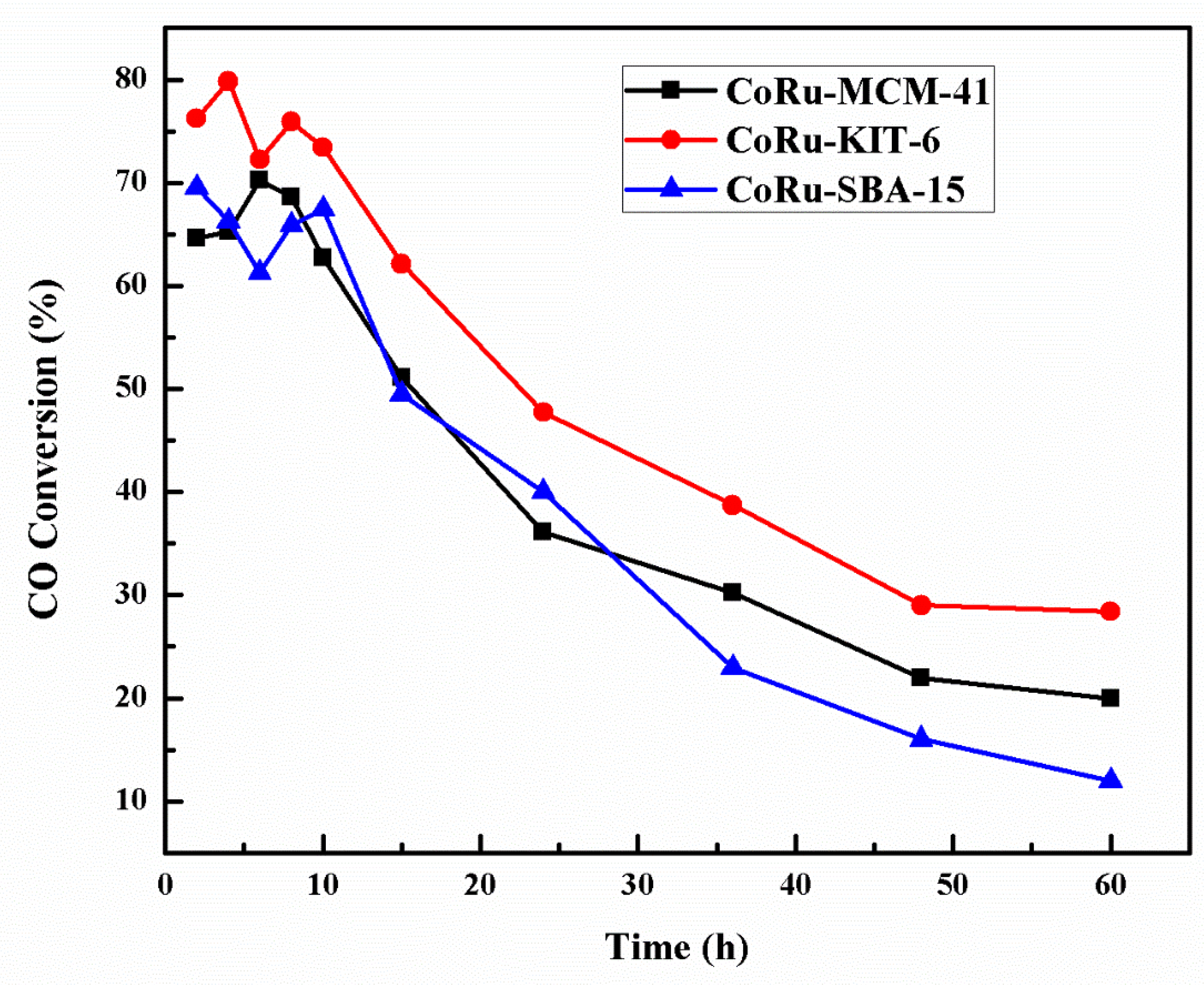
2.5. Deactivation Studies
3. Materials and Methods
3.1. Materials
3.2. Fabrication of Microreactor
3.3. Catalyst Synthesis and Loading
3.4. Catalyst Characterization
3.5. Fischer–Tropsch Synthesis in Microreactor and Kinetic Data Collection
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
References
- Fischer, F.; Tropsch, H. The synthesis of petroleum at atmospheric pressures from gasification products of coal. Brennstoff-Chemie 1926, 7, 97–104. [Google Scholar]
- Steynberg, A.P. Chapter 1—Introduction to Fischer-Tropsch Technology. In Studies in Surface Science and Catalysis; Steynberg, A., Dry, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2004; Volume 152, pp. 1–63. [Google Scholar]
- Davis, B.H. Overview of reactors for liquid phase Fischer—Tropsch synthesis. Catal. Today 2002, 71, 249–300. [Google Scholar] [CrossRef]
- Wood, D.A.; Nwaoha, C.; Towler, B.F. Gas-to-liquids (GTL): A review of an industry offering several routes for monetizing natural gas. J. Nat. Gas Sci. Eng. 2012, 9, 196–208. [Google Scholar] [CrossRef]
- Pohar, A. Process Intensification through Microreactor Application. Chem. Biochem. Eng. Q. 2009, 23, 537–544. [Google Scholar]
- Lerou, J.J.; Tonkovich, A.L.; Silva, L.; Perry, S.; McDaniel, J. Microchannel reactor architecture enables greener processes. Chem. Eng. Sci. 2010, 65, 380–385. [Google Scholar] [CrossRef]
- Hessel, V.; Löb, P.; Holger, L. Industrial and Real-Life Applications of Micro-Reactor Process Engineering for Fine and Functional Chemistry; Elsevier: Amsterdam, The Netherlands, 2006; Volume 159, pp. 35–46. [Google Scholar]
- Ouyang, X.; Besser, R.S. Development of a microreactor-based parallel catalyst analysis system for synthesis gas conversion. Catal. Today 2003, 84, 33–41. [Google Scholar] [CrossRef]
- Gavriilidis, A.; Angeli, P.; Cao, E.; Yeong, K.K.; Wan, Y.S.S. Technology and Applications of Microengineered Reactors. Chem. Eng. Res. Design 2002, 80, 3–30. [Google Scholar] [CrossRef]
- Myrstad, R.; Eri, S.; Pfeifer, P.; Rytter, E.; Holmen, A. Fischer-Tropsch synthesis in a microstructured reactor. Catal. Today 2009, 147, S301–S304. [Google Scholar] [CrossRef]
- LeViness, S.; Deshmukh, S.R.; Richard, L.A.; Robota, H.J. Velocys Fischer—Tropsch synthesis technology—New advances on state-of-the-art. Top. Catal. 2014, 57, 518–525. [Google Scholar] [CrossRef]
- Mazanec, T.; Perry, S.; Tonkovich, L.; Wang, Y. Microchannel gas-to-liquids conversion-thinking big by thinking small. In Studies in Surface Science and Catalysis; Elsevier: Amsterdam, The Netherlands, 2004; Volume 147, pp. 169–174. [Google Scholar]
- Deshmukh, S.R.; Tonkovich, A.L.Y.; Jarosch, K.T.; Schrader, L.; Fitzgerald, S.P.; Kilanowski, D.R.; Lerou, J.J.; Mazanec, T.J. Scale-up of microchannel reactors for Fischer—Tropsch synthesis. Ind. Eng. Chem. Res. 2010, 49, 10883–10888. [Google Scholar] [CrossRef]
- Almeida, L.C.; Sanz, O.; D’olhaberriague, J.; Yunes, S.; Montes, M. Microchannel reactor for Fischer—Tropsch synthesis: Adaptation of a commercial unit for testing microchannel blocks. Fuel 2013, 110, 171–177. [Google Scholar] [CrossRef]
- Schulz, H. Short history and present trends of Fischer—Tropsch synthesis. Appl. Catal. A Gen. 1999, 186, 3–12. [Google Scholar] [CrossRef]
- Borg, Ø.; Eri, S.; Blekkan, E.A.; Storsæter, S.; Wigum, H.; Rytter, E.; Holmen, A. Fischer—Tropsch synthesis over γ-alumina-supported cobalt catalysts: Effect of support variables. J. Catal. 2007, 248, 89–100. [Google Scholar] [CrossRef]
- Chu, W.; Chernavskii, P.A.; Gengembre, L.; Pankina, G.A.; Fongarland, P.; Khodakov, A.Y. Cobalt species in promoted cobalt alumina-supported Fischer—Tropsch catalysts. J. Catal. 2007, 252, 215–230. [Google Scholar] [CrossRef]
