Electrochemical Promotion of Nanostructured Palladium Catalyst for Complete Methane Oxidation



Abstract
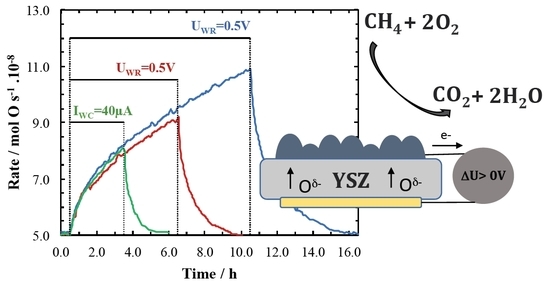
:
1. Introduction
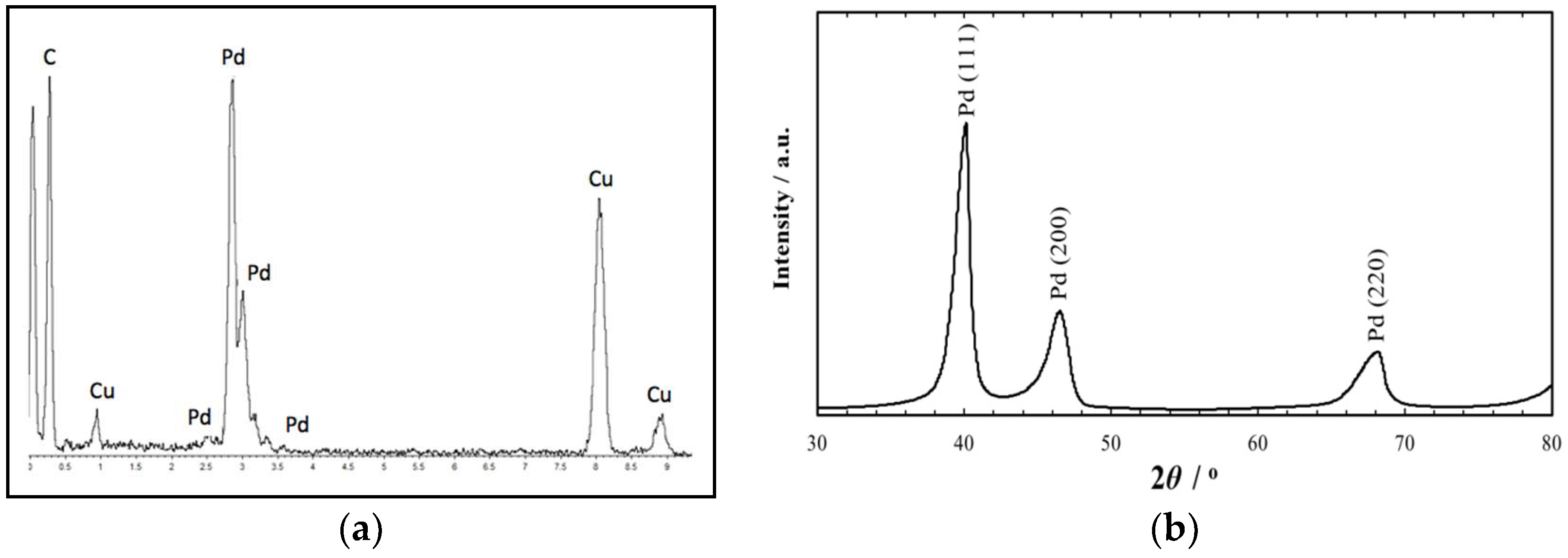
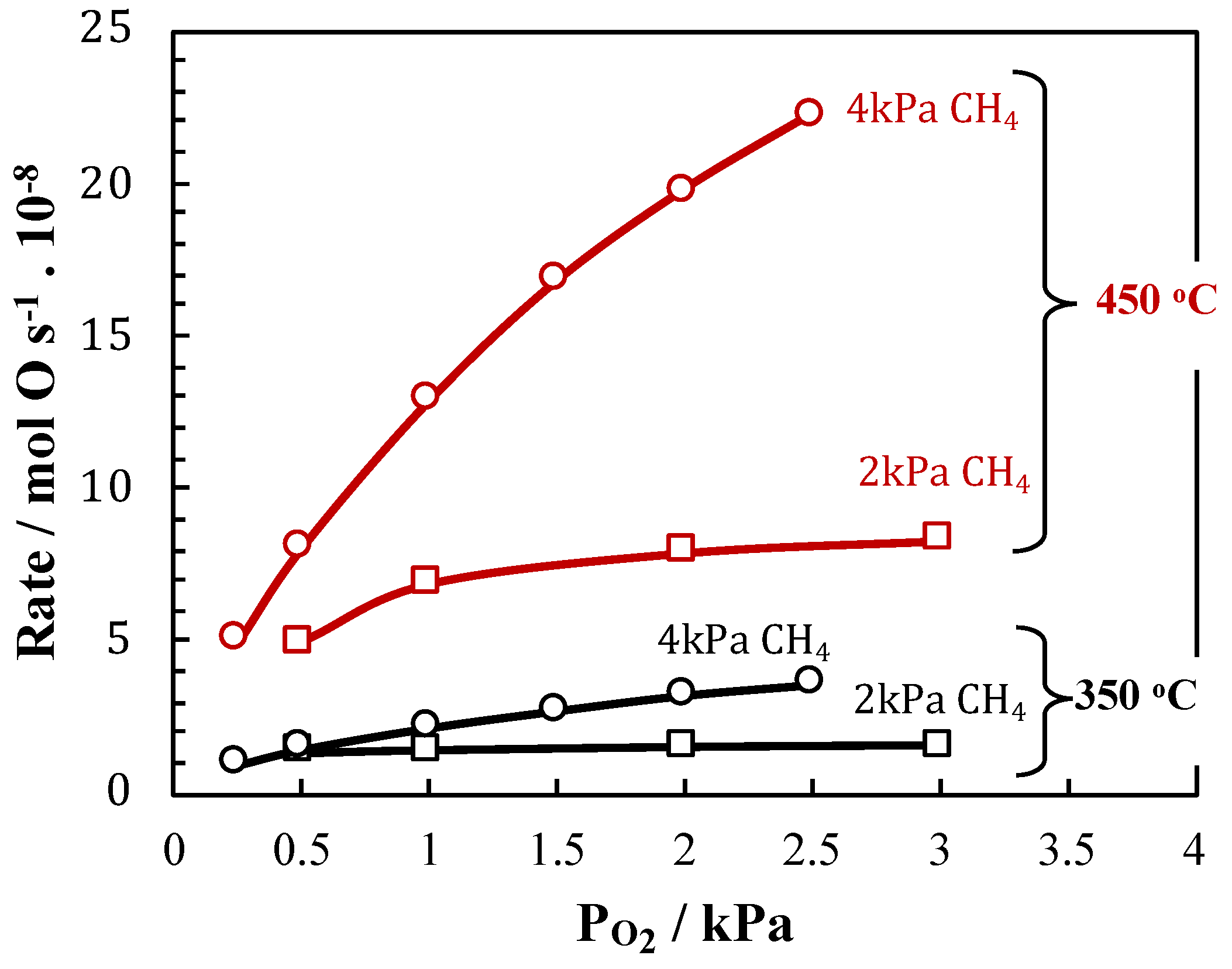
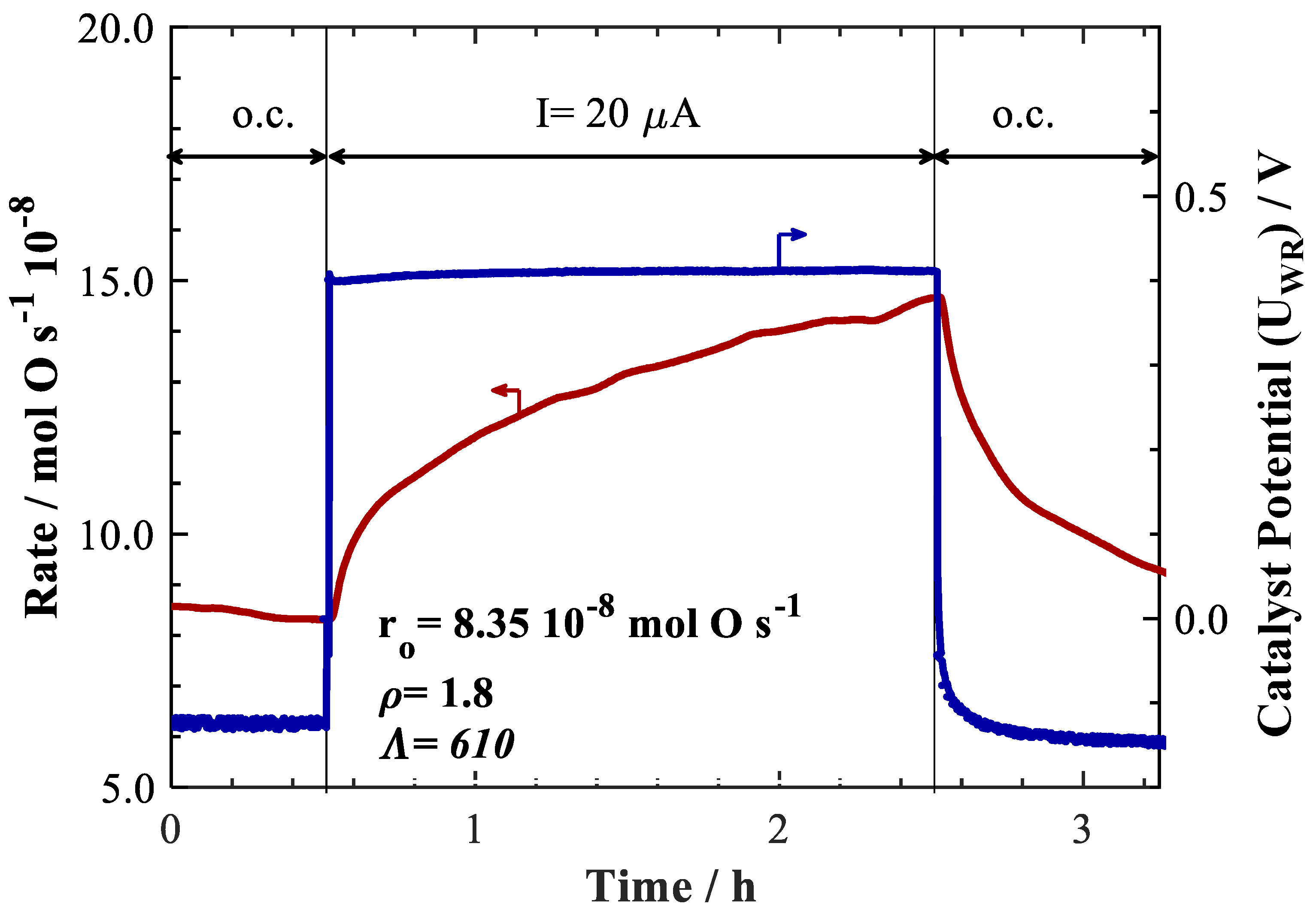
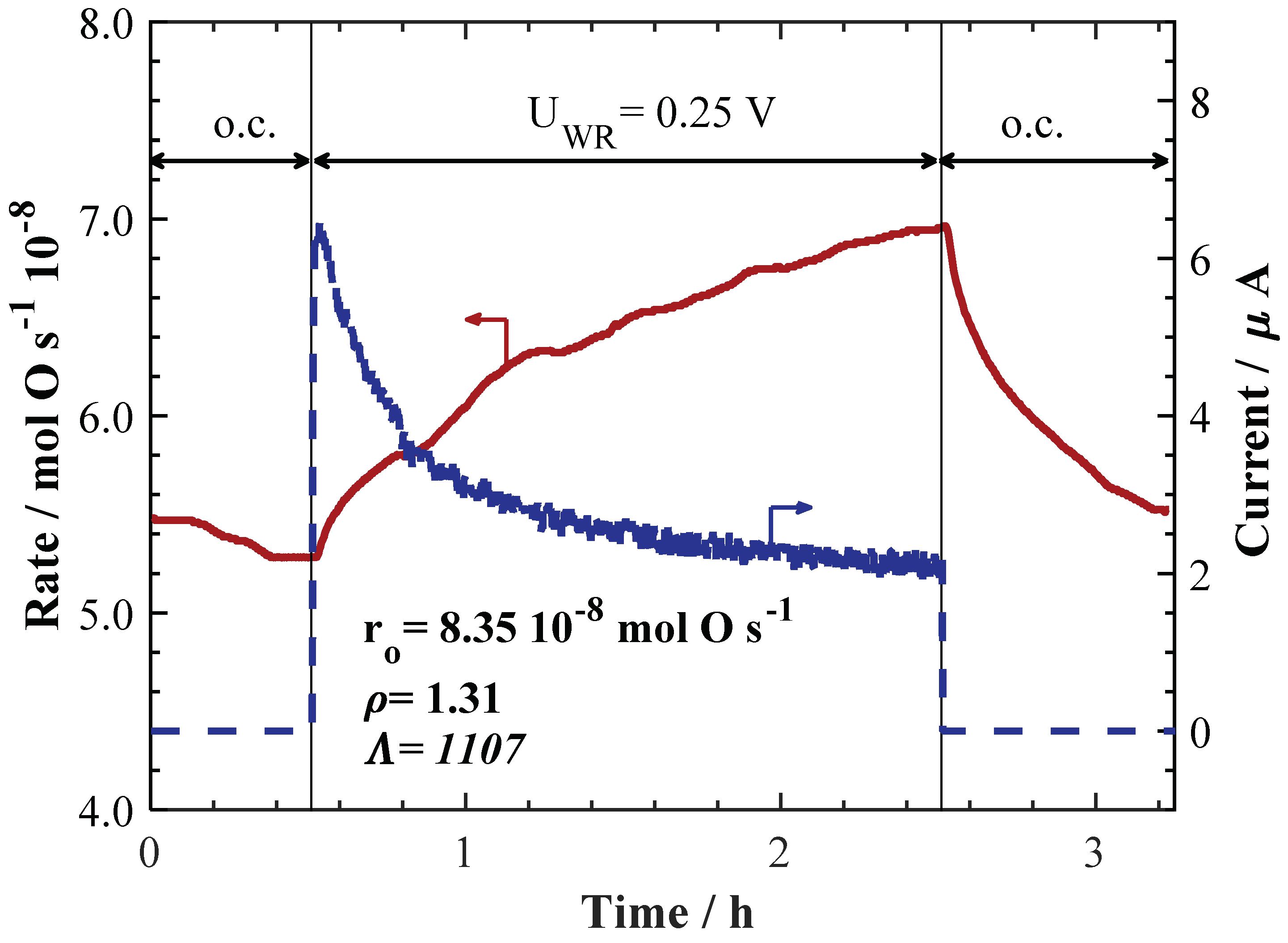
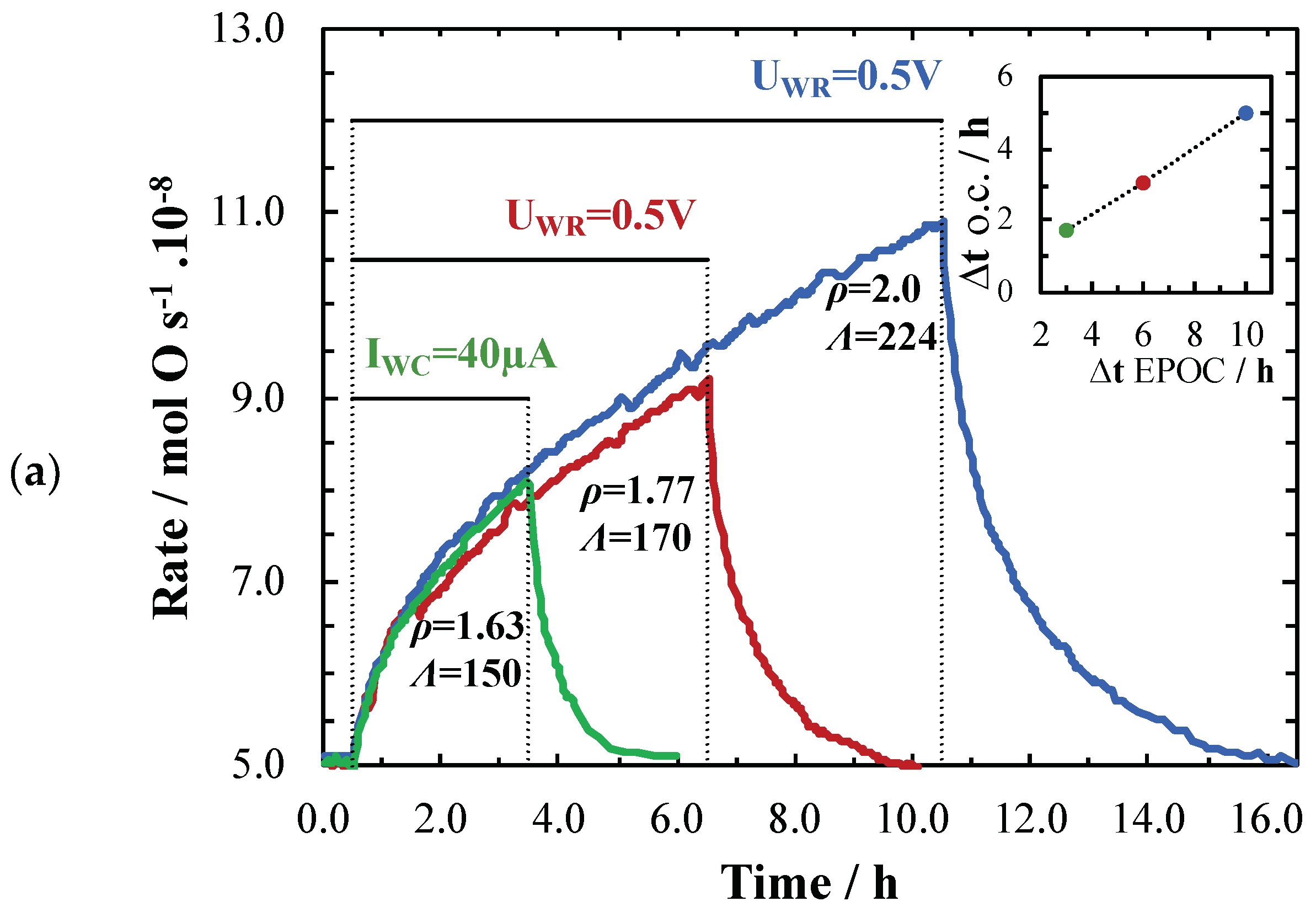
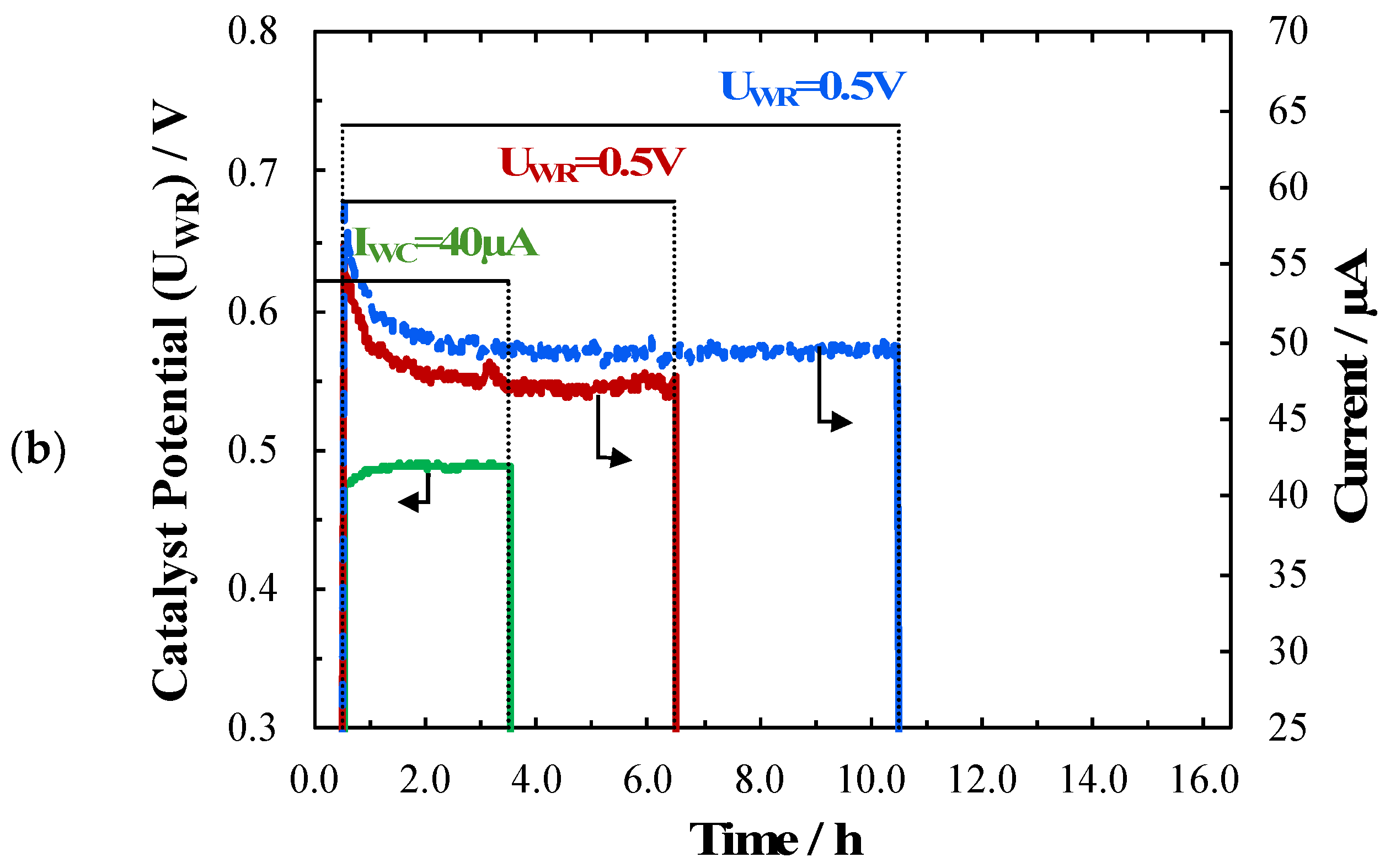
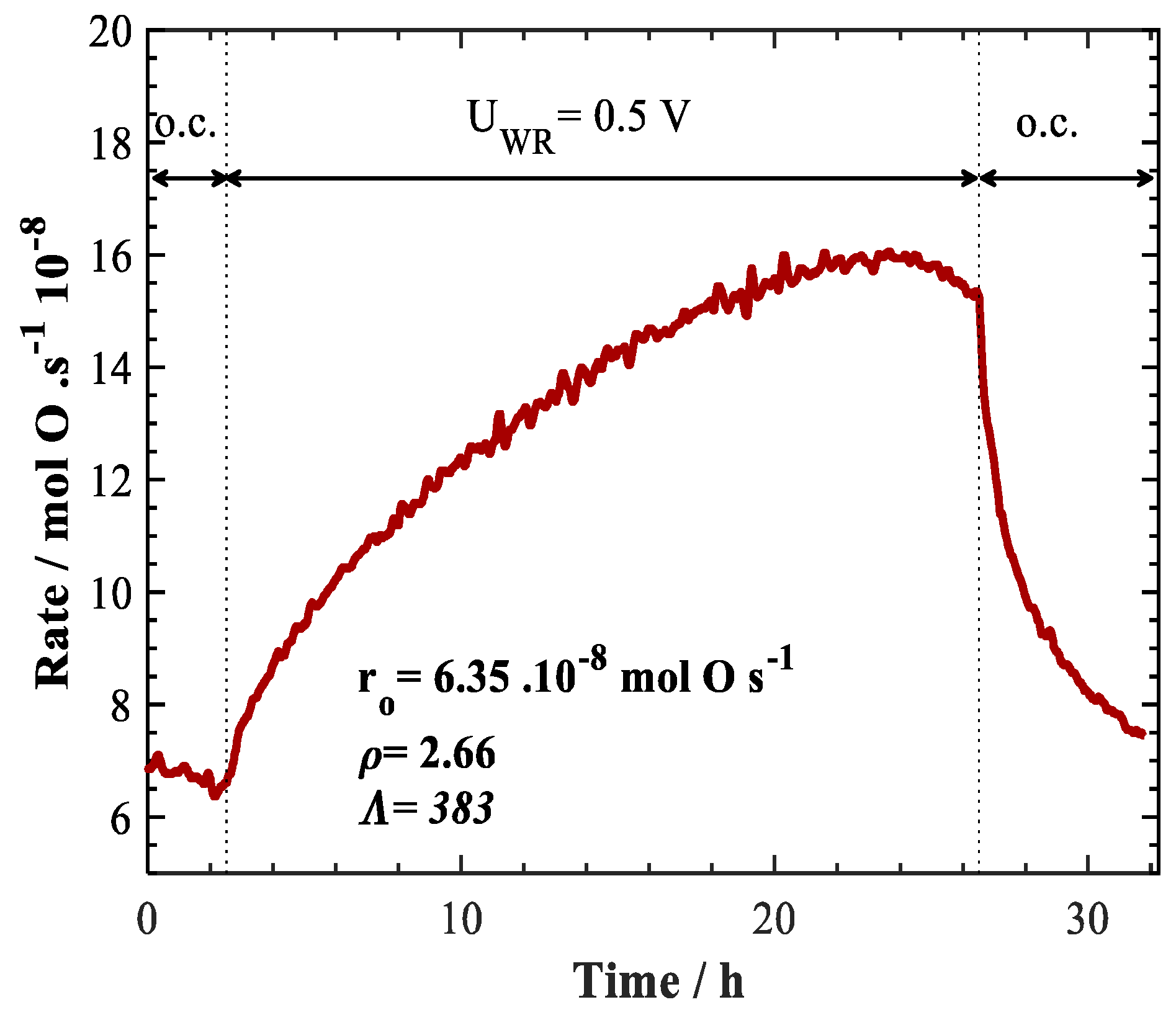
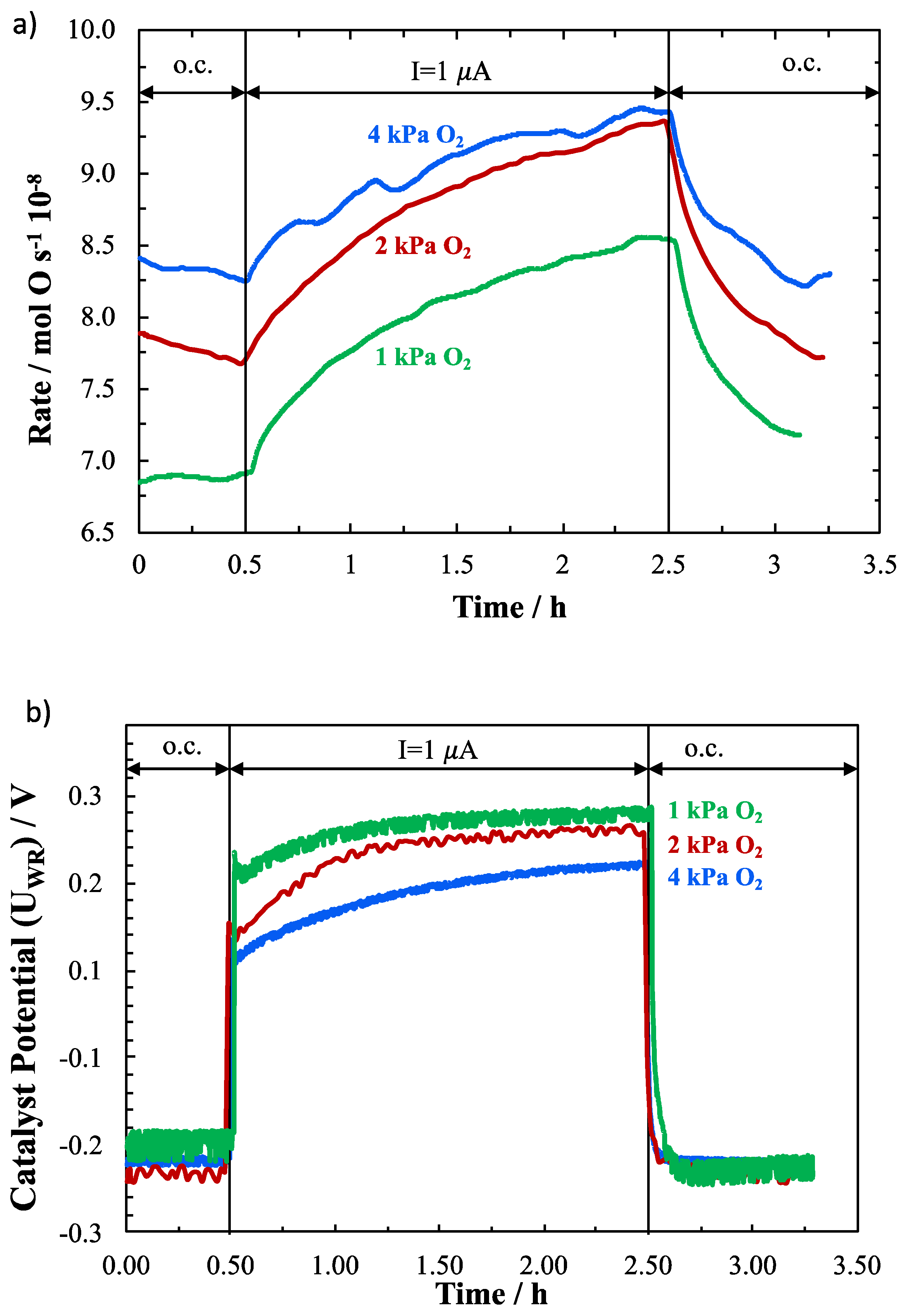
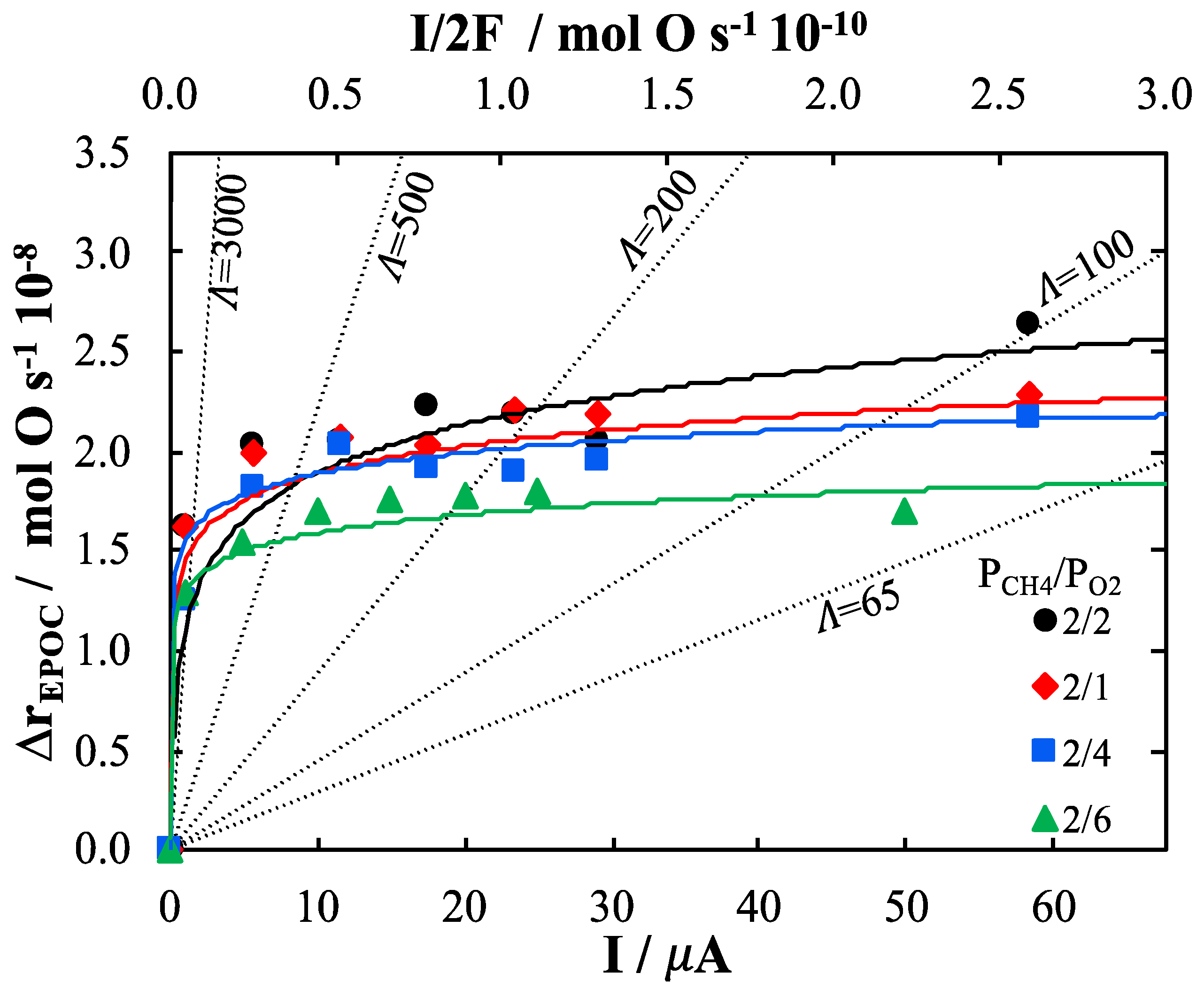
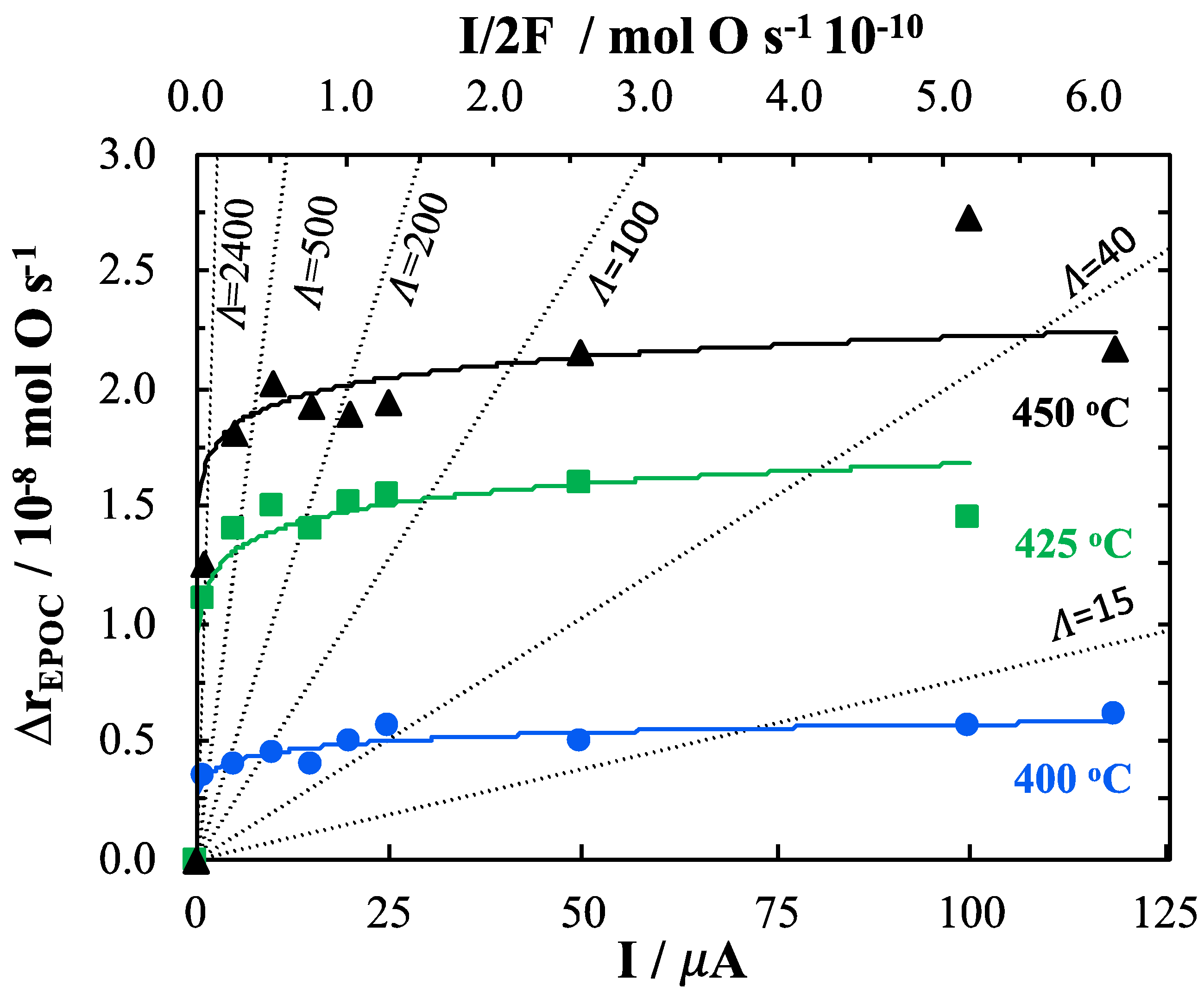
2. Results and Discussion
3. Experimental
3.1. Synthesis of Pd Nanoparticles
3.2. Catalyst Characterization
3.3. Electrochemical Cell and Reactor
3.4. Catalytic and Electrochemical Measurements
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Demirbaş, A. Biomass resource facilities and biomass conversion processing for fuels and chemicals. Energy Convers. Manag. 2001, 42, 1357–1378. [Google Scholar] [CrossRef]
- Cheng, H.; Hu, Y. Municipal solid waste (MSW) as a renewable source of energy: Current and future practices in China. Bioresour. Technol. 2010, 101, 3816–3824. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, M.; Sahu, J.N.; Ganesan, P. Effect of process parameters on production of biochar from biomass waste through pyrolysis: A review. Renew. Sustain. Energy Rev. 2016, 55, 467–481. [Google Scholar] [CrossRef]
