Co-Immobilization of Ketoreductase and Glucose Dehydrogenase

 ,
,
Abstract
:1. Introduction
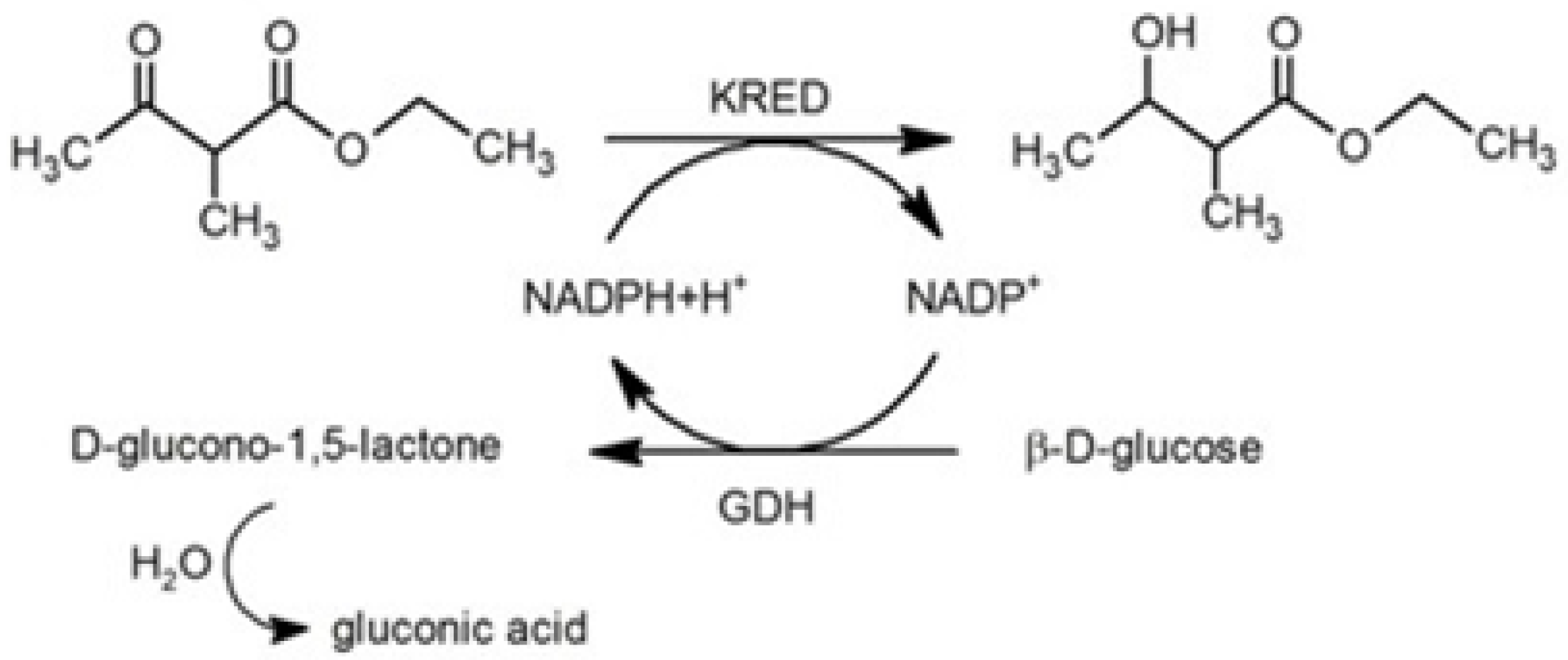
2. Results and Discussion
2.1. KRED and GDH Expression and Purification
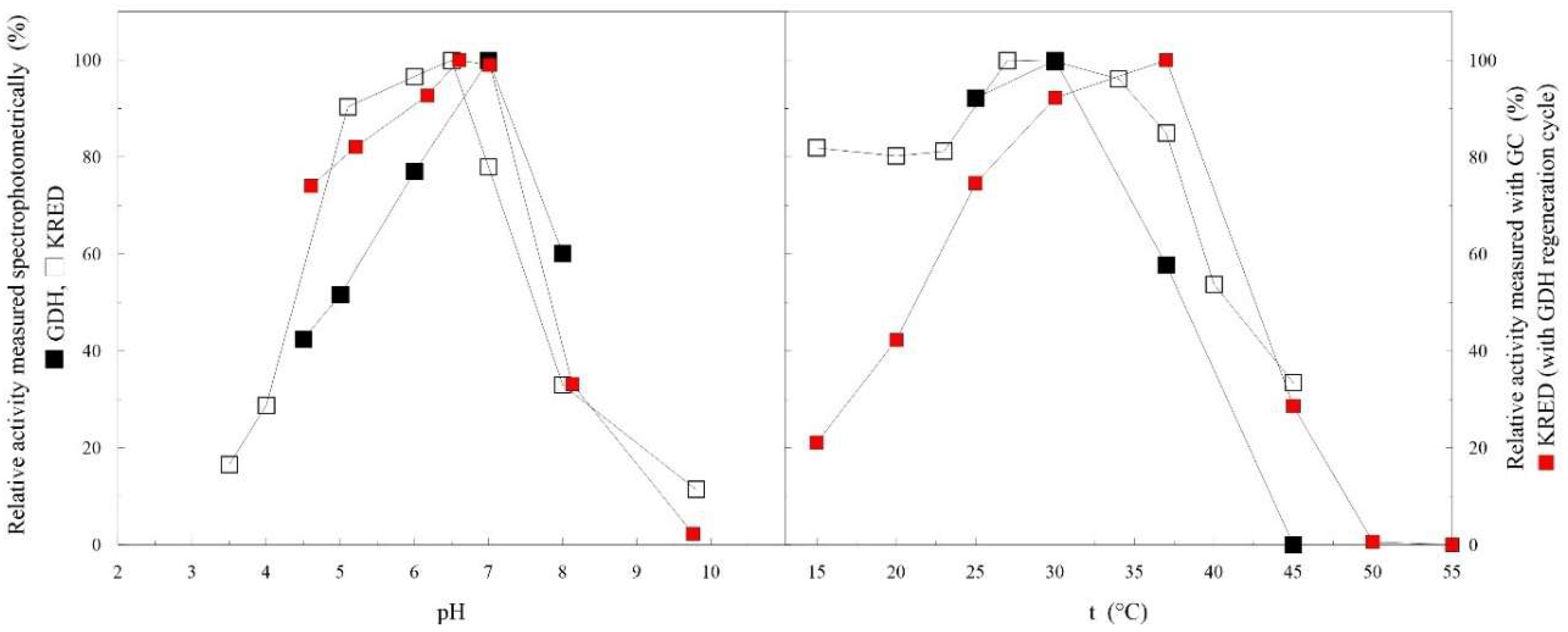
2.2. Free-Enzyme Biotransformations
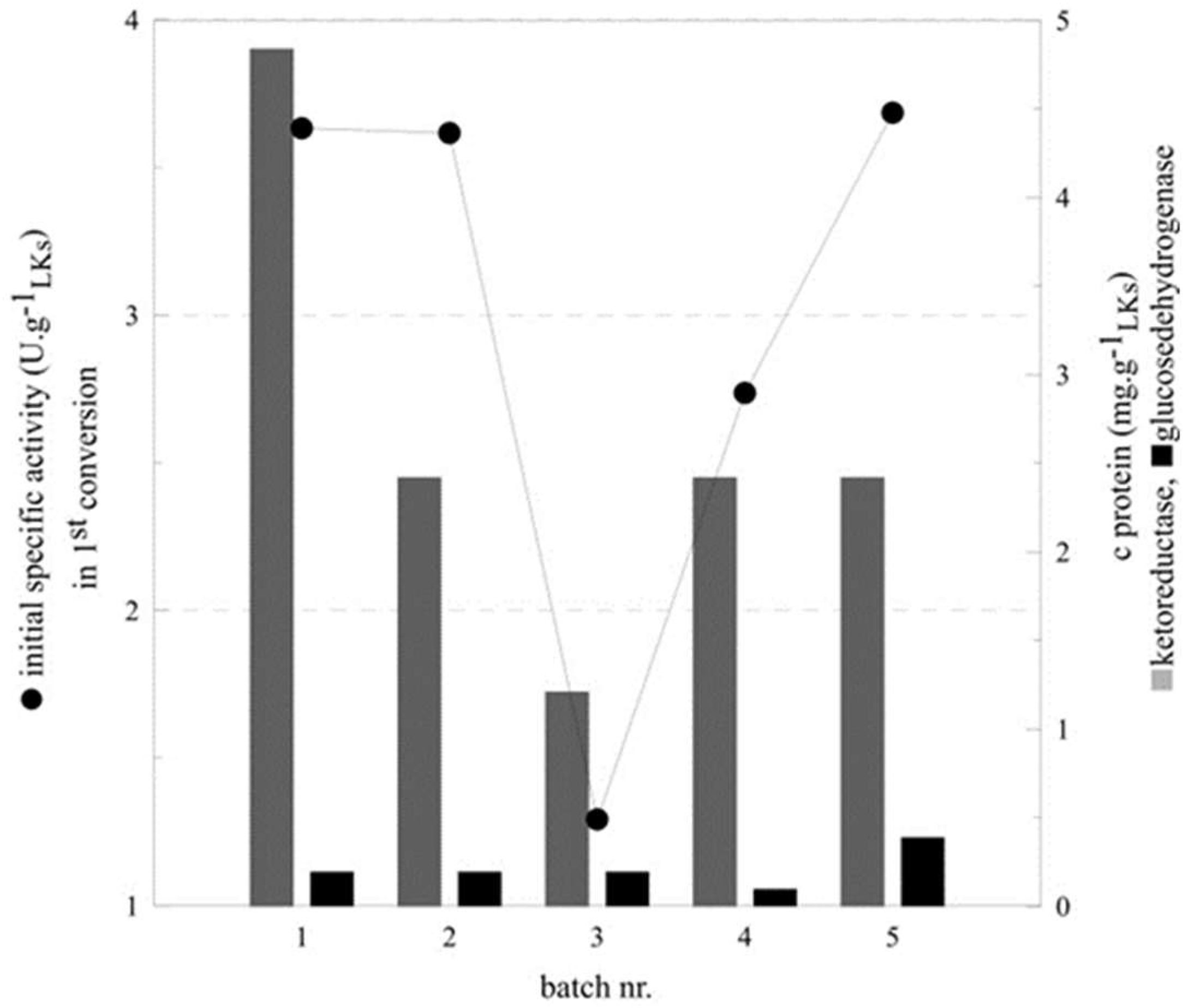
2.3. Optimization of Co-Immobilization
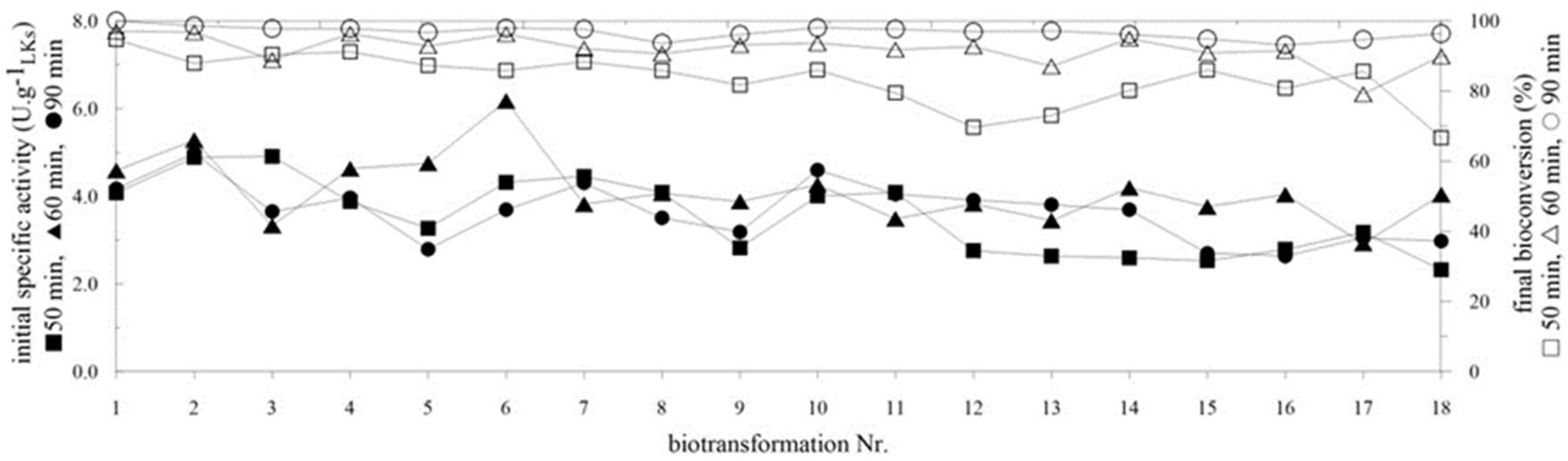
2.4. Repeated Biotransformations
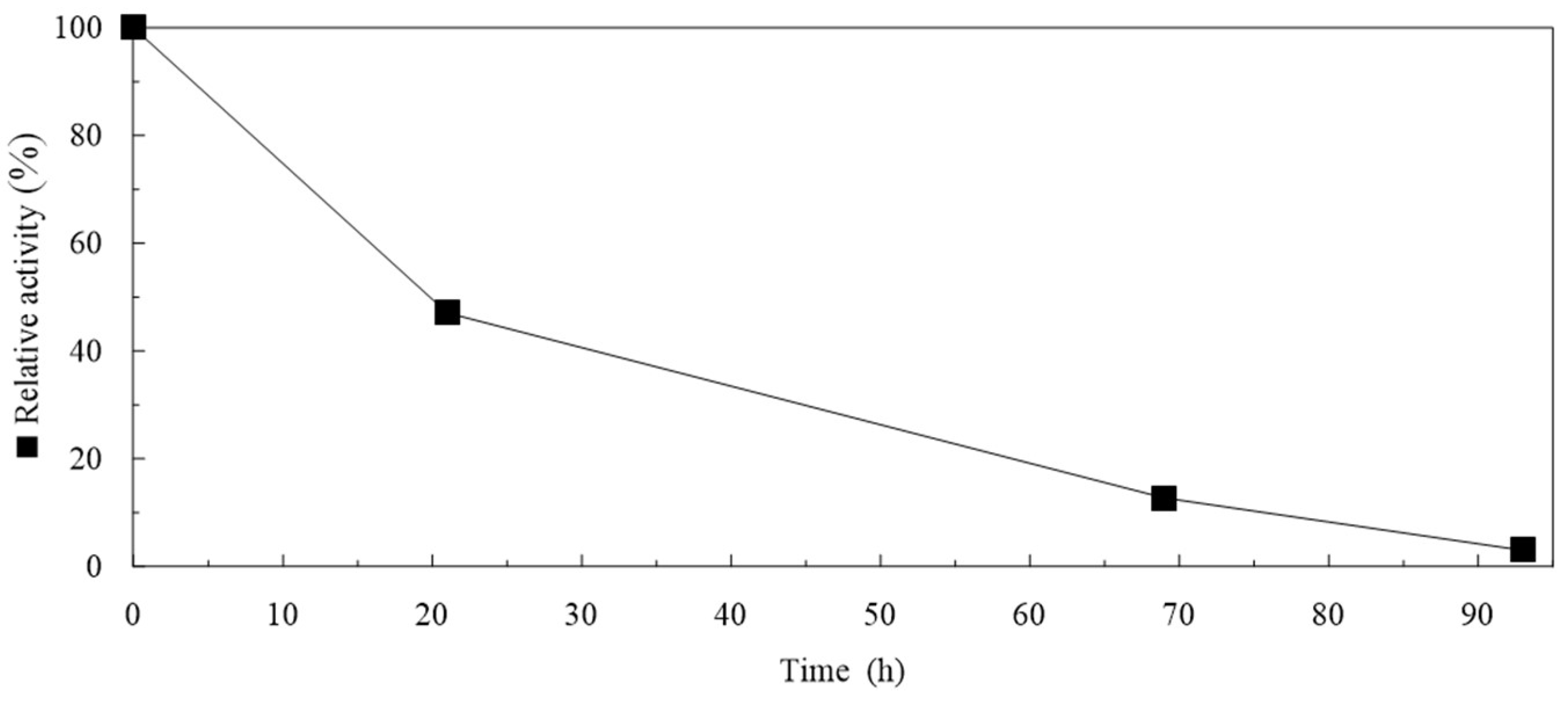
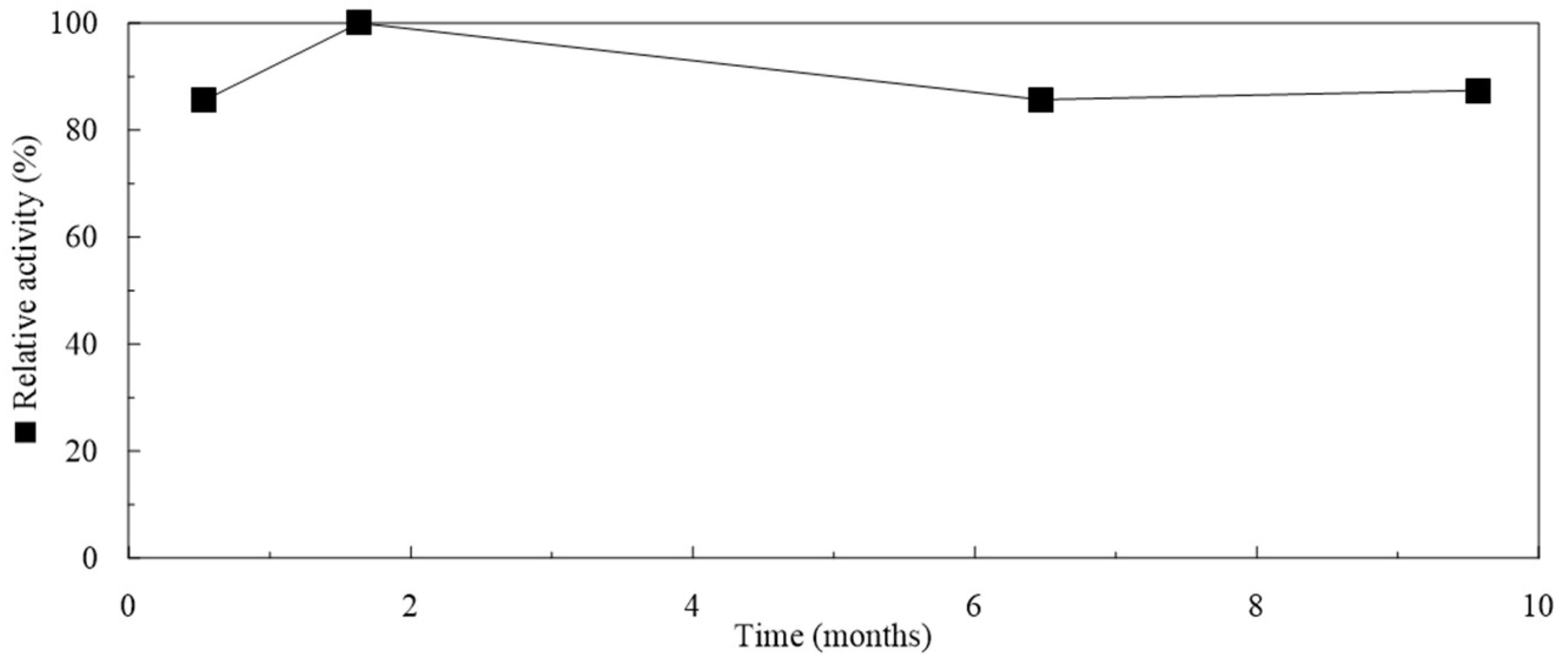
2.5. Storage of Particles
3. Materials and Methods
3.1. Chemicals and Media
3.2. Cloning
3.3. Preparation and Purification of KRED and GDH
3.4. Enzyme Assays
3.5. Co-Immobilization
3.6. Biotransformation
3.7. Analytics
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Li, H.M.; Moncecchi, J.; Truppo, M.D. Development of an immobilized ketoreductase for enzymatic (r)-1-(3,5-bis(trifluoromethyl)phenyl)ethanol production. Organ. Process Res. Dev. 2015, 19, 695–700. [Google Scholar] [CrossRef]