- Hilmen, A.; Schanke, D.; Hanssen, K.; Holmen, A. Study of the effect of water on alumina supported cobalt Fischer—Tropsch catalysts. Appl. Catal. A Gen. 1999, 186, 169–188. [Google Scholar] [CrossRef]
- Storsæter, S.; Tøtdal, B.; Walmsley, J.C.; Tanem, B.S.; Holmen, A. Characterization of alumina-, silica-, and titania-supported cobalt Fischer—Tropsch catalysts. J. Catal. 2005, 236, 139–152. [Google Scholar] [CrossRef]
- Hilmen, A.; Schanke, D.; Holmen, A. TPR study of the mechanism of rhenium promotion of alumina-supported cobalt Fischer-Tropsch catalysts. Catal. Lett. 1996, 38, 143–147. [Google Scholar] [CrossRef]
- Morales, F.; De Groot, F.M.; Gijzeman, O.L.; Mens, A.; Stephan, O.; Weckhuysen, B.M. Mn promotion effects in Co/TiO2 Fischer—Tropsch catalysts as investigated by XPS and STEM-EELS. J. Catal. 2005, 230, 301–308. [Google Scholar] [CrossRef]
- Iglesia, E.; Soled, S.L.; Fiato, R.A. Fischer-Tropsch synthesis on cobalt and ruthenium. Metal dispersion and support effects on reaction rate and selectivity. J. Catal. 1992, 137, 212–224. [Google Scholar] [CrossRef]
- Li, J.; Coville, N.J. The effect of boron on the catalyst reducibility and activity of Co/TiO2 Fischer—Tropsch catalysts. Appl. Catal. A Gen. 1999, 181, 201–208. [Google Scholar] [CrossRef]
- Feltes, T.E.; Espinosa-Alonso, L.; De Smit, E.; D’Souza, L.; Meyer, R.J.; Weckhuysen, B.M.; Regalbuto, J.R. Selective adsorption of manganese onto cobalt for optimized Mn/Co/TiO2 Fischer—Tropsch catalysts. J. Catal. 2010, 270, 95–102. [Google Scholar] [CrossRef]
- Li, J.; Jacobs, G.; Zhang, Y.; Das, T.; Davis, B.H. Fischer—Tropsch synthesis: Effect of small amounts of boron, ruthenium and rhenium on Co/TiO2 catalysts. Appl. Catal. A Gen. 2002, 223, 195–203. [Google Scholar] [CrossRef]
- Arai, H.; Mitsuishi, K.; Seiyama, T. TiO2-supported fe–co, co–ni, and ni–fe alloy catalysts for fischer-tropsch synthesis. Chem. Lett. 1984, 13, 1291–1294. [Google Scholar] [CrossRef]
- Duvenhage, D.; Coville, N. Fe: Co/TiO2 bimetallic catalysts for the Fischer—Tropsch reaction: Part 2. The effect of calcination and reduction temperature. Appl. Catal. A Gen. 2002, 233, 63–75. [Google Scholar] [CrossRef]
- Ying, X.; Zhang, L.; Xu, H.; Ren, Y.-L.; Luo, Q.; Zhu, H.-W.; Qu, H.; Xuan, J. Efficient Fischer—Tropsch microreactor with innovative aluminizing pretreatment on stainless steel substrate for Co/Al2O3 catalyst coating. Fuel Process. Technol. 2016, 143, 51–59. [Google Scholar] [CrossRef]
- Ernst, B.; Libs, S.; Chaumette, P.; Kiennemann, A. Preparation and characterization of Fischer—Tropsch active Co/SiO2 catalysts. Appl. Catal. A Gen. 1999, 186, 145–168. [Google Scholar] [CrossRef]
- Li, J.; Jacobs, G.; Das, T.; Zhang, Y.; Davis, B. Fischer—Tropsch synthesis: Effect of water on the catalytic properties of a Co/SiO2 catalyst. Appl. Catal. A Gen. 2002, 236, 67–76. [Google Scholar] [CrossRef]
- Barbier, A.; Tuel, A.; Arcon, I.; Kodre, A.; Martin, G.A. Characterization and catalytic behavior of Co/SiO2 catalysts: Influence of dispersion in the Fischer—Tropsch reaction. J. Catal. 2001, 200, 106–116. [Google Scholar] [CrossRef]
- Yang, Y.; Xiang, H.-W.; Tian, L.; Wang, H.; Zhang, C.-H.; Tao, Z.-C.; Xu, Y.-Y.; Zhong, B.; Li, Y.-W. Structure and Fischer—Tropsch performance of iron—Manganese catalyst incorporated with SiO2. Appl. Catal. A Gen. 2005, 284, 105–122. [Google Scholar] [CrossRef]
- Moradi, G.; Basir, M.; Taeb, A.; Kiennemann, A. Promotion of Co/SiO2 Fischer—Tropsch catalysts with zirconium. Catal. Commun. 2003, 4, 27–32. [Google Scholar] [CrossRef]
- Chen, W.; Fan, Z.; Pan, X.; Bao, X. Effect of confinement in carbon nanotubes on the activity of Fischer—Tropsch iron catalyst. J. Am. Chem. Soc. 2008, 130, 9414–9419. [Google Scholar] [CrossRef] [PubMed]