- Xiao, Y.; He, P.; Cheng, W.; Liu, J.; Shan, W.; Song, H. Converting solid wastes into liquid fuel using a novel methanolysis process. Waste Manag. 2016, 49, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Farrauto, R.J.; Hobson, M.C.; Kennelly, T.; Waterman, E.M. Catalytic chemistry of supported palladium for combustion of methane. Appl. Catal. A Gen. 1992, 81, 227–237. [Google Scholar] [CrossRef]
- Ciuparu, D.; Lyubovsky, M.R.; Altman, E.; Pfefferle, L.D.; Datye, A. Catalytic combustion of methane over palladium-based catalysts. Catal. Rev. Sci. Eng. 2002, 44, 593–649. [Google Scholar] [CrossRef]
- Burch, R.; Urbano, F.J. Investigation of the active state of supported palladium catalysts in the combustion of methane. Appl. Catal. A Gen. 1995, 124, 121–138. [Google Scholar] [CrossRef]
- Boudart, M.; Djega-Mariadassou, G. Kinetics of Heterogeneous Catalytic Reactions; Princeton University Press: Princeton, NJ, USA, 1984. [Google Scholar]
- Campbell, I.M. Catalysis at Surfaces; Springer Science & Business Media: New York, NY, USA, 1988. [Google Scholar]
