Recent Progress in the Transition Metal Catalyzed Synthesis of Indoles


Abstract
: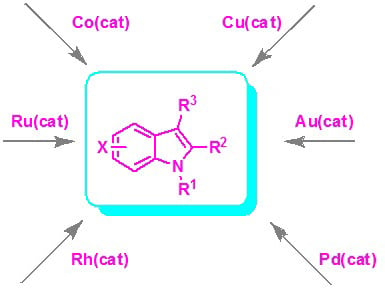
1. Introduction
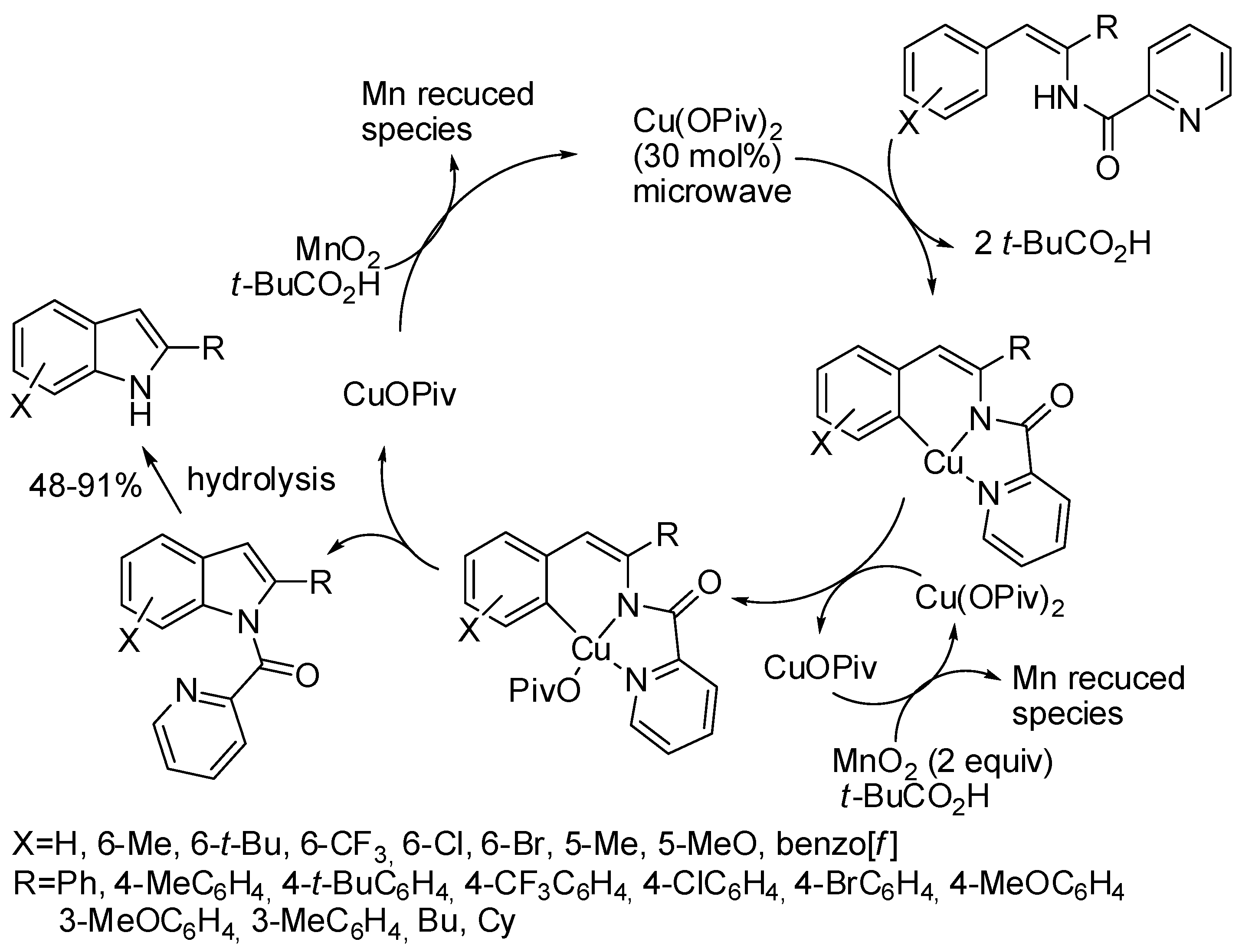