- Bahome, M.C.; Jewell, L.L.; Hildebrandt, D.; Glasser, D.; Coville, N.J. Fischer—Tropsch synthesis over iron catalysts supported on carbon nanotubes. Appl. Catal. A Gen. 2005, 287, 60–67. [Google Scholar] [CrossRef]
- Abbaslou, R.M.M.; Tavassoli, A.; Soltan, J.; Dalai, A.K. Iron catalysts supported on carbon nanotubes for Fischer—Tropsch synthesis: Effect of catalytic site position. Appl. Catal. A Gen. 2009, 367, 47–52. [Google Scholar] [CrossRef]
- Nagineni, V.S.; Zhao, S.; Potluri, A.; Liang, Y.; Siriwardane, U.; Seetala, N.V.; Fang, J.; Palmer, J.; Kuila, D. Microreactors for Syngas Conversion to Higher Alkanes: Characterization of Sol−Gel-Encapsulated Nanoscale Fe−Co Catalysts in the Microchannels. Ind. Eng. Chem. Res. 2005, 44, 5602–5607. [Google Scholar] [CrossRef]
- Mehta, S.; Deshmane, V.; Zhao, S.; Kuila, D. Comparative Studies of Silica-Encapsulated Iron, Cobalt, and Ruthenium Nanocatalysts for Fischer—Tropsch Synthesis in Silicon-Microchannel Microreactors. Ind. Eng. Chem. Res. 2014, 53, 16245–16253. [Google Scholar] [CrossRef]
- Zhao, S.; Nagineni, V.S.; Seetala, N.V.; Kuila, D. Microreactors for Syngas Conversion to Higher Alkanes: Effect of Ruthenium on Silica-Supported Iron−Cobalt Nanocatalysts. Ind. Eng. Chem. Res. 2008, 47, 1684–1688. [Google Scholar] [CrossRef]
- Abrokwah, R.Y.; Rahman, M.M.; Deshmane, V.G.; Kuila, D. Effect of titania support on Fischer-Tropsch synthesis using cobalt, iron, and ruthenium catalysts in silicon-microchannel microreactor. Mol. Catal. 2019, 478, 110566. [Google Scholar] [CrossRef]
- Gutmann, B.; Koeckinger, M.; Glotz, G.; Ciaglia, T.; Slama, E.; Zadravec, M.; Pfanner, S.; Gruber-Woelfler, H.; Maier, M.; Kappe, C.O. Design and 3D Printing of a Stainless Steel Reactor for Continuous Difluoromethylations Using Fluoroform. React. Chem. Eng. 2017, 2. [Google Scholar] [CrossRef]
- Monaghan, T.; Harding, M.J.; Harris, R.A.; Friel, R.J.; Christie, S.D. Customisable 3D printed microfluidics for integrated analysis and optimisation. Lab Chip 2016, 16, 3362–3373. [Google Scholar] [CrossRef] [Green Version]
- Scotti, G.; Matilainen, V.; Kanninen, P.; Piili, H.; Salminen, A.; Kallio, T.; Franssila, S. Laser additive manufacturing of stainless steel micro fuel cells. J. Power Sources 2014, 272, 356–361. [Google Scholar] [CrossRef]
- Scotti, G.; Kanninen, P.; Matilainen, V.-P.; Salminen, A.; Kallio, T. Stainless steel micro fuel cells with enclosed channels by laser additive manufacturing. Energy 2016, 106, 475–481. [Google Scholar] [CrossRef]
- Capel, A.J.; Edmondson, S.; Christie, S.D.; Goodridge, R.D.; Bibb, R.J.; Thurstans, M. Design and additive manufacture for flow chemistry. Lab Chip 2013, 13, 4583–4590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amin, R.; Knowlton, S.; Hart, A.; Yenilmez, B.; Ghaderinezhad, F.; Katebifar, S.; Messina, M.; Khademhosseini, A.; Tasoglu, S. 3D-printed microfluidic devices. Biofabrication 2016, 8, 022001. [Google Scholar] [CrossRef] [PubMed]
- Reintjens, R.; Ager, D.J.; De Vries, A.H. Flow chemistry, how to bring it to industrial scale? Chim. Oggi 2015, 33, 21–24. [Google Scholar]
- Abrokwah, R.Y.; Deshmane, V.G.; Kuila, D. Comparative performance of M-MCM-41 (M: Cu, Co, Ni, Pd, Zn and Sn) catalysts for steam reforming of methanol. J. Mol. Catal. A Chem. 2016, 425, 10–20. [Google Scholar] [CrossRef] [Green Version]
- Deshmane, V.G.; Abrokwah, R.Y.; Kuila, D. Synthesis of stable Cu-MCM-41 nanocatalysts for H2 production with high selectivity via steam reforming of methanol. Int. J. Hydrogen Energy 2015, 40, 10439–10452. [Google Scholar] [CrossRef]
- Abrokwah, R.Y.; Deshmane, V.G.; Owen, S.L.; Kuila, D. Cu-Ni Nanocatalysts in Mesoporous MCM-41 and TiO2 to Produce Hydrogen for Fuel Cells via Steam Reforming Reactions. Adv. Mater. Res. 2015, 1096, 161–168. [Google Scholar] [CrossRef]