- Vayenas, C.G.; Bebelis, S.; Neophytides, S. Non-Faradaic Electrochemical Modification of Catalytic Activity. J. Phys. Chem. 1988, 92, 5083–5085. [Google Scholar] [CrossRef]
- Ladas, S.; Kennou, S.; Bebelis, S.; Vayenas, C.G. Origin of non-faradaic electrochemical modification of catalytic activity. J. Phys. Chem. 1993, 97, 8845–8848. [Google Scholar] [CrossRef]
- Nicole, J.; Tsiplakides, D.; Pliangos, C.; Verykios, X.E.E.; Comninellis, C.; Vayenas, C.G.G. Electrochemical Promotion and Metal–Support Interactions. J. Catal. 2001, 204, 23–34. [Google Scholar] [CrossRef]
- Vayenas, C.G.; Bebelis, S.; Ladas, S. Dependence of catalytic rates on catalyst work function. Nature 1990, 343, 625–627. [Google Scholar] [CrossRef]
- Vayenas, C.G.; Bebelis, S.; Pliangos, C.; Brosda, S.; Tsiplakides, D. Electrochemical Activation of Catalysis: Promotion, Electrochemical Promotion, and Metal-Support Interactions; Springer: New York, NY, USA, 2001. [Google Scholar] [CrossRef]
- Giannikos, A.; Frantzis, A.D.; Pliangos, C.; Bebelis, S.; Vayenas, C.G. Electrochemical promotion of CH4 oxidation on Pd. Ionics (Kiel) 1998, 4, 53–60. [Google Scholar] [CrossRef]