- Dai, Z.; Guillemette, K.; Green, T.K. Stereoselective synthesis of aryl γ,δ-unsaturated β-hydroxyesters by ketoreductases. J. Mol. Catal. B Enzym. 2013, 97, 264–269. [Google Scholar] [CrossRef]
- Kalaitzakis, D.; David Rozzell, J.; Kambourakis, S.; Smonou, I. Highly stereoselective reductions of α-alkyl-1,3-diketones and α-alkyl-β-keto esters catalyzed by isolated nadph-dependent ketoreductases. Org. Lett. 2005, 7, 4799–4801. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Chen, Y.; Goswami, A.; Hanson, R.L.; Patel, R.N. Synthesis of ethyl and t-butyl (3r,5s)-dihydroxy-6-benzyloxy hexanoates via diastereo- and enantioselective microbial reduction. Tetrahedron Asymmetry 2006, 17, 1589–1602. [Google Scholar] [CrossRef]
- Zhu, D.; Mukherjee, C.; Rozzell, J.D.; Kambourakis, S.; Hua, L. A recombinant ketoreductase tool-box. Assessing the substrate selectivity and stereoselectivity toward the reduction of β-ketoesters. Tetrahedron 2006, 62, 901–905. [Google Scholar] [CrossRef]
- Kaluzna, I.A.; David Rozzell, J.; Kambourakis, S. Ketoreductases: Stereoselective catalysts for the facile synthesis of chiral alcohols. Tetrahedron Asymmetry 2005, 16, 3682–3689. [Google Scholar] [CrossRef]
