Hydrolysis of Glycosyl Thioimidates by Glycoside Hydrolase Requires Remote Activation for Efficient Activity


 , and
, and
Abstract
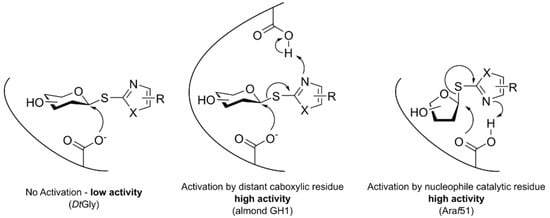
:
1. Introduction
2. Results
2.1. DtGly Can Hydrolyze Thioglycosides
2.2. Identification of Residues Surrounding the Thioglycoside Substrates in DtGly Active Site
2.3. DtGly Hydrolysis of S-Glycosides Does Not Involve General Acid/Base Catalysis
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Production of WT and E159Q DtGly
4.3. pNP Release Quantification Assay
4.4. Glucose Release Assay
4.5. Computational Studies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sheldon, R.A.; Woodley, J.M. Role of Biocatalysis in Sustainable Chemistry. Chem. Rev. 2018, 118, 801–838. [Google Scholar] [CrossRef] [PubMed]
- Quin, M.B.; Schmidt-Dannert, C. Engineering of Biocatalysts: From Evolution to Creation. ACS Catal. 2011, 1, 1017–1021. [Google Scholar] [CrossRef] [PubMed]
- Hancock, S.M.; Vaughan, M.D.; Withers, S.G. Engineering of glycosidases and glycosyltransferases. Curr. Opin. Chem. Biol. 2006, 10, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Li, T.-L.; Liu, Y.-C.; Lyu, S.-Y. Combining biocatalysis and chemoselective chemistries for glycopeptide antibiotics modification. Curr. Opin. Chem. Biol. 2012, 16, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Ati, J.; Lafite, P.; Daniellou, R. Enzymatic synthesis of glycosides: From natural O- and N-glycosides to rare C- and S-glycosides. Beilstein J. Org. Chem. 2017, 13, 1857–1865. [Google Scholar] [CrossRef]
- Guillotin, L.; Lafite, P.; Daniellou, R. Chapter 10 Enzymatic thioglycosylation: Current knowledge and challenges. In Carbohydrate Chemistry; The Royal Society of Chemistry: London, UK, 2014; Volume 40, pp. 178–194. ISBN 978-1-84973-965-8. [Google Scholar]
- Driguez, H. Thiooligosaccharides as Tools for Structural Biology. Chembiochem 2001, 2, 311–318. [Google Scholar] [CrossRef]
- Wardrop, D.J.; Waidyarachchi, S.L. Synthesis and biological activity of naturally occurring α-glucosidase inhibitors. Nat. Prod. Rep. 2010, 27, 1431–1468. [Google Scholar] [CrossRef]
- O’Neill, E.C.; Field, R.A. Enzymatic synthesis using glycoside phosphorylases. Carbohydr. Res. 2015, 403, 23–37. [Google Scholar] [CrossRef]
- Lairson, L.L.; Henrissat, B.; Davies, G.J.; Withers, S.G. Glycosyltransferases: Structures, Functions and Mechanisms. Annu. Rev. Biochem. 2008, 77, 521–555. [Google Scholar] [CrossRef]
- De Bruyn, F.; Maertens, J.; Beauprez, J.; Soetaert, W.; De Mey, M. Biotechnological advances in UDP-sugar based glycosylation of small molecules. Biotechnol. Adv. 2015, 33, 288–302. [Google Scholar] [CrossRef]
- Rye, C.S.; Withers, S.G. Glycosidase mechanisms. Curr. Opin. Chem. Biol. 2000, 4, 573–580. [Google Scholar] [CrossRef]
- Wang, L.-X.; Huang, W. Enzymatic transglycosylation for glycoconjugate synthesis. Curr. Opin. Chem. Biol. 2009, 13, 592–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahn, M.; Marles, J.; Warren, R.A.J.; Withers, S.G. Thioglycoligases: Mutant Glycosidases for Thioglycoside Synthesis. Angew. Chem. Int. Ed. 2003, 42, 352–354. [Google Scholar] [CrossRef] [PubMed]
- Almendros, M.; Danalev, D.; François-Heude, M.; Loyer, P.; Legentil, L.; Nugier-Chauvin, C.; Daniellou, R.; Ferrières, V. Exploring the synthetic potency of the first furanothioglycoligase through original remote activation. Org. Biomol. Chem. 2011, 9, 8371–8378. [Google Scholar] [CrossRef] [Green Version]