- Frantzis, A.D.; Bebelis, S.; Vayenas, C.G. Electrochemical promotion (NEMCA) of CH4 and C2H4 oxidation on Pd/YSZ and investigation of the origin of NEMCA via AC impedance spectroscopy. Solid State Ionics 2000, 136–137, 863–872. [Google Scholar] [CrossRef]
- Roche, V.; Karoum, R.; Billard, A.; Revel, R.; Vernoux, P. Electrochemical promotion of deep oxidation of methane on Pd/YSZ. J. Appl. Electrochem. 2008, 38, 1111–1119. [Google Scholar] [CrossRef]
- Jiménez-Borja, C.; Dorado, F.; de Lucas-Consuegra, A.; García-Vargas, J.M.; Valverde, J.L.J.L. Complete oxidation of methane on Pd/YSZ and Pd/CeO2/YSZ by electrochemical promotion. Catal. Today 2009, 146, 326–329. [Google Scholar] [CrossRef]
- Roche, V.; Revel, R.; Vernoux, P. Electrochemical promotion of YSZ monolith honeycomb for deep oxidation of methane. Catal. Commun. 2010, 11, 1076–1080. [Google Scholar] [CrossRef]
- Jiménez-Borja, C.; Dorado, F.; Consuegra, A.D.; Vargas, J.M.G.; Valverde, J.L. Electrochemical Promotion of CH4 Combustion over a Pd/CeO2-YSZ Catalyst. Fuel Cells 2011, 11, 131–139. [Google Scholar] [CrossRef]
- Matei, F.; Ciuparu, D.; Jiménez-Borja, C.; Dorado, F.; Valverde, J.L.; Brosda, S. Electrochemical promotion of methane oxidation on impregnated and sputtered Pd catalyst-electrodes deposited on YSZ. Appl. Catal. B Environ. 2012, 127, 18–27. [Google Scholar] [CrossRef]