- Tatineni, B.; Basova, Y.; Rahman, A.; Islam, S.; Rahman, M.; Islam, A.; Perkins, J.; King, J.; Taylor, J.; Kumar, D.; et al. Development of Mesoporous Silica Encapsulated Pd-Ni Nanocatalyst for Hydrogen Production. In Production and Purification of Ultraclean Transportation Fuels; American Chemical Society: Washington, DC, USA, 2011; Volume 1088, pp. 177–190. [Google Scholar]
- Taghizadeh, M.; Akhoundzadeh, H.; Rezayan, A.; Sadeghian, M. Excellent catalytic performance of 3D-mesoporous KIT-6 supported Cu and Ce nanoparticles in methanol steam reforming. Int. J. Hydrogen Energy 2018, 43, 10926–10937. [Google Scholar] [CrossRef]
- Lanzafame, P.; Perathoner, S.; Centi, G.; Frusteri, F. Synthesis and characterization of Co-containing SBA-15 catalysts. J. Porous Mater. 2007, 14, 305–313. [Google Scholar] [CrossRef]
- Sing, K.S. Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity (Recommendations 1984). Pure Appl. Chem. 1985, 57, 603–619. [Google Scholar] [CrossRef]
- Biz, S.; Occelli, M.L. Synthesis and characterization of mesostructured materials. Catal. Rev. 1998, 40, 329–407. [Google Scholar] [CrossRef]
- Zhao, D.; Feng, J.; Huo, Q.; Melosh, N.; Fredrickson, G.H.; Chmelka, B.F.; Stucky, G.D. Triblock copolymer syntheses of mesoporous silica with periodic 50 to 300 angstrom pores. Science 1998, 279, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Karthikeyan, G.; Pandurangan, A. Post synthesis alumination of KIT-6 materials with Ia3d symmetry and their catalytic efficiency towards multicomponent synthesis of 1H-pyrazolo[1,2-]phthalazine-5,10-dione carbonitriles and carboxylates. J. Mol. Catal. A Chem. 2012, 361, 58–67. [Google Scholar] [CrossRef]
- Schwertmann, U.; Cambier, P.; Murad, E. Properties of Goethites of Varying Crystallinity. Clays Clay Miner. 1985, 33, 369–378. [Google Scholar] [CrossRef]
- Li, G.; Zhang, C.; Wang, Z.; Huang, H.; Peng, H.; Li, X. Fabrication of mesoporous Co3O4 oxides by acid treatment and their catalytic performances for toluene oxidation. Appl. Catal. A Gen. 2018, 550, 67–76. [Google Scholar] [CrossRef]
- Liu, X.; He, J.; Yang, L.; Wang, Y.; Zhang, S.; Wang, W.; Wang, J. Liquid-phase oxidation of cyclohexane to cyclohexanone over cobalt-doped SBA-3. Catal. Commun. 2010, 11, 710–714. [Google Scholar] [CrossRef]
- Li, Z.; Miao, Z.; Wang, X.; Zhao, J.; Zhou, J.; Si, W.; Zhuo, S. One-pot synthesis of ZrMo-KIT-6 solid acid catalyst for solvent-free conversion of glycerol to solketal. Fuel 2018, 233, 377–387. [Google Scholar] [CrossRef]
- Hess, C.; Tzolova-Müller, G.; Herbert, R. The Influence of Water on the Dispersion of Vanadia Supported on Silica SBA-15: A Combined XPS and Raman Study. J. Phys. Chem. C 2007, 111, 9471–9479. [Google Scholar] [CrossRef]
- Bhoware, S.S.; Singh, A.P. Characterization and catalytic activity of cobalt containing MCM-41 prepared by direct hydrothermal, grafting and immobilization methods. J. Mol. Catal. A Chem. 2007, 266, 118–130. [Google Scholar] [CrossRef]
- Yao, Q.; Lu, Z.-H.; Yang, K.; Chen, X.; Zhu, M. Ruthenium nanoparticles confined in SBA-15 as highly efficient catalyst for hydrolytic dehydrogenation of ammonia borane and hydrazine borane. Sci. Rep. 2015, 5, 15186. [Google Scholar] [CrossRef]
- Panpranot, J.; Goodwin, J.G., Jr.; Sayari, A. Synthesis and characteristics of MCM-41 supported CoRu catalysts. Catal. Today 2002, 77, 269–284. [Google Scholar] [CrossRef]
- Martínez, A.; López, C.; Márquez, F.; Díaz, I. Fischer-Tropsch synthesis of hydrocarbons over mesoporous Co/SBA-15 catalysts: The influence of metal loading, cobalt precursor, and promoters. J. Catal. 2003, 220, 486–499. [Google Scholar] [CrossRef]