- Avegno, E.A.-B.; Hasty, S.J.; Parameswar, A.R.; Howarth, G.S.; Demchenko, A.V.; Byers, L.D. Reactive thioglucoside substrates for β-glucosidase. Arch. Biochem. Biophys. 2013, 537, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Goodman, I.; Fouts, J.R.; Bresnick, E.; Menegas, R.; Hitchings, G.H. A Mammalian Thioglycosidase. Science 1959, 130, 450–451. [Google Scholar] [CrossRef]
- Meulenbeld, G.H.; Hartmans, S. Thioglucosidase activity from Sphingobacterium sp. strain OTG1. Appl. Microbiol. Biotechnol. 2001, 56, 700–706. [Google Scholar] [CrossRef] [PubMed]
- Macauley, M.S.; Stubbs, K.A.; Vocadlo, D.J. O-GlcNAcase catalyzes cleavage of thioglycosides without general acid catalysis. J. Am. Chem. Soc. 2005, 127, 17202–17203. [Google Scholar] [CrossRef] [PubMed]
- Cetinbaş, N.; Macauley, M.S.; Stubbs, K.A.; Drapala, R.; Vocadlo, D.J. Identification of Asp174 and Asp175 as the key catalytic residues of human O-GlcNAcase by functional analysis of site-directed mutants. Biochemistry 2006, 45, 3835–3844. [Google Scholar] [CrossRef]
- Narine, A.A.; Watson, J.N.; Bennet, A.J. Mechanistic requirements for the efficient enzyme-catalyzed hydrolysis of thiosialosides. Biochemistry 2006, 45, 9319–9326. [Google Scholar] [CrossRef]
- Shen, H.; Byers, L.D. Thioglycoside hydrolysis catalyzed by beta-glucosidase. Biochem. Biophys. Res. Commun. 2007, 362, 717–720. [Google Scholar] [CrossRef] [PubMed]
- Sansenya, S.; Opassiri, R.; Kuaprasert, B.; Chen, C.J.; Cairns, J.R.K. The crystal structure of rice (Oryza sativa L.) Os4BGlu12, an oligosaccharide and tuberonic acid glucoside-hydrolyzing beta-glucosidase with significant thioglucohydrolase activity. Arch. Biochem. Biophys. 2011, 510, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Wathelet, J.-P.; Iori, R.; Leoni, O.; Rollin, P.; Mabon, N.; Marlier, M.; Palmieri, S. A recombinant β-O-glucosidase from Caldocellum saccharolyticum to hydrolyse desulfo-glucosinolates. Biotechnol. Lett. 2001, 23, 443–446. [Google Scholar] [CrossRef]
- Reese, E.T.; Clapp, R.C.; Mandels, M. A thioglucosidase in fungi. Arch. Biochem. Biophys. 1958, 75, 228–242. [Google Scholar] [CrossRef]
- Yip, V.L.Y.; Withers, S.G. Family 4 glycosidases carry out efficient hydrolysis of thioglycosides by an alpha,beta-elimination mechanism. Angew. Chem. Int. Ed. 2006, 45, 6179–6182. [Google Scholar] [CrossRef] [PubMed]
- Niemec-Cyganek, A.; Szeja, W. Heteroaryl thioglycosides, a new class of substrated for glycosidases. Pol. J. Chem. 2003, 77, 969–973. [Google Scholar]
- Jensen, J.L.; Jencks, W.P. Hydrolysis of benzaldehyde O,S-acetals. J. Am. Chem. Soc. 1979, 101, 1476–1488. [Google Scholar] [CrossRef]
- Hanessian, S.; Bacquet, C.; Lehong, N. Chemistry of the glycosidic linkage. Exceptionally fast and efficient formation of glycosides by remote activation. Carbohydr. Res. 1980, 80, C17–C22. [Google Scholar] [CrossRef]
- Ferrières, V.; Blanchard, S.; Fischer, D.; Plusquellec, D. A novel synthesis of D-galactofuranosyl, D-glucofuranosyl and D-mannofuranosyl 1-phosphates based on remote activation of new and free hexofuranosyl donors. Bioorg. Med. Chem. Lett. 2002, 12, 3515–3518. [Google Scholar] [CrossRef]
- Demchenko, A.V.; Malysheva, N.N.; De Meo, C. S-Benzoxazolyl (SBox) Glycosides as Novel, Versatile Glycosyl Donors for Stereoselective 1,2-Cis Glycosylation. Org. Lett. 2003, 5, 455–458. [Google Scholar] [CrossRef]
- Demchenko, A.V.; Pornsuriyasak, P.; De Meo, C.; Malysheva, N.N. Potent, Versatile and Stable: Thiazolyl Thioglycosides as Glycosyl Donors. Angew. Chem. Int. Ed. 2004, 43, 3069–3072. [Google Scholar] [CrossRef] [PubMed]
- Euzen, R.; Guégan, J.-P.; Ferrières, V.; Plusquellec, D. First O-Glycosylation from Unprotected 1-Thioimidoyl Hexofuranosides Assisted by Divalent Cations. J. Org. Chem. 2007, 72, 5743–5747. [Google Scholar] [CrossRef] [PubMed]