- Jimenez-Borja, C.; Brosda, S.; Matei, F.; Makri, M.; Delgado, B.; Sapountzi, F.; Ciuparu, D.; Dorado, F.; Valverde, J.L.; Vayenas, C.G. Electrochemical promotion of methane oxidation on Pd catalyst-electrodes deposited on Y2O3-stabilized-ZrO2. Appl. Catal. B Environ. 2012, 128, 48–54. [Google Scholar] [CrossRef]
- Matei, F.; Jimenez-Borja, C.; Canales-Vazquez, J.; Brosda, S.; Dorado, F.; Valverde, J.L.; Ciuparu, D. Enhanced electropromotion of methane combustion on palladium catalysts deposited on highly porous supports. Appl. Catal. B Environ. 2013, 132–133, 80–89. [Google Scholar] [CrossRef]
- Jimenez-Borja, C.; Matei, F.; Dorado, F.; Valverde, J.L. Characterization of Pd catalyst-electrodes deposited on YSZ: Influence of the preparation technique and the presence of a ceria interlayer. Appl. Surf. Sci. 2012, 261, 671–678. [Google Scholar] [CrossRef]
- Garbowski, E.; Feumi-Jantou, C.; Mouaddib, N.; Primet, M. Catalytic combustion of methane over palladium supported on alumina catalysts: Evidence for reconstruction of particles. Appl. Catal. A Gen. 1994, 109, 277–292. [Google Scholar] [CrossRef]
- Vayenas, C.G.; Bebelis, S.; Yentekakis, I.V.; Lintz, H.-G. Non-faradaic electrochemical modification of catalytic activity: A status report. Catal. Today 1992, 11, 303–442. [Google Scholar] [CrossRef]
- Nicole, J.; Comninellis, C.H. Electrochemical promotion of IrO2 catalyst activity for the gas phase combustion of ethylene. J. Appl. Electrochem. 1998, 28, 223–226. [Google Scholar] [CrossRef]
- Vayenas, C.; Brosda, S.; Pliangos, C. The double-layer approach to promotion, electrocatalysis, electrochemical promotion, and metal–support interactions. J. Catal. 2003, 216, 487–504. [Google Scholar] [CrossRef]
- Reuter, K.; Scheffler, M. Composition, structure, and stability of RuO2 (110) as a function of oxygen pressure. Phys. Rev. B 2001, 65, 035406. [Google Scholar] [CrossRef]
- Gibson, I.R.; Dransfield, G.P.; Irvine, J.T.S. Sinterability of commercial 8 mol % yttria-stabilized zirconia powders and the effect of sintered density on the ionic conductivity. J. Mater. Sci. 1998, 33, 4297–4305. [Google Scholar] [CrossRef]
- Hajar, Y.M.; Dole, H.A.; Couillard, M.; Baranova, E.A. Investigation of heterogeneous catalysts by electrochemical method: Ceria and titania supported iridium for ethylene oxidation. ECS Trans. 2016, 72, 161–172. [Google Scholar] [CrossRef]
- Hajar, Y.M.; Patel, K.D.; Tariq, U.; Baranova, E.A. Functional equivalence of electrochemical promotion and metal support interaction for Pt and RuO2 nanoparticles. J. Catal. 2017, 352, 42–51. [Google Scholar] [CrossRef]
- Hajar, Y.M.; Houache, M.S.; Tariq, U.; Vernoux, P.; Baranova, E.A. Nanoscopic Ni interfaced with oxygen conductive supports: Link between electrochemical and catalytic studies. Electrochem. Soc. 2017, 77, 51–66. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst Synthesis Method | Loading | I or U Applied | T/°C | PCH4/kPa | PO2/kPa | Total Flow/ccm | Rate Change/mol O.s−1 10−8 | ρ | Ʌ | Authors, Year & Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| Paste coating | n/a | +300 µA | 400 | 2.75 | 1.55 | n/a | 4.52 to 20.5 | 4.5 | 103 | a Giannikos et al. (1998) [15] |
| Paste coating | n/a | +1 V | 400 | 2.6 | 1.9 | n/a | 0.295 to 20 | 68 | 153 | a Frantzis et al. (2000) [16] |
| PVD | 24 µg | +100 µA | 500 | 2 | 10 | 166 | 7.3 to 20.4 | 2.8 | 258 | Roche et al. (2008) [17] |
| Thermal decomposition | 1.1 mg/cm2 | +10 mA | 600 | 0.4 | 1 | 150 | 0.47 to 0.68 | 2.6 | <1 | a,b Jimenez-Borja et al. (2009) [18] |
| Electroless deposition | 5 mg total | +100 µA | 400 | 2 | 10 | 166 | 136.4 to 135.2 | 0.99 | −23 | Roche et al. (2010) [19] |
| Paste coating | 7 mg/cm2 | +25 µA | 560 | 0.4 | 1.2 | 150 | 1.6 to 9.1 | 5.6 | 579 | a Jimenez-Borja et al. (2011) [20] |
| Sputtered | 0.4 mg/cm2 | +1 mA | 350 | 1.3 | 4.5 | 200 | 11.4 to 18.2 | 1.6 | 12 | Matei et al. (2012) [21] |
| Impregnation | 0.4 mg/cm2 | +300 µA | 350 | 1.3 | 4.5 | 200 | 22 to 26 | 1.18 | 25 | Jimenez-Borja et al. (2012) [22] |
| Impregnation | 0.4 mg/cm2 | +5 mA | 400 | 1.4 | 2.8 | 200 | 135 to 158 | 1.2 | 17 | Matei et al. (2013) [23] |
| Polyol method | 0.3 mg/cm2 | +0.5 V | 425 | 2 | 4 | 100 | 6 to 16 | 2.66 | 383 | a This work |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hajar, Y.M.; Venkatesh, B.; Baranova, E.A. Electrochemical Promotion of Nanostructured Palladium Catalyst for Complete Methane Oxidation. Catalysts 2019, 9, 48. https://doi.org/10.3390/catal9010048
Hajar YM, Venkatesh B, Baranova EA. Electrochemical Promotion of Nanostructured Palladium Catalyst for Complete Methane Oxidation. Catalysts. 2019; 9(1):48. https://doi.org/10.3390/catal9010048
Chicago/Turabian StyleHajar, Yasmine M., Balaji Venkatesh, and Elena A. Baranova. 2019. "Electrochemical Promotion of Nanostructured Palladium Catalyst for Complete Methane Oxidation" Catalysts 9, no. 1: 48. https://doi.org/10.3390/catal9010048