Photo-Induced Charge Separation vs. Degradation of a BODIPY-Based Photosensitizer Assessed by TDDFT and RASPT2


Abstract
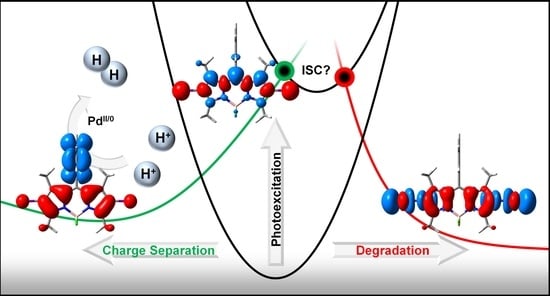
:
1. Introduction
2. Results and Discussion
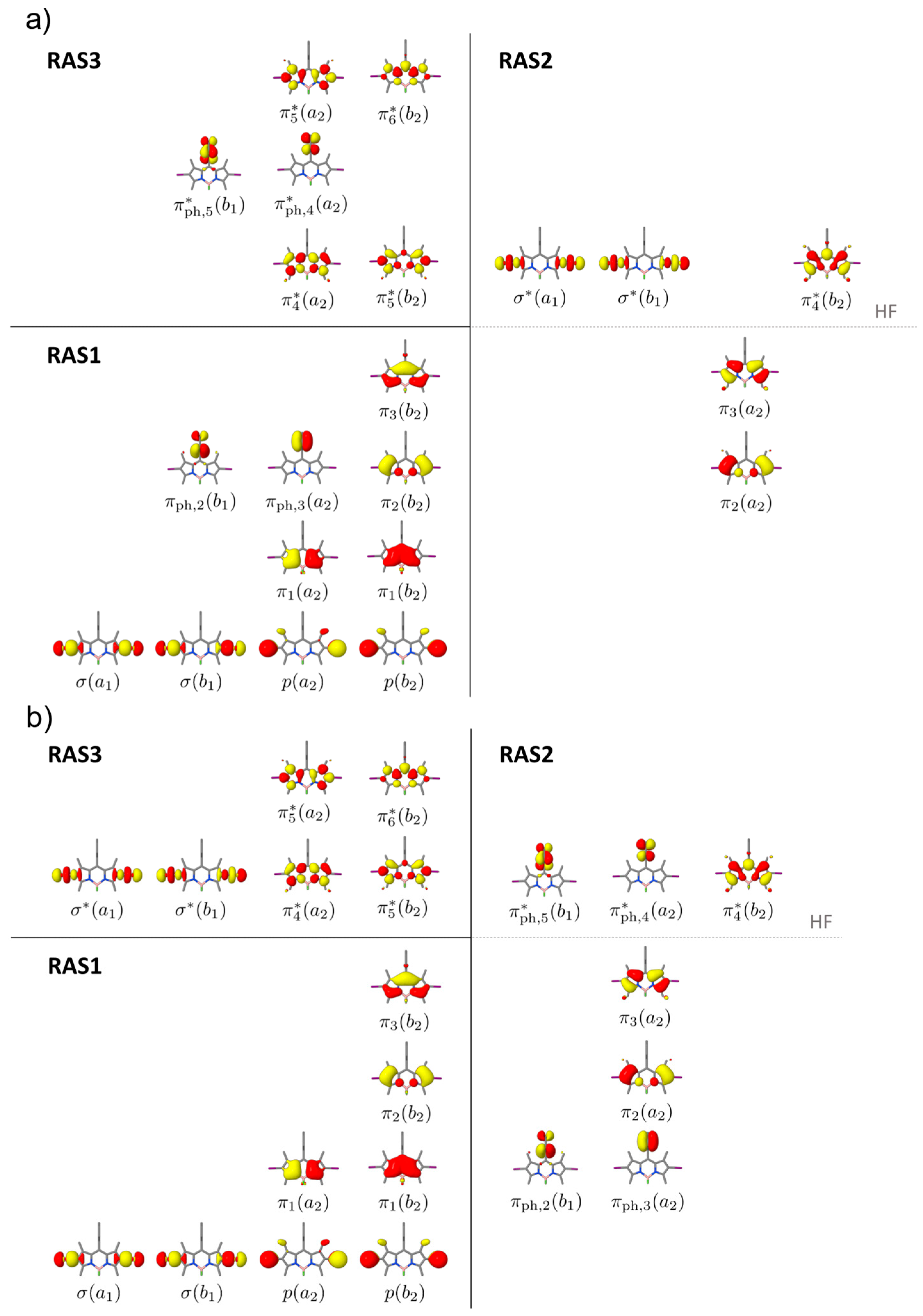
2.1. Preliminary Benchmark
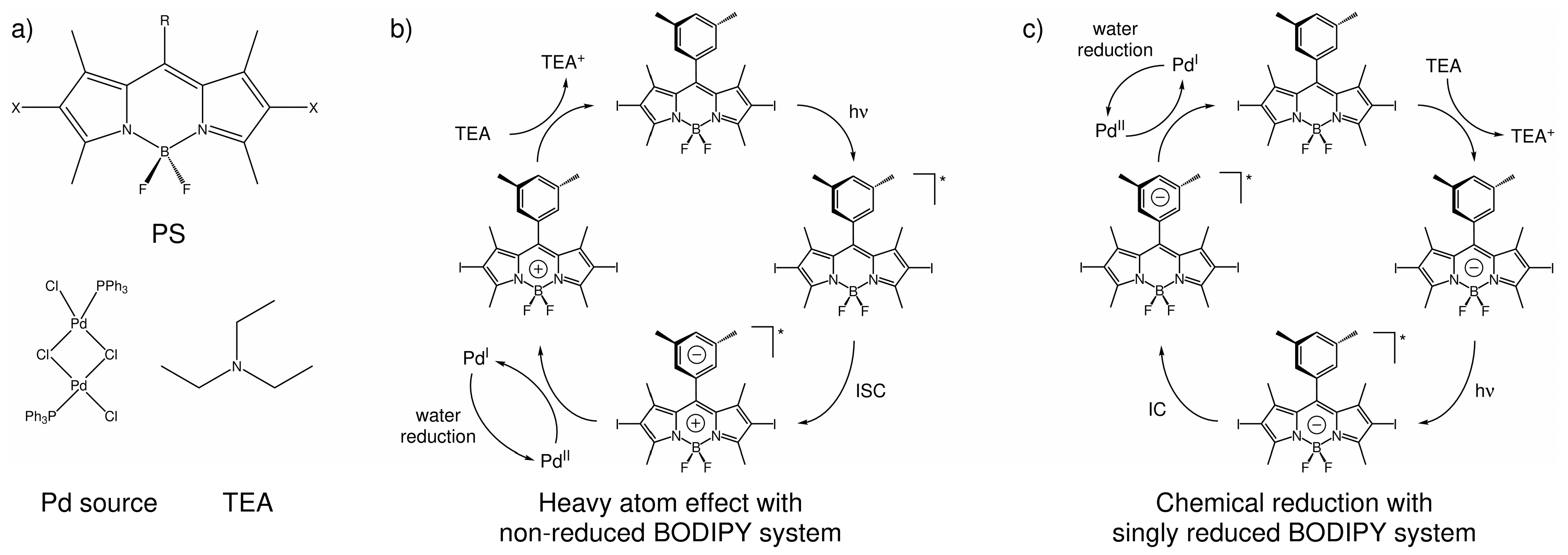
2.2. Light-Induced Charging of the PS
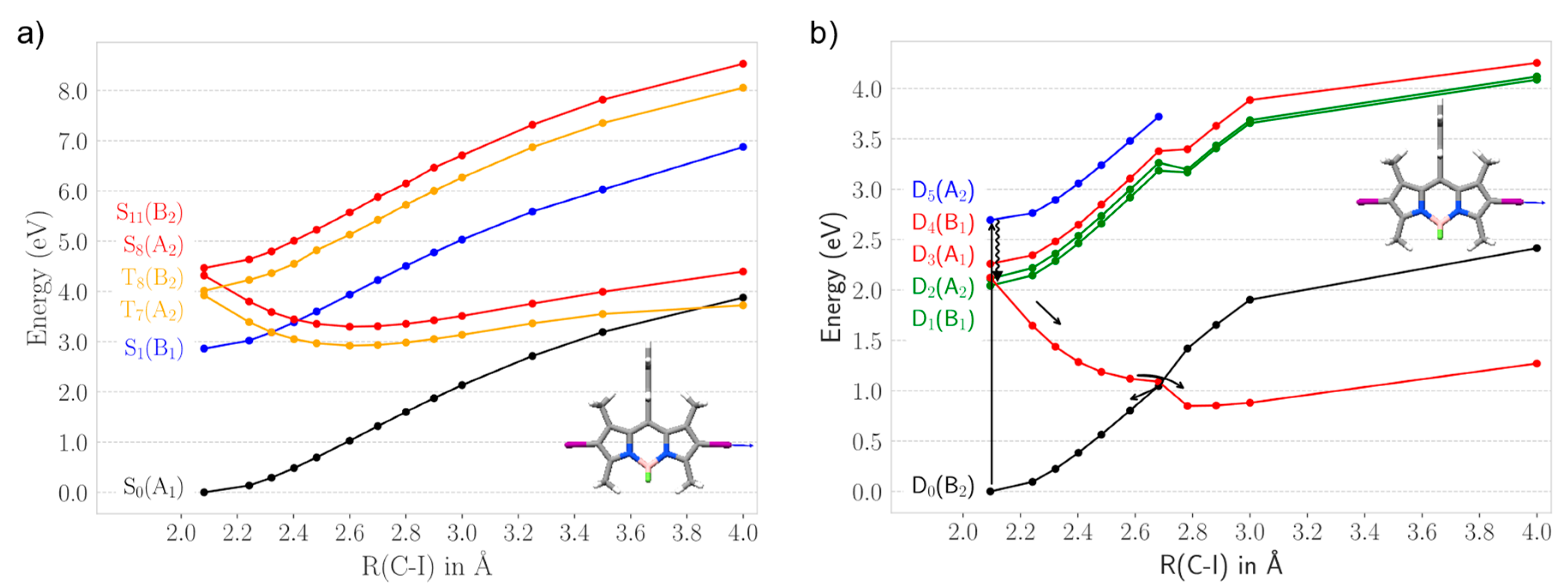
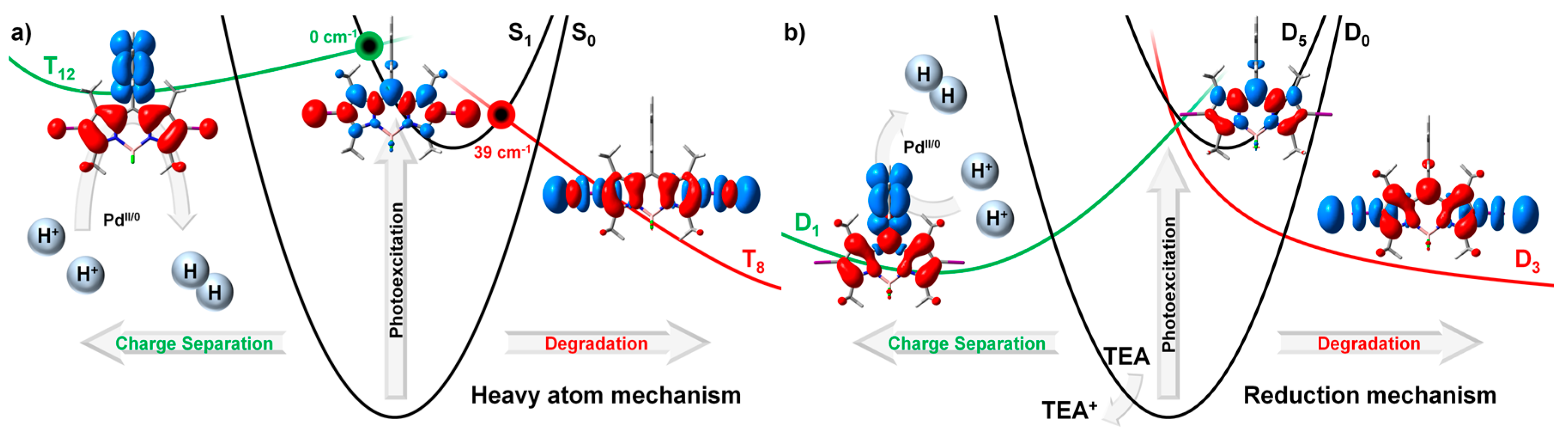
2.2.1. Heavy Atom Mechanism
2.2.2. Chemical Reduction Mechanism
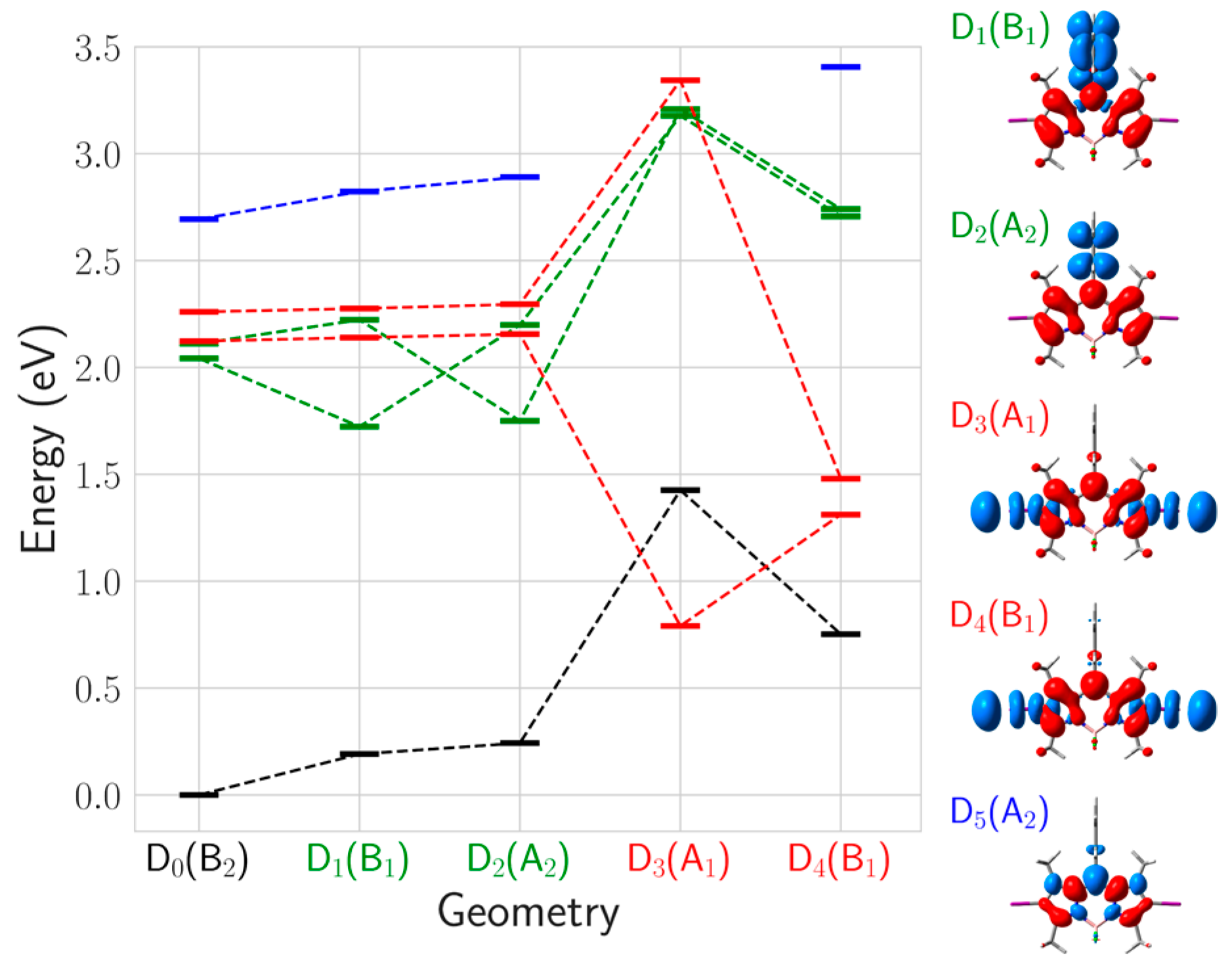
2.3. Photodegradation
3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lewis, N.S.; Nocera, D.G. Powering the planet: Chemical challenges in solar energy utilization. Proc. Natl. Acad. Sci. USA 2006, 103, 15729–15735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Sakimoto, K.K.; Hong, D.; Yang, P. Artificial photosynthesis for sustainable fuel and chemical production. Angew. Chem. Int. Ed. 2015, 54, 3259–3266. [Google Scholar] [CrossRef] [PubMed]
- Esswein, A.J.; Nocera, D.G. Hydrogen production by molecular photocatalysis. Chem. Rev. 2007, 107, 4022–4047. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.; Majumdar, A. Opportunities and challenges for a sustainable energy future. Nature 2012, 488, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.-J.; Yu, Z.-T.; Chen, D.-Q.; Zou, Z.-G. Metal-complex chromophores for solar hydrogen generation. Chem. Soc. Rev. 2017, 46, 603–631. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.-M.; Zhang, J.-H.; Hou, Y.-J.; Wang, H.-P.; Pan, M. Visible-light-driven CO2 photo-catalytic reduction of Ru(II) and Ir(III) coordination complexes. Inorg. Chem. Commun. 2016, 73, 80–89. [Google Scholar] [CrossRef]
- Barber, J. Photosynthetic energy conversion: Natural and artificial. Chem. Soc. Rev. 2009, 38, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Maeda, K.; Chen, X.; Takanabe, K.; Domen, K.; Hou, Y.; Fu, X.; Antonietti, M. Polymer semiconductors for artificial photosynthesis: Hydrogen evolution by mesoporous graphitic carbon nitride with visible light. J. Am. Chem. Soc. 2009, 131, 1680–1681. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zhang, J.; Gong, J. Tantalum-based semiconductors for solar water splitting. Chem. Soc. Rev. 2014, 43, 4395–4422. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wang, D.; Han, H.; Li, C. Roles of cocatalysts in photocatalysis and photoelectrocatalysis. Acc. Chem. Res. 2013, 46, 1900–1909. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.; Mei, Z.; Wang, T.; Kang, Q.; Ouyang, S.; Ye, J. MoS2/graphene cocatalyst for efficient photocatalytic H2 evolution under visible light irradiation. ACS Nano 2014, 8, 7078–7087. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Liu, D.; Mubeen, S.; Chuong, T.T.; Moskovits, M.; Stucky, G.D. Anisotropic growth of TiO2 onto gold nanorods for plasmon-enhanced hydrogen production from water reduction. J. Am. Chem. Soc. 2016, 138, 1114–1117. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Jin, Z.; Li, Y.; Li, S.; Lu, G. Efficient Photocatalytic Hydrogen Evolution from Water without an Electron Mediator over Pt–Rose Bengal Catalysts. J. Phys. Chem. C 2009, 113, 2630–2635. [Google Scholar] [CrossRef]
- Gong, L.; Wang, J.; Li, H.; Wang, L.; Zhao, J.; Zhu, Z. Acriflavine–cobaloxime–triethanolamine homogeneous photocatalytic system for water splitting and the multiple effects of cobaloxime and triethanolamine. Catal. Commun. 2011, 12, 1099–1103. [Google Scholar] [CrossRef]
- Zhang, W.; Hong, J.; Zheng, J.; Huang, Z.; Zhou, J.; Xu, R. Nickel–thiolate complex catalyst assembled in one step in water for solar H2 production. J. Am. Chem. Soc. 2011, 133, 20680–20683. [Google Scholar] [CrossRef] [PubMed]
- Lazarides, T.; McCormick, T.; Du, P.; Luo, G.; Lindley, B.; Eisenberg, R. Making hydrogen from water using a homogeneous system without noble metals. J. Am. Chem. Soc. 2009, 131, 9192–9194. [Google Scholar] [CrossRef] [PubMed]
- McCormick, T.M.; Calitree, B.D.; Orchard, A.; Kraut, N.D.; Bright, F.V.; Detty, M.R.; Eisenberg, R. Reductive side of water splitting in artificial photosynthesis: New homogeneous photosystems of great activity and mechanistic insight. J. Am. Chem. Soc. 2010, 132, 15480–15483. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.-Y.; Xia, L.-M.; Lennox, A.J.; Sun, Y.-Y.; Chen, H.; Jin, H.-M.; Junge, H.; Wu, Q.-A.; Jia, J.-H.; Beller, M.; et al. Structure-Activated Copper Photosensitisers for Photocatalytic Water Reduction. Chem. Eur. J. 2017, 23, 3631–3636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rau, S.; Schäfer, B.; Gleich, D.; Anders, E.; Rudolph, M.; Friedrich, M.; Görls, H.; Henry, W.; Vos, J.G. Ein supramolekularer Photokatalysator zur Erzeugung von Wasserstoff und zur selektiven Hydrierung von Tolan. Angew. Chem. 2006, 118, 6361–6364. [Google Scholar] [CrossRef]
- Kirch, M.; Lehn, J.-M.; Sauvage, J.-P. Hydrogen generation by visible light irradiation of aqueous solutions of metal complexes. An approach to the photochemical conversion and storage of solar energy. Helv. Chim. Acta 1979, 62, 1345–1384. [Google Scholar] [CrossRef]