- Lim, S.; Ciuparu, D.; Yang, Y.; Du, G.; Pfefferle, L.D.; Haller, G.L. Improved synthesis of highly ordered Co-MCM-41. Microporous Mesoporous Mater. 2007, 101, 200–206. [Google Scholar] [CrossRef]
- Jiang, T.; Shen, W.; Zhao, Q.; Li, M.; Chu, J.; Yin, H. Characterization of CoMCM-41 mesoporous molecular sieves obtained by the microwave irradiation method. J. Solid State Chem. 2008, 181, 2298–2305. [Google Scholar] [CrossRef]
- Haddad, G.J.; Goodwin, J.G., Jr. The impact of aqueous impregnation on the properties of prereduced vs. precalcined Co/SiO2. J. Catal. 1995, 157, 25–34. [Google Scholar] [CrossRef]
- Cai, Q.; Li, J. Catalytic properties of the Ru promoted Co/SBA-15 catalysts for Fischer—Tropsch synthesis. Catal. Commun. 2008, 9, 2003–2006. [Google Scholar] [CrossRef]
- Van Der Laan, G.P.; Beenackers, A. Kinetics and selectivity of the Fischer—Tropsch synthesis: A literature review. Catal. Rev. 1999, 41, 255–318. [Google Scholar] [CrossRef]
- Yates, I.C.; Satterfield, C.N. Intrinsic kinetics of the Fischer-Tropsch synthesis on a cobalt cνatalyst. Energy Fuels 1991, 5, 168–173. [Google Scholar] [CrossRef]
- Iglesia, E.; Reyes, S.C.; Soled, S.L. Reaction-transport selectivity models and the design of Fischer-Tropsch catalysts. Chem. Ind. N. Y. Marcel Dekker 1993, 51, 199–257. [Google Scholar]
- Zennaro, R.; Tagliabue, M.; Bartholomew, C.H. Kinetics of Fischer—Tropsch synthesis on titania-supported cobalt. Catal. Today 2000, 58, 309–319. [Google Scholar] [CrossRef]
- Das, T.K.; Conner, W.A.; Li, J.; Jacobs, G.; Dry, M.E.; Davis, B.H. Fischer−Tropsch synthesis: Kinetics and effect of water for a Co/SiO2 catalyst. Energy Fuels 2005, 19, 1430–1439. [Google Scholar] [CrossRef]
- Almeida, L.; Sanz, O.; Merino, D.; Arzamendi, G.; Gandía, L.; Montes, M. Kinetic analysis and microstructured reactors modeling for the Fischer—Tropsch synthesis over a Co–Re/Al2O3 catalyst. Catal. Today 2013, 215, 103–111. [Google Scholar] [CrossRef]
- Yang, C.-H.; Massoth, F.; Oblad, A. Kinetics of CO+ H2 reaction over Co–Cu–Al2O3 catalyst. Hydrocarbon synthesis from carbon monoxide and hydrogen. J. Am. Chem. Soc. 1979, 178, 35–46. [Google Scholar]
- Pannell, R.B.; Kibby, C.L.; Kobylinski, T.P. A Steady-State Study of Fischer-Tropsch Product Distributions Over Cobalt, Iron and Ruthenium. In Studies in Surface Science and Catalysis; Seivama, T., Tanabe, K., Eds.; Elsevier: Amsterdam, The Netherlands, 1981; Volume 7, pp. 447–459. [Google Scholar]
- Outi, A.; Rautavuoma, I.; Van der Baan, H.S. Kinetics and mechanism of the fischer tropsch hydrocarbon synthesis on a cobalt on alumina catalyst. Appl. Catal. 1981, 1, 247–272. [Google Scholar] [CrossRef]
- Sun, Y.; Jia, Z.; Yang, G.; Zhang, L.; Sun, Z. Fischer-Tropsch synthesis using iron based catalyst in a microchannel reactor: Performance evaluation and kinetic modeling. Int. J. Hydrogen Energy 2017, 42, 29222–29235. [Google Scholar] [CrossRef]
- Mirzaei, A.A.; Kiai, R.M.; Atashi, H.; Arsalanfar, M.; Shahriari, S. Kinetic study of CO hydrogenation over co-precipitated iron–nickel catalyst. J. Ind. Eng. Chem. 2012, 18, 1242–1251. [Google Scholar] [CrossRef]
- Sarup, B.; Wojciechowski, B.W. Studies of the fischer-tropsch synthesis on a cobalt catalyst II. Kinetics of carbon monoxide conversion to methane and to higher hydrocarbons. Can. J. Chem. Eng. 1989, 67, 62–74. [Google Scholar] [CrossRef]
- Bepari, S.; Pradhan, N.C.; Dalai, A.K. Selective production of hydrogen by steam reforming of glycerol over Ni/Fly ash catalyst. Catal. Today 2017, 291, 36–46. [Google Scholar] [CrossRef]
- Fogler, H.S. Elements of Chemical Reaction Engineering, 3rd ed.; Prentice Hall PTR: Upper Saddle River, NJ, USA, 1999. [Google Scholar]
- Van Santen, R.A.; Ciobîcâ, I.M.; Van Steen, E.; Ghouri, M.M. Mechanistic Issues in Fischer-Tropsch Catalysis. In Advances in Catalysis; Academic Press: Cambridge, MA, USA, 2011; Volume 54, pp. 127–187. [Google Scholar]