- Kamat, M.N.; De Meo, C.; Demchenko, A.V. S-Benzoxazolyl as a Stable Protecting Moiety and a Potent Anomeric Leaving Group in Oligosaccharide Synthesis. J. Org. Chem. 2007, 72, 6947–6955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasty, S.J.; Demchenko, A.V. Glycosyl Thioimidates as Versatile Building Blocks for Organic Synthesis. Chem. Heterocycl. Compd. 2012, 48, 220–240. [Google Scholar] [CrossRef] [PubMed]
- Guillotin, L.; Cancellieri, P.; Lafite, P.; Landemarre, L.; Daniellou, R. Chemo-enzymatic synthesis of 3-O-(β-d-glycopyranosyl)-sn-glycerols and their evaluation as preservative in cosmetics. Pure Appl. Chem. 2017, 89, 11302. [Google Scholar] [CrossRef]
- Fukusumi, S.; Kamizono, A.; Horinouchi, S.; Beppu, T. Cloning and nucleotide sequence of a heat-stable amylase gene from an anaerobic thermophile, Dictyoglomus thermophilum. Eur. J. Biochem. 1988, 174, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Li, Z.; Ye, Q.; Xu, J.; Liu, Y. Heterologous expression and characterization of a novel thermo-halotolerant endoglucanase Cel5H from Dictyoglomus thermophilum. Bioresour. Technol. 2013, 142, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, M.; Imamura, H.; Shoun, H.; Horinouchi, S.; Wakagi, T. Transglycosylation Activity of Dictyoglomus thermophilum Amylase A. Biosci. Biotechnol. Biochem. 2004, 68, 2369–2373. [Google Scholar] [CrossRef]
- Guillotin, L.; Richet, N.; Lafite, P.; Daniellou, R. Is the acid/base catalytic residue mutation in β-d-mannosidase Dt Man from Dictyoglomus thermophilum sufficient enough to provide thioglycoligase activity? Biochimie 2017, 137, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Guillotin, L.; Kim, H.; Traore, Y.; Moreau, P.; Lafite, P.; Coquoin, V.; Nuccio, S.; De Vaumas, R.; Daniellou, R. Biochemical Characterization of the α-L-Rhamnosidase Dt Rha from Dictyoglomus thermophilum: Application to the Selective Derhamnosylation of Natural Flavonoids. ACS Omega 2019, 4, 1916–1922. [Google Scholar] [CrossRef]
- Huggett, A.S.G.; Nixon, D.A. Use of glucose oxidase, peroxidase and o-dianisidine in determination of blood and urinary glucose. Lancet 1957, 270, 368–370. [Google Scholar] [CrossRef]
- Guillotin, L.; Lafite, P.; Daniellou, R. Unraveling the Substrate Recognition Mechanism and Specificity of the Unusual Glycosyl Hydrolase Family 29 BT2192 from Bacteroides thetaiotaomicron. Biochemistry 2014, 53, 1447–1455. [Google Scholar] [CrossRef] [PubMed]
- Akiba, T.; Nishio, M.; Matsui, I.; Harata, K. X-ray structure of a membrane-bound β-glycosidase from the hyperthermophilic archaeon Pyrococcus horikoshii. Proteins Struct. Funct. Bioinform. 2004, 57, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Zechel, D.L.; Boraston, A.B.; Gloster, T.; Boraston, C.M.; Macdonald, J.M.; Tilbrook, D.M.G.; Stick, R.V.; Davies, G.J. Iminosugar Glycosidase Inhibitors: Structural and Thermodynamic Dissection of the Binding of Isofagomine and 1-Deoxynojirimycin to β-Glucosidases. J. Am. Chem. Soc. 2003, 125, 14313–14323. [Google Scholar] [CrossRef] [PubMed]
- Zechel, D.L.; Withers, S.G. Dissection of nucleophilic and acid-base catalysis in glycosidases. Curr. Opin. Chem. Biol. 2001, 5, 643–649. [Google Scholar] [CrossRef]
- Zhu, X.; Schmidt, R.R. New Principles for Glycoside-Bond Formation. Angew. Chem. Int. Ed. 2009, 48, 1900–1934. [Google Scholar] [CrossRef] [PubMed]
- Codée, J.D.C.; Litjens, R.E.J.N.; Van den Bos, L.J.; Overkleeft, H.S.; Van der Marel, G.A. Thioglycosides in sequential glycosylation strategies. Chem. Soc. Rev. 2005, 34, 769–782. [Google Scholar] [CrossRef] [PubMed]