- Du, P.; Knowles, K.; Eisenberg, R. A homogeneous system for the photogeneration of hydrogen from water based on a platinum(II) terpyridyl acetylide chromophore and a molecular cobalt catalyst. J. Am. Chem. Soc. 2008, 130, 12576–12577. [Google Scholar] [CrossRef] [PubMed]
- Metz, S.; Bernhard, S. Robust photocatalytic water reduction with cyclometalated Ir(III) 4-vinyl-2,2′-bipyridine complexes. Chem. Commun. 2010, 46, 7551–7553. [Google Scholar] [CrossRef] [PubMed]
- DiSalle, B.F.; Bernhard, S. Orchestrated photocatalytic water reduction using surface-adsorbing iridium photosensitizers. J. Am. Chem. Soc. 2011, 133, 11819–11821. [Google Scholar] [CrossRef] [PubMed]
- Whang, D.R.; Sakai, K.; Park, S.Y. Highly efficient photocatalytic water reduction with robust iridium(III) photosensitizers containing arylsilyl substituents. Angew. Chem. Int. Ed. 2013, 52, 11612–11615. [Google Scholar] [CrossRef] [PubMed]
- Mak, C.S.; Wong, H.L.; Leung, Q.Y.; Tam, W.Y.; Chan, W.K.; Djurišić, A.B. The use of sublimable chlorotricarbonyl bis(phenylimino) acenaphthene rhenium(I) complexes as photosensitizers in bulk-heterojunction photovoltaic devices. J. Organomet. Chem. 2009, 694, 2770–2776. [Google Scholar] [CrossRef] [Green Version]
- Eckenhoff, W.T.; Eisenberg, R. Molecular systems for light driven hydrogen production. Dalton Trans. 2012, 41, 13004–13021. [Google Scholar] [CrossRef] [PubMed]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Lakadamyali, F.; Kato, M.; Muresan, N.M.; Reisner, E. Selective reduction of aqueous protons to hydrogen with a synthetic cobaloxime catalyst in the presence of atmospheric oxygen. Angew. Chem. Int. Ed. 2012, 51, 9381–9384. [Google Scholar] [CrossRef] [PubMed]
- Sakai, T.; Mersch, D.; Reisner, E. Photocatalytic Hydrogen Evolution with a Hydrogenase in a Mediator-Free System under High Levels of Oxygen. Angew. Chem. Int. Ed. 2013, 52, 12313–12316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, M.; Yang, X.; Li, J.; Zhang, F.; Sun, L. Co-sensitization of Organic Dyes for Efficient Dye-Sensitized Solar Cells. ChemSusChem 2013, 6, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wang, M.; Li, C.; Li, X.; Dong, J.; Sun, L. Photochemical H2 production with noble-metal-free molecular devices comprising a porphyrin photosensitizer and a cobaloxime catalyst. Chem. Commun. 2010, 46, 8806–8808. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; McNamara, W.R.; Eum, M.-S.; Holland, P.L.; Eisenberg, R. A Nickel Thiolate Catalyst for the Long-Lived Photocatalytic Production of Hydrogen in a Noble-Metal-Free System. Angew. Chem. Int. Ed. 2012, 51, 1667–1670. [Google Scholar] [CrossRef] [PubMed]