- Van Santen, R.A.; Ghouri, M.M.; Shetty, S.; Hensen, E.M.H. Structure sensitivity of the Fischer-Tropsch reaction; Molecular kinetics simulations. Catal. Sci. Technol. 2011, 1, 891–911. [Google Scholar] [CrossRef]
- Markvoort, A.J.; Van Santen, R.A.; Hilbers, P.A.J.; Hensen, E.J.M. Kinetics of the Fischer-Tropsch reaction. Angew. Chem.-Int. Ed. 2012, 51, 9015–9019. [Google Scholar] [CrossRef]
- Mansouri, M.; Atashi, H.; Mirzaei, A.A.; Jangi, R. Kinetics of the Fischer-Tropsch synthesis on silica-supported cobalt-cerium catalyst. Int. J. Ind. Chem. 2013, 4, 1. [Google Scholar] [CrossRef] [Green Version]
- Pant, K.K.; Upadhyayula, S. Detailed kinetics of Fischer Tropsch synthesis over Fe-Co bimetallic catalyst considering chain length dependent olefin desorption. Fuel 2019, 236, 1263–1272. [Google Scholar] [CrossRef]
- Mosayebi, A.; Abedini, R. Detailed kinetic study of Fischer—Tropsch synthesis for gasoline production over CoNi/HZSM-5 nano-structure catalyst. Int. J. Hydrogen Energy 2017, 42, 27013–27023. [Google Scholar] [CrossRef]
- Abbasi, M.; Mirzaei, A.A.; Atashi, H. The mechanism and kinetics study of Fischer‒Tropsch reaction over iron-nickel-cerium nano-structure catalyst. Int. J. Hydrogen Energy 2019, 44, 24667–24679. [Google Scholar] [CrossRef]
- Sun, Y.; Wei, J.; Zhang, J.P.; Yang, G. Optimization using response surface methodology and kinetic study of Fischer—Tropsch synthesis using SiO2 supported bimetallic Co–Ni catalyst. J. Nat. Gas Sci. Eng. 2016, 28, 173–183. [Google Scholar] [CrossRef]
- Sun, Y.; Yang, G.; Zhang, L.; Sun, Z. Fischer-Tropsch synthesis in a microchannel reactor using mesoporous silica supported bimetallic Co-Ni catalyst: Process optimization and kinetic modeling. Chem. Eng. Proc. Process Intensif. 2017, 119, 44–61. [Google Scholar] [CrossRef]
- Moazami, N.; Wyszynski, M.L.; Rahbar, K.; Tsolakis, A.; Mahmoudi, H. A comprehensive study of kinetics mechanism of Fischer-Tropsch synthesis over cobalt-based catalyst. Chem. Eng. Sci. 2017, 171, 32–60. [Google Scholar] [CrossRef] [Green Version]
- Moazami, N.; Wyszynski, M.L.; Mahmoudi, H.; Tsolakis, A.; Zou, Z.; Panahifar, P.; Rahbar, K. Modelling of a fixed bed reactor for Fischer—Tropsch synthesis of simulated N2-rich syngas over Co/SiO2: Hydrocarbon production. Fuel 2015, 154, 140–151. [Google Scholar] [CrossRef]
- Marchese, M.; Heikkinen, N.; Giglio, E.; Lanzini, A.; Lehtonen, J.; Reinikainen, M. Kinetic Study Based on the Carbide Mechanism of a Co-Pt/γ-Al2O3 Fischer—Tropsch Catalyst Tested in a Laboratory-Scale Tubular Reactor. Catalysts 2019, 9, 717. [Google Scholar] [CrossRef]
- Bhatelia, T.; Li, C.e.; Sun, Y.; Hazewinkel, P.; Burke, N.; Sage, V. Chain length dependent olefin re-adsorption model for Fischer—Tropsch synthesis over Co-Al2O3 catalyst. Fuel Process. Technol. 2014, 125, 277–289. [Google Scholar] [CrossRef]
- Todic, B.; Bhatelia, T.; Froment, G.F.; Ma, W.; Jacobs, G.; Davis, B.H.; Bukur, D.B. Kinetic Model of Fischer—Tropsch Synthesis in a Slurry Reactor on Co–Re/Al2O3 Catalyst. Ind. Eng. Chem. Res. 2013, 52, 669–679. [Google Scholar] [CrossRef]
- Jahangiri, H.; Bennett, J.; Mahjoubi, P.; Wilson, K.; Gu, S. A review of advanced catalyst development for Fischer—Tropsch synthesis of hydrocarbons from biomass derived syn-gas. Catal. Sci. Technol. 2014, 4, 2210–2229. [Google Scholar] [CrossRef]
- Iglesia, E. Design, synthesis, and use of cobalt-based Fischer-Tropsch synthesis catalysts. Appl. Catal. A Gen. 1997, 161, 59–78. [Google Scholar] [CrossRef]
- Iglesia, E.; Soled, S.L.; Fiato, R.A.; Via, G.H. Bimetallic Synergy in Cobalt Ruthenium Fischer-Tropsch Synthesis Catalysts. J. Catal. 1993, 143, 345–368. [Google Scholar] [CrossRef]
- Kleitz, F.; Choi, S.H.; Ryoo, R. Cubic Ia 3 d large mesoporous silica: Synthesis and replication to platinum nanowires, carbon nanorods and carbon nanotubes. Chem. Commun. 2003, 17, 2136–2137. [Google Scholar] [CrossRef]
- Barrett, E.P.; Joyner, L.G.; Halenda, P.P. The Determination of Pore Volume and Area Distributions in Porous Substances. I. Computations from Nitrogen Isotherms. J. Am. Chem. Soc. 1951, 73, 373–380. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mesoporous Silica Supported Catalyst with Intended Metal Loadings | Surface Area a (m2/g) | Pore Volume b (cm3/g) | Pore Size c (nm) | Metal Loadings Obtained from SEM-EDX (wt %) |
|---|---|---|---|---|
| 10% Co5%Ru-MCM-41 | 1025 | 0.77 | 3.2 | 9%Co3.9%Ru-MCM-41 |
| 10% Co5%Ru-SBA-15 | 691 | 0.73 | 4.2 | 8.4%Co4.5%Ru-KIT-6 |
| 10%Co5%Ru-KIT-6 | 690 | 0.92 | 5.3 | 11.1%Co5.6%Ru-SBA-15 |
| Model | No | Elementary Reaction |
|---|---|---|
| FT-1 | 1 | |
| 2 | ||
| FT-2 | 1 | |
| 2 | ||
| FT-3 | 1 | |
| 2 | ||
| 3 | ||
| FT-4 | 1 | |
| 2 | ||
| 3 | ||
| FT-5 | 1 | |
| 2 | ||
| 3 | ||
| FT-6 | 1 | |
| 2 | ||
| 3 | ||
| 4 |
| Model | Rate Controlling Step (RCS) | Kinetic Equation |
|---|---|---|
| FT-1 | (2) | |
| FT-2 | (2) | |
| FT-3 | (3) | |
| FT-4 | (3) | |
| FT-5 | (3) | |
| FT-6 | (4) |
| Temperature | W/F (Kgcat.h/kmol) | CoRu-MCM-41 | CoRu-SBA-15 | CoRu-KIT-6 | |||
|---|---|---|---|---|---|---|---|
| Reaction Rate (Kmol/Kg.h) | Conversion (%) | Reaction Rate (Kmol/Kg.h) | Conversion (%) | Reaction Rate (Kmol/Kg.h) | Conversion (%) | ||
| 210 °C | 25.2 | 0.035 | 89.53 | 0.035 | 88.35 | 0.033 | 84.12 |
| 12.6 | 0.070 | 88.43 | 0.068 | 86.08 | 0.065 | 83.03 | |
| 8.39 | 0.100 | 84.29 | 0.1 | 84.29 | 0.098 | 82.41 | |
| 6.30 | 0.130 | 81.97 | 0.13 | 81.97 | 0.13 | 81.97 | |
| 5.04 | 0.156 | 78.64 | 0.162 | 81.52 | 0.158 | 79.34 | |
| 4.20 | 0.177 | 74.45 | 0.188 | 79.11 | 0.186 | 78.15 | |
| 3.60 | 0.181 | 65.46 | 0.208 | 74.74 | 0.21 | 75.45 | |
| 3.15 | 0.196 | 61.98 | 0.231 | 72.79 | 0.224 | 70.43 | |
| 240 °C | 25.2 | 0.036 | 93.09 | 0.037 | 94.43 | 0.034 | 87.39 |
| 12.6 | 0.071 | 90.09 | 0.071 | 90.36 | 0.067 | 85.27 | |
| 8.39 | 0.105 | 88.84 | 0.01 | 85.18 | 0.101 | 84.79 | |
| 6.30 | 0.127 | 80.14 | 0.013 | 82.07 | 0.126 | 79.63 | |
| 5.04 | 0.153 | 77.34 | 0.157 | 79.24 | 0.154 | 77.48 | |
| 4.20 | 0.160 | 67.23 | 0.182 | 76.32 | 0.176 | 74.02 | |
| 3.60 | 0.176 | 63.42 | 0.209 | 75.25 | 0.201 | 72.35 | |
| 3.15 | 0.187 | 59.02 | 0.218 | 68.76 | 0.224 | 70.44 | |
| 270 °C | 25.2 | 0.037 | 94.95 | 0.038 | 95.98 | 0.036 | 92.40 |
| 12.6 | 0.070 | 88.39 | 0.072 | 91.55 | 0.069 | 87.39 | |
| 8.39 | 0.100 | 83.96 | 0.107 | 89.90 | 0.101 | 85.01 | |
| 6.30 | 0.115 | 72.70 | 0.135 | 85.22 | 0.130 | 81.80 | |
| 5.04 | 0.133 | 67.48 | 0.162 | 81.42 | 0.153 | 77.08 | |
| 4.20 | 0.158 | 66.53 | 0.182 | 76.32 | 0.177 | 74.18 | |
| 3.60 | 0.175 | 63.20 | 0.206 | 74.19 | 0.2 | 71.93 | |
| 3.15 | 0.194 | 61.34 | 0.222 | 69.90 | 0.222 | 70.00 | |
| Model | Temperature | |||||
|---|---|---|---|---|---|---|
| 210 °C | R2 | 240 °C | R2 | 270 °C | R2 | |
| FT-1 | k = 0.872 ± 0.0037 K1 = 21.19 ± 1.79 | 0.88 | k = 0.23 ± 0.027 K1 = 16.49 ± 7.66 | 0.98 | k = 0.38 ± 0.118 K1 = 3.92 ± 2.46 | 0.98 |
| FT-2 | k = 0.87 ± 0.0037 K1 = 21.19 ± 1.79 | 0.88 | k = 0.74 ± 0.0014 K1 = 21.19 ± 0.84 | 0.72 | k = 0.69 ± 0.0067 K1 = 6.05 ± 0.34 | 0.93 |
| FT-3 | k = 1.32 ± 0.000029 K1 = 5.04 ± 0.000194 K2 = 2.877 ± 0.000468 | 0.97 | k = 2.21 ± 0.017 K1 = 4.02 ± 0.103 K2 = 0.51 ± 0.0064 | 0.96 | k = 3.22 ± 0.068 K1 = 1.41 ± 0.05 K2 = 0.5 ± 0.019 | 0.97 |
| FT-4 | k = 0.443 ± 1.69 K1 = 1.88 ± 19.53 K2 = 2.49 ± 34.08 | 0.98 | k = 0.85 ± 0.015 K1 = 11.09 ± 0.686 K2 = 0.365 ± 0.0077 | 0.98 | k = 2.01 ± 0.037 K1 = 3.13 ± 0.11 K2 = 0.228 ± 0.0045 | 0.98 |
| FT-5 | k = 0.536 ± 0.000355 K1 = 21.19 ± 0.403 K2 = 2.42 ± 0.0023 | 0.97 | k = 0.48 ± 0.000205 K1 = 21.19 ± 0.265 K2 = 2.33 ± 0.0015 | 0.90 | k = 0.41 ± 0.009 K1 = 0.33 ± 0.014 K2 = 11.09 ± 0.396 | 0.97 |
| FT-6 | k = 0.704 ± 0.0022 K1 = 0.931 ± 0.0039 K2 = 31.29 ± 3.21 K3 = 1.77 ± 0.0074 | 0.96 | k = 0.67 ± 0.0012 K1 = 0.897 ± 0.0021 K2 = 31.29 ± 1.83 K3 = 1.69 ± 0.0041 | 0.87 | k = 0.243 ± 0.00018 K1 = 3.14 ± 0.0052 K2 = 31.29 ± 0.76 K3 = 1.67 ± 0.0027 | 0.83 |
| Model | Temperature | |||||
|---|---|---|---|---|---|---|
| 210 °C | R2 | 240 °C | R2 | 270 °C | R2 | |
| FT-1 | k = 3.02 ± 0.092 K1 = 0.46 ± 0.015 | 0.94 | k = 2.01 ± 0.059 K1 = 0.655 ± 0.021 | 0.94 | k = 2.01 ± 0.026 K1 = 0.727 ± 0.01 | 0.98 |
| FT-2 | k = 9.08 ± 0.266 K1 = 0.166 ± 0.005 | 0.94 | k = 11.09 ± 0.281 K1 = 0.118 ± 0.0033 | 0.95 | k = 13.12 ± 0.005 K1 = 0.109 ± 0.000046 | 0.99 |
| FT-3 | k = 11.09 ± 0.698 K1 = 2.23 ± 0.145 K2 = 20.62 ± 1.49 | 0.82 | k = 2.01 ± 0.043 K1 = 10.79 ± 0.285 K2 = 17.51 ± 0.536 | 0.79 | k = 11.09 ± 0.337 K1 = 2.01 ± 0.063 K2 = 18.10 ± 0.645 | 0.95 |
| FT-4 | k = 11.09 ± 0.379 K1 = 0.161 ± 0.0057 K2 = 0.761 ± 0.026 | 0.94 | k = 2.01 ± 0.0026 K1 = 0.283 ± 0.00042 K2 = 2.31 ± 0.0032 | 0.93 | k = 2.01 ± 0.037 K1 = 3.13 ± 0.11 K2 = 0.228 ± 0.0045 | 0.98 |
| FT-5 | k = 2.01 ± 0.026 K1 = 0.189 ± 0.0031 K2 = 4.41 ± 0.062 | 0.92 | k = 2.01 ± 0.0237 K1 = 0.193 ± 0.0029 K2 = 3.92 ± 0.050 | 0.94 | k = 2.01 ± 0.032 K1 = 0.205 ± 0.0036 K2 = 3.62 ± 0.063 | 0.98 |
| FT-6 | k = 2.01 ± 0.037 K1 = 0.988 ± 0.021 K2 = 0.894 ± 0.031 K3 = 1.57 ± 0.032 | 0.90 | k = 6.05 ± 0.253 K1 = 0.273 ± 0.012 K2 = 0.266 ± 0.014 K3 = 3.56 ± 0.153 | 0.94 | k = 2.01 ± 0.018 K1 = 0.937 ± 0.0094 K2 = 2.18 ± 0.057 K3 = 1.09 ± 0.011 | 0.97 |
| Model | Temperature | |||||
|---|---|---|---|---|---|---|
| 210 °C | R2 | 240 °C | R2 | 270 °C | R2 | |
| FT-1 | k = 2.18 ± 8.89 K1 = 0.73 ± 3.28 | 0.97 | k = 0.79 ± 0.543 K1 = 2.13 ± 1.91 | 0.98 | k = 1.38 ± 1.42 K1 = 1.19 ± 1.44 | 0.99 |
| FT-2 | k = 1.42 ± 0.00044 K1 = 11.09±0.038 | 0.95 | k = 1.29±0.022 K1 = 11.09±2.09 | 0.98 | k = 2.02±3.30 K1 = 1.94±8.71 | 0.98 |
| FT-3 | k = 11.09±0.316 K1 = 0.892±0.031 K2 = 0.279±0.013 | 0.96 | k = 10.09±0.166 K1 = 0.882±0.018 K2 = 0.279±0.0073 | 0.98 | k = 11.09±0.088 K1 = 1.94±0.016 K2 = 15.64±0.15 | 0.97 |
| FT-4 | k = 2.01±0.052 K1 = 0.122±0.0038 K2 = 7.27±0.203 | 0.97 | k = 2.01±0.088 K1 = 1.195±0.067 K2 = 0.694±0.033 | 0.98 | k = 2.01±0.072 K1 = 0.355±0.015 K2 = 2.29±0.089 | 0.99 |
| FT-5 | k = 2.01±0.051 K1 = 0.121±0.0038 K2 = 7.27±0.203 | 0.97 | k = 1.46±0.00069 K1 = 10.08±0.055 K2 = 0.91±0.00049 | 0.92 | k = 2.01±0.028 K1 = 0.842±0.022 K2 = 1.62±0.024 | 0.99 |
| FT-6 | k = 1.71±0.02 K1 = 1.39±0.02 K2 = 0.828±0.018 K3 = 1.61±0.021 | 0.95 | k = 2.01±0.039 K1 = 1.11±0.027 K2 = 0.454±0.013 K3 = 2.21±0.048 | 0.98 | k = 2.41±0.040 K1 = 0.91±0.017 K2 = 0.315±0.0071 K3 = 3.12±0.056 | 0.99 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohammad, N.; Bepari, S.; Aravamudhan, S.; Kuila, D. Kinetics of Fischer–Tropsch Synthesis in a 3-D Printed Stainless Steel Microreactor Using Different Mesoporous Silica Supported Co-Ru Catalysts. Catalysts 2019, 9, 872. https://doi.org/10.3390/catal9100872
Mohammad N, Bepari S, Aravamudhan S, Kuila D. Kinetics of Fischer–Tropsch Synthesis in a 3-D Printed Stainless Steel Microreactor Using Different Mesoporous Silica Supported Co-Ru Catalysts. Catalysts. 2019; 9(10):872. https://doi.org/10.3390/catal9100872
Chicago/Turabian StyleMohammad, Nafeezuddin, Sujoy Bepari, Shyam Aravamudhan, and Debasish Kuila. 2019. "Kinetics of Fischer–Tropsch Synthesis in a 3-D Printed Stainless Steel Microreactor Using Different Mesoporous Silica Supported Co-Ru Catalysts" Catalysts 9, no. 10: 872. https://doi.org/10.3390/catal9100872