- Pornsuriyasak, P.; Demchenko, A.V. S-Thiazolinyl (STaz) Glycosides as Versatile Building Blocks for Convergent Selective, Chemoselective and Orthogonal Oligosaccharide Synthesis. Chem.-Eur J. 2006, 12, 6630–6646. [Google Scholar] [CrossRef] [PubMed]
- Jerez, G.; Kaufman, G.; Prystai, M.; Schenkeveld, S.; Donkor, K.K. Determination of thermodynamic p K a values of benzimidazole and benzimidazole derivatives by capillary electrophoresis. J. Sep. Sci. 2009, 32, 1087–1095. [Google Scholar] [CrossRef]
- Akhond, M.; Ghaedi, M.; Tashkhourian, J. Development of a New Copper(II) Ion-selective Poly(vinyl chloride) Membrane Electrode Based on 2-Mercaptobenzoxazole. Bull. Korean Chem. Soc. 2005, 26, 882–886. [Google Scholar] [Green Version]
- Kamat, M.N.; Rath, N.P.; Demchenko, A. V Versatile Synthesis and Mechanism of Activation of S-Benzoxazolyl Glycosides. J. Org. Chem. 2007, 72, 6938–6946. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Darden, T.A.; III, T.E.C.; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Walker, R.C.; Zhang, W.; Merz, K.M.; et al. AMBER 12 2012; University of California: San Francisco, CA, USA, 2012. [Google Scholar]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.-P.; McKiernan, K.A.; Gomes, J.; Beauchamp, K.A.; Head-Gordon, T.; Rice, J.E.; Swope, W.C.; Martínez, T.J.; Pande, V.S. Building a More Predictive Protein Force Field: A Systematic and Reproducible Route to AMBER-FB15. J. Phys. Chem. B 2017, 121, 4023–4039. [Google Scholar] [CrossRef] [PubMed]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef] [PubMed]
- Lafite, P.; André, F.; Zeldin, D.C.; Dansette, P.M.; Mansuy, D. Unusual Regioselectivity and Active Site Topology of Human Cytochrome P450 2J2. Biochemistry 2007, 46, 10237–10247. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Organism | Substrates Tested | Relative Activity S-vs. O-(%) | Ref |
|---|---|---|---|---|
| β-D-Glucosidase | Sweet almond | pNPSGlc pNPSGal pNPSFuc GlcSBiz GlcS(N-Me)Biz GlcSBox | 0.13 a 0.07 a 0.06 a 80 a 10 a 5 a | [22] [16] |
| β-D-Glucosidase | A. niger | GlcSBiz GlcS(N-Me)Biz | 5 a 1 a | [16] |
| Sialidase | M. viridifaciens | Substituted pNPSNeuAc | 0.01–60 | [21] |
| Os4BGlu12 | O. sativa | pNPSGlc OctylSGlc | 0.5 a 0.1 b | [23] |
| Enzyme | Substrate | KM (µM) | kcat (s−1) | kcat/KM (s−1.mM−1) |
|---|---|---|---|---|
| WT | pNP-Glc a | 460 ± 40 | 31 ± 0.7 | 67 |
| WT | GlcSBiz | 1533 ± 114 | 0.23 ± 0.01 | 0.15 |
| WT | GlcSBox | 2246 ± 289 | 0.38 ± 0.03 | 0.17 |
| WT | GlcSTaz | 880 ± 52 | 0.31 ± 0.01 | 0.35 |
| E159Q | pNP-Glc | 200 ± 20 | 0.20 ± 0.01 | 1.0 |
| E159Q | GlcSBox | 445 ± 40 | 0.06 ± 0.01 | 0.13 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guillotin, L.; Assaf, Z.; Pistorio, S.G.; Lafite, P.; Demchenko, A.V.; Daniellou, R. Hydrolysis of Glycosyl Thioimidates by Glycoside Hydrolase Requires Remote Activation for Efficient Activity. Catalysts 2019, 9, 826. https://doi.org/10.3390/catal9100826
Guillotin L, Assaf Z, Pistorio SG, Lafite P, Demchenko AV, Daniellou R. Hydrolysis of Glycosyl Thioimidates by Glycoside Hydrolase Requires Remote Activation for Efficient Activity. Catalysts. 2019; 9(10):826. https://doi.org/10.3390/catal9100826
Chicago/Turabian StyleGuillotin, Laure, Zeinab Assaf, Salvatore G. Pistorio, Pierre Lafite, Alexei V. Demchenko, and Richard Daniellou. 2019. "Hydrolysis of Glycosyl Thioimidates by Glycoside Hydrolase Requires Remote Activation for Efficient Activity" Catalysts 9, no. 10: 826. https://doi.org/10.3390/catal9100826