- Hartley, C.L.; DiRisio, R.J.; Screen, M.E.; Mayer, K.J.; McNamara, W.R. Iron polypyridyl complexes for photocatalytic hydrogen generation. Inorg. Chem. 2016, 55, 8865–8870. [Google Scholar] [CrossRef] [PubMed]
- Sabatini, R.P.; McCormick, T.M.; Lazarides, T.; Wilson, K.C.; Eisenberg, R.; McCamant, D.W. Intersystem crossing in halogenated Bodipy chromophores used for solar hydrogen production. J. Phys. Chem. Lett. 2011, 2, 223–227. [Google Scholar] [CrossRef]
- Bartelmess, J.; Francis, A.J.; El Roz, K.A.; Castellano, F.N.; Weare, W.W.; Sommer, R.D. Light-driven hydrogen evolution by bodipy-sensitized cobaloxime catalysts. Inorg. Chem. 2014, 53, 4527–4534. [Google Scholar] [CrossRef] [PubMed]
- Manton, J.C.; Long, C.; Vos, J.G.; Pryce, M.T. A photo-and electrochemical investigation of BODIPY–cobaloxime complexes for hydrogen production, coupled with quantum chemical calculations. Phys. Chem. Chem. Phys. 2014, 16, 5229–5236. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.-G.; Fang, K.; Wu, J.-H.; Dai, J.-C.; Zhao, Q.-H. Noble-metal-free BODIPY–cobaloxime photocatalysts for visible-light-driven hydrogen production. Phys. Chem. Chem. Phys. 2014, 16, 23884–23894. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.-G.; Fang, K.; Wu, J.-H.; Mo, J. Photocatalytic water reduction from a noble-metal-free molecular dyad based on a thienyl-expanded BODIPY photosensitizer. Chem. Commun. 2015, 51, 12361–12364. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.-G.; Lu, H.; Zhang, X.-L.; Dai, J.-C.; Wu, J.-H.; Wu, J.-J. The relationship between the boron dipyrromethene (BODIPY) structure and the effectiveness of homogeneous and heterogeneous solar hydrogen-generating systems as well as DSSCs. Phys. Chem. Chem. Phys. 2015, 17, 9716–9729. [Google Scholar] [CrossRef] [PubMed]
- Loudet, A.; Burgess, K. BODIPY dyes and their derivatives: Syntheses and spectroscopic properties. Chem. Rev. 2007, 107, 4891–4932. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, G.; Ziessel, R.; Harriman, A. The chemistry of fluorescent bodipy dyes: Versatility unsurpassed. Angew. Chem. Int. Ed. 2008, 47, 1184–1201. [Google Scholar] [CrossRef] [PubMed]
- Momeni, M.R.; Brown, A. Why do TD-DFT excitation energies of BODIPY/aza-BODIPY families largely deviate from experiment? Answers from electron correlated and multireference methods. J. Chem. Theory Comput. 2015, 11, 2619–2632. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Zeng, W.; Huang, K.-W.; Wu, J. Benzene-fused BODIPYs: Synthesis and the impact of fusion mode. Chem. Commun. 2013, 49, 1217–1219. [Google Scholar] [CrossRef] [PubMed]