- Li, H.; Zhu, D.; Hua, L.; Biehl, E.R. Enantioselective reduction of diaryl ketones catalyzed by a carbonyl reductase from sporobolomyces salmonicolor and its mutant enzymes. Adv. Synth. Catal. 2009, 351, 583–588. [Google Scholar] [CrossRef]
- Truppo, M.D.; Kim, J.; Brower, M.; Madin, A.; Sturr, M.G.; Moore, J.C. A novel resolution of a pharmaceutically important bridged bicyclic ketone intermediate via selective enzymatic reduction with a commercially available ketoreductase. J. Mol. Catal. B Enzym. 2006, 38, 158–162. [Google Scholar] [CrossRef]
- Milner, S.E.; Maguire, A.R. Recent trends in whole cell and isolated enzymes in enantioselective synthesis. Arkivoc 2012, 2012, 321–382. [Google Scholar]
- Nagayama, K.; Spiess, A.C.; Büchs, J. Gas phase enantioselective reduction catalyzed by immobilized ketoreductase: Effects of water activity and reaction temperature. Biochem. Eng. J. 2010, 52, 301–303. [Google Scholar] [CrossRef]
- Dall'Oglio, F.; Contente, M.L.; Conti, P.; Molinari, F.; Monfredi, D.; Pinto, A.; Romano, D.; Ubiali, D.; Tamborini, L.; Serra, I. Flow-based stereoselective reduction of ketones using an immobilized ketoreductase/glucose dehydrogenase mixed bed system. Catal. Commun. 2017, 93, 29–32. [Google Scholar] [CrossRef]
- Ning, C.; Su, E.; Tian, Y.; Wei, D. Combined cross-linked enzyme aggregates (combi-cleas) for efficient integration of a ketoreductase and a cofactor regeneration system. J. Biotechnol. 2014, 184, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Rebroš, M.; Rosenberg, M.; Mlichová, Z.; Krištofíková, L.; Paluch, M. A simple entrapment of glucoamylase into lentikats® as an efficient catalyst for maltodextrin hydrolysis. Enzym. Microb. Technol. 2006, 39, 800–804. [Google Scholar] [CrossRef]
- Rebroš, M.; Rosenberg, M.; Stloukal, R.; Krištofíková, L. High efficiency ethanol fermentation by entrapment of zymomonas mobilis into lentikats®. Lett. Appl. Microbiol. 2005, 41, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, M.; Rebroš, M.; Krištofíková, L.; Malátová, K. High temperature lactic acid production by bacillus coagulans immobilized in lentikats. Biotechnol. Lett. 2005, 27, 1943–1947. [Google Scholar] [CrossRef] [PubMed]
- Dolejš, I.; Krasňan, V.; Stloukal, R.; Rosenberg, M.; Rebroš, M. Butanol production by immobilised clostridium acetobutylicum in repeated batch, fed-batch, and continuous modes of fermentation. Bioresour. Technol. 2014, 169, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Rebroš, M.; Dolejš, I.; Stloukal, R.; Rosenberg, M. Butyric acid production with clostridium tyrobutyricum immobilised to pva gel. Process Biochem. 2015, 51, 704–708. [Google Scholar] [CrossRef]
- Cerreti, M.; Markošová, K.; Esti, M.; Rosenberg, M.; Rebroš, M. Immobilisation of pectinases into pva gel for fruit juice application. Int. J. Food Sci. Technol. 2017, 52, 531–539. [Google Scholar] [CrossRef]
- Rebroš, M.; Rosenberg, M.; Mlichová, Z.; Krištofíková, L. Hydrolysis of sucrose by invertase entrapped in polyvinyl alcohol hydrogel capsules. Food Chem. 2007, 102, 784–787. [Google Scholar] [CrossRef]
- Grosová, Z.; Rosenberg, M.; Rebroš, M.; Šipocz, M.; Sedláčková, B. Entrapment of β-galactosidase in polyvinylalcohol hydrogel. Biotechnol. Lett. 2008, 30, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Grosová, Z.; Rosenberg, M.; Gdovin, M.; Sláviková, L.; Rebroš, M. Production of d-galactose using β-galactosidase and saccharomyces cerevisiae entrapped in poly(vinylalcohol) hydrogel. Food Chem. 2009, 116, 96–100. [Google Scholar] [CrossRef]
- Krasnan, V.; Stloukal, R.; Rosenberg, M.; Rebros, M. Immobilization of cells and enzymes to lentikats(r). Appl. Microbiol. Biotechnol. 2016, 100, 2535–2553. [Google Scholar] [CrossRef] [PubMed]
- Hanson, R.L.; Goldberg, S.; Goswami, A.; Tully, T.P.; Patel, R.N. Purification and cloning of a ketoreductase used for the preparation of chiral alcohols. Adv. Synth. Catal. 2005, 347, 1073–1080. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KRED:GDH | C KRED (µg·mL−1) | C GDH (µg·mL−1) | Initial Activity (U) | Final Conversion (%) |
|---|---|---|---|---|
| 1:1.7 | 9.21 | 15.79 | 0.27 | 84 |
| 5:1.7 | 46.04 | 15.79 | 1.51 | >99 |
| 5:6.9 | 46.04 | 63.16 | 1.85 | >99 |
| Batch Nr. | KRED:GDH | C KRED (mg g−1LKs) | c GDH (mg g−1LKs) | Initial Specific Activity (U g−1LKs) | Conversion a (%) |
|---|---|---|---|---|---|
| 1 | 25:1 | 4.84 | 0.19 | 3.63 | >99 |
| 2 | 12.5:1 | 2.42 | 0.19 | 3.62 | 92 |
| 3 | 6.25:1 | 1.21 | 0.19 | 1.29 | 50 |
| 4 | 12.5:0.5 | 2.42 | 0.1 | 2.74 | 92 |
| 5 | 12.5:2 | 2.42 | 0.39 | 3.69 | 95 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrovičová, T.; Markošová, K.; Hegyi, Z.; Smonou, I.; Rosenberg, M.; Rebroš, M. Co-Immobilization of Ketoreductase and Glucose Dehydrogenase. Catalysts 2018, 8, 168. https://doi.org/10.3390/catal8040168
Petrovičová T, Markošová K, Hegyi Z, Smonou I, Rosenberg M, Rebroš M. Co-Immobilization of Ketoreductase and Glucose Dehydrogenase. Catalysts. 2018; 8(4):168. https://doi.org/10.3390/catal8040168
Chicago/Turabian StylePetrovičová, Tatiana, Kristína Markošová, Zuzana Hegyi, Ioulia Smonou, Michal Rosenberg, and Martin Rebroš. 2018. "Co-Immobilization of Ketoreductase and Glucose Dehydrogenase" Catalysts 8, no. 4: 168. https://doi.org/10.3390/catal8040168