- Gresser, R.; Hartmann, H.; Wrackmeyer, M.; Leo, K.; Riede, M. Synthesis of thiophene-substituted aza-BODIPYs and their optical and electrochemical properties. Tetrahedron 2011, 67, 7148–7155. [Google Scholar] [CrossRef]
- Hayashi, Y.; Obata, N.; Tamaru, M.; Yamaguchi, S.; Matsuo, Y.; Saeki, A.; Seki, S.; Kureishi, Y.; Saito, S.; Yamaguchi, S.; et al. Facile synthesis of biphenyl-fused BODIPY and its property. Org. Lett. 2012, 14, 866–869. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.-D.; Fu, Y.; Zhang, T.; Zhao, W. Synthesis and properties of NIR aza-BODIPYs with aryl and alkynyl substituents on the boron center. Tetrahedron Lett. 2012, 53, 5703–5706. [Google Scholar] [CrossRef]
- Yu, C.; Xu, Y.; Jiao, L.; Zhou, J.; Wang, Z.; Hao, E. Isoindole-BODIPY Dyes as Red to Near-Infrared Fluorophores. Chem. Eur. J. 2012, 18, 6437–6442. [Google Scholar] [CrossRef] [PubMed]
- Berhe, S.A.; Rodriguez, M.T.; Park, E.; Nesterov, V.N.; Pan, H.; Youngblood, W.J. Optoelectronic Tuning of Organoborylazadipyrromethenes via Effective Electronegativity at the Metalloid Center. Inorg. Chem. 2014, 53, 2346–2348. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.K.; Mukherjee, S.; Thilagar, P. Going beyond red with a tri-and tetracoordinate boron conjugate: Intriguing near-IR optical properties and applications in anion sensing. Inorg. Chem. 2014, 53, 2343–2345. [Google Scholar] [CrossRef] [PubMed]
- Dura, L.; Wächtler, M.; Kupfer, S.; Kübel, J.; Ahrens, J.; Höfler, S.; Bröring, M.; Dietzek, B.; Beweries, T. Photophysics of BODIPY Dyes as Readily-Designable Photosensitisers in Light-Driven Proton Reduction. Inorganics 2017, 5, 21. [Google Scholar] [CrossRef]
- Dura, L.; Ahrens, J.; Pohl, M.-M.; Höfler, S.; Bröring, M.; Beweries, T. Design of BODIPY Dyes as Photosensitisers in Multicomponent Catalyst Systems for Light-Driven Hydrogen Production. Chem. Eur. J. 2015, 21, 13549–13552. [Google Scholar] [CrossRef] [PubMed]
- Staniszewska, M.; Kupfer, S.; Łabuda, M.; Guthmuller, J. Theoretical Assessment of Excited State Gradients and Resonance Raman Intensities for the Azobenzene Molecule. J. Chem. Theory Comput. 2017, 13, 1263–1274. [Google Scholar] [CrossRef] [PubMed]
- Caricato, M.; Lipparini, F.; Scalmani, G.; Cappelli, C.; Barone, V. Vertical Electronic Excitations in Solution with the EOM-CCSD Method Combined with a Polarizable Explicit/Implicit Solvent Model. J. Chem. Theory Comput. 2013, 9, 3035–3042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kállay, M.; Gauss, J. Calculation of excited-state properties using general coupled-cluster and configuration-interaction models. J. Chem. Phys. 2004, 121, 9257–9269. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, J.D.; Kleinschmidt, M.; Larbig, A.; Tatchen, J.; Marian, C.M. Quantum-Chemical Studies on Excitation Energy Transfer Processes in BODIPY-Based Donor–Acceptor Systems. J. Chem. Theory Comput. 2015, 11, 4316–4327. [Google Scholar] [CrossRef] [PubMed]
- Chibani, S.; Laurent, A.D.; Le Guennic, B.; Jacquemin, D. Improving the Accuracy of Excited-State Simulations of BODIPY and Aza-BODIPY Dyes with a Joint SOS-CIS(D) and TD-DFT Approach. J. Chem. Theory Comput. 2014, 10, 4574–4582. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Ge, J.; Escudero, D.; Wang, Z.; Zhao, J.; Jacquemin, D. Molecular Structure–Intersystem Crossing Relationship of Heavy-Atom-Free BODIPY Triplet Photosensitizers. J. Org. Chem. 2015, 80, 5958–5963. [Google Scholar] [CrossRef] [PubMed]
- Escudero, D.; Happ, B.; Winter, A.; Hager, M.D.; Schubert, U.S.; González, L. The Radiative Decay Rates Tune the Emissive Properties of Ruthenium(II) Polypyridyl Complexes: A Computational Study. Chem. Asian J. 2012, 7, 667–671. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.; Trucks, G.; Schlegel, H.B.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.; et al. Gaussian 09; Revision A. 02; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Aquilante, F.; Autschbach, J.; Carlson, R.K.; Chibotaru, L.F.; Delcey, M.G.; De Vico, L.; Ferré, N.; Frutos, L.M.; Gagliardi, L.; Garavelli, M.; et al. Molcas 8: New capabilities for multiconfigurational quantum chemical calculations across the periodic table. J. Comput. Chem. 2016, 37, 506–541. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Godbout, N.; Salahub, D.R.; Andzelm, J.; Wimmer, E. Optimization of Gaussian-type basis sets for local spin density functional calculations. Part I. Boron through neon, optimization technique and validation. Can. J. Chem. 1992, 70, 560–571. [Google Scholar] [CrossRef] [Green Version]
- Figgen, D.; Rauhut, G.; Dolg, M.; Stoll, H. Energy-consistent pseudopotentials for group 11 and 12 atoms: Adjustment to multi-configuration Dirac–Hartree–Fock data. Chem. Phys. 2005, 311, 227–244. [Google Scholar] [CrossRef]
- Mennucci, B.; Tomasi, J.; Cammi, R.; Cheeseman, J.; Frisch, M.; Devlin, F.; Gabriel, S.; Stephens, P. Polarizable continuum model (PCM) calculations of solvent effects on optical rotations of chiral molecules. J. Phys. Chem. A 2002, 106, 6102–6113. [Google Scholar] [CrossRef]
- Malmqvist, P.-Å.; Pierloot, K.; Shahi, A.R.M.; Cramer, C.J.; Gagliardi, L. The restricted active space followed by second-order perturbation theory method: Theory and application to the study of CuO2 and Cu2O2 systems. J. Chem. Phys. 2008, 128, 204109. [Google Scholar] [CrossRef] [PubMed]
- Li Manni, G.; Aquilante, F.; Gagliardi, L. Strong correlation treated via effective hamiltonians and perturbation theory. J. Chem. Phys. 2011, 134, 034114. [Google Scholar] [CrossRef] [PubMed]
- Malmqvist, P.-Å.; Rendell, A.; Roos, B.O. The restricted active space self-consistent-field method, implemented with a split graph unitary group approach. J. Phys. Chem. 1990, 94, 5477–5482. [Google Scholar] [CrossRef]
- Olsen, J.; Roos, B.O.; Jorgensen, P.; Jensen, H.J.A. Determinant based configuration interaction algorithms for complete and restricted configuration interaction spaces. J. Chem. Phys. 1988, 89, 2185–2192. [Google Scholar] [CrossRef]
- Pierloot, K.; Dumez, B.; Widmark, P.-O.; Roos, B.O. Density matrix averaged atomic natural orbital (ANO) basis sets for correlated molecular wave functions. Theor. Chim. Acta 1995, 90, 87–114. [Google Scholar] [CrossRef]
- Roos, B.O.; Lindh, R.; Malmqvist, P.-Å.; Veryazov, V.; Widmark, P.-O. New relativistic ANO basis sets for transition metal atoms. J. Phys. Chem. A 2005, 109, 6575–6579. [Google Scholar] [CrossRef] [PubMed]
- Aquilante, F.; Lindh, R.; Bondo Pedersen, T. Unbiased auxiliary basis sets for accurate two-electron integral approximations. J. Chem. Phys. 2007, 127, 114107. [Google Scholar] [CrossRef] [PubMed]
- Sauri, V.; Serrano-Andrés, L.; Shahi, A.R.M.; Gagliardi, L.; Vancoillie, S.; Pierloot, K. Multiconfigurational Second-Order Perturbation Theory Restricted Active Space (RASPT2) Method for Electronic Excited States: A Benchmark Study. J. Chem. Theory Comput. 2011, 7, 153–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finley, J.; Malmqvist, P.-Å.; Roos, B.O.; Serrano-Andrés, L. The multi-state CASPT2 method. Chem. Phys. Lett. 1998, 288, 299–306. [Google Scholar] [CrossRef]
- Roos, B.O.; Andersson, K. Multiconfigurational perturbation theory with level shift—The Cr2 potential revisited. Chem. Phys. Lett. 1995, 245, 215–223. [Google Scholar] [CrossRef]
- Malmqvist, P.-Å.; Roos, B.O. The CASSCF state interaction method. Chem. Phys. Lett. 1989, 155, 189–194. [Google Scholar] [CrossRef]
- Malmqvist, P.-Å.; Roos, B.O.; Schimmelpfennig, B. The restricted active space (RAS) state interaction approach with spin–orbit coupling. Chem. Phys. Lett. 2002, 357, 230–240. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 110 (Non-Reduced Singlet Structure) | |||||||||
| Transition | TDDFT | RASPT2 | |||||||
| State | E/eV | f | State | E/eV | f | RAS | |||
| π3(a2) | → | π4*(b2) (IL) | S1 (B1) | 2.88 | 0.518 | S1 (B1) | 2.71 | 0.938 | Diss |
| π2(a2) | → | π4*(b2) (IL) | S3 (B1) | 3.45 | 0.311 | S2 (B1) | 3.65 | 0.094 | Diss |
| π3(a2) | → | σ*(a1) (Diss) | S8 (A2) | 4.41 | 0.000 | S3 (A2) | 5.06 | 0.000 | Diss |
| π3(a2) | → | σ*(b1) (Diss) | S11 (B2) | 4.55 | 0.000 | S5 (B2) | 5.42 | 0.001 | Diss |
| π3(a2) | → | π4*(b2) (IL) | T1 (B1) | 1.55 | - | T1 (B1) | 1.88 | - | Diss |
| π2(a2) | → | π4*(b2) (IL) | T2 (B1) | 2.58 | - | T2 (B1) | 3.04 | - | Diss |
| πph,2(b1) | → | π4*(b2) (CT) | T6 (A2) | 3.90 | - | T7 (A2) | 4.93 | - | CT |
| π3(a2) | → | σ*(a1) (Diss) | T7 (A2) | 4.02 | - | T5 (A2) | 4.83 | - | Diss |
| π3(a2) | → | σ*(b1) (Diss) | T8 (B2) | 4.11 | - | T6 (B2) | 5.09 | - | Diss |
| πph,3(a2) | → | π4*(b2) (CT) | T11 (B1) | 4.29 | - | T8 (B1) | 5.09 | - | CT |
| π3(a2) | → | πph,5*(b1) (CT) | T12 (B2) | 4.39 | - | T10 (B2) | 5.51 | - | CT |
| π3(a2) | → | πph,4*(a2) (CT) | T13 (A1) | 4.46 | - | T11 (A1) | 5.52 | - | CT |
| 21−1 (Singly Reduced Doublet Structure) | |||||||||
| Transition | TDDFT | RASPT2 | |||||||
| State | E/eV | f | State | E/eV | f | RAS | |||
| π4*(b2) | → | πph,5*(b1) (CT) | D1 (B1) | 2.04 | 0.000 | D1 (B1) | 2.11 | 0.000 | CT |
| π4*(b2) | → | πph,4*(a2) (CT) | D2 (A2) | 2.11 | 0.000 | D5 (A2) | 3.54 | 0.150 | CT |
| π4*(b2) | → | σ*(a1) (Diss) | D3 (A1) | 2.12 | 0.000 | D1 (A1) | 2.58 | 0.000 | Diss |
| π4*(b2) | → | σ*(b1) (Diss) | D4 (B1) | 2.26 | 0.000 | D3 (B1) | 2.83 | 0.000 | Diss |
| π3(a2) | → | π4*(b2) (IL) | D5 (A2) | 2.69 | 0.194 | D2 (A2) | 2.49 | 0.183 | CT |
| π2(a2) | → | π4*(b2) (IL) | D6 (A2) | 3.38 | 0.003 | D4 (A2) | 3.35 | 0.010 | CT |
| π3(a2) | → | σ*(a1) (Diss) | Q1 (B1) | 3.55 | - | Q1 (B1) | 4.26 | - | Diss |
| π3(a2) | → | σ*(b1) (Diss) | Q3 (A1) | 3.64 | - | Q2 (A1) | 4.43 | - | Diss |
| π3(a2) | → | πph,5*(b1) (CT) | - | - | - | Q3 (A1) | 4.72 | - | CT |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ziems, K.M.; Gräfe, S.; Kupfer, S. Photo-Induced Charge Separation vs. Degradation of a BODIPY-Based Photosensitizer Assessed by TDDFT and RASPT2. Catalysts 2018, 8, 520. https://doi.org/10.3390/catal8110520
Ziems KM, Gräfe S, Kupfer S. Photo-Induced Charge Separation vs. Degradation of a BODIPY-Based Photosensitizer Assessed by TDDFT and RASPT2. Catalysts. 2018; 8(11):520. https://doi.org/10.3390/catal8110520
Chicago/Turabian StyleZiems, Karl Michael, Stefanie Gräfe, and Stephan Kupfer. 2018. "Photo-Induced Charge Separation vs. Degradation of a BODIPY-Based Photosensitizer Assessed by TDDFT and RASPT2" Catalysts 8, no. 11: 520. https://doi.org/10.3390/catal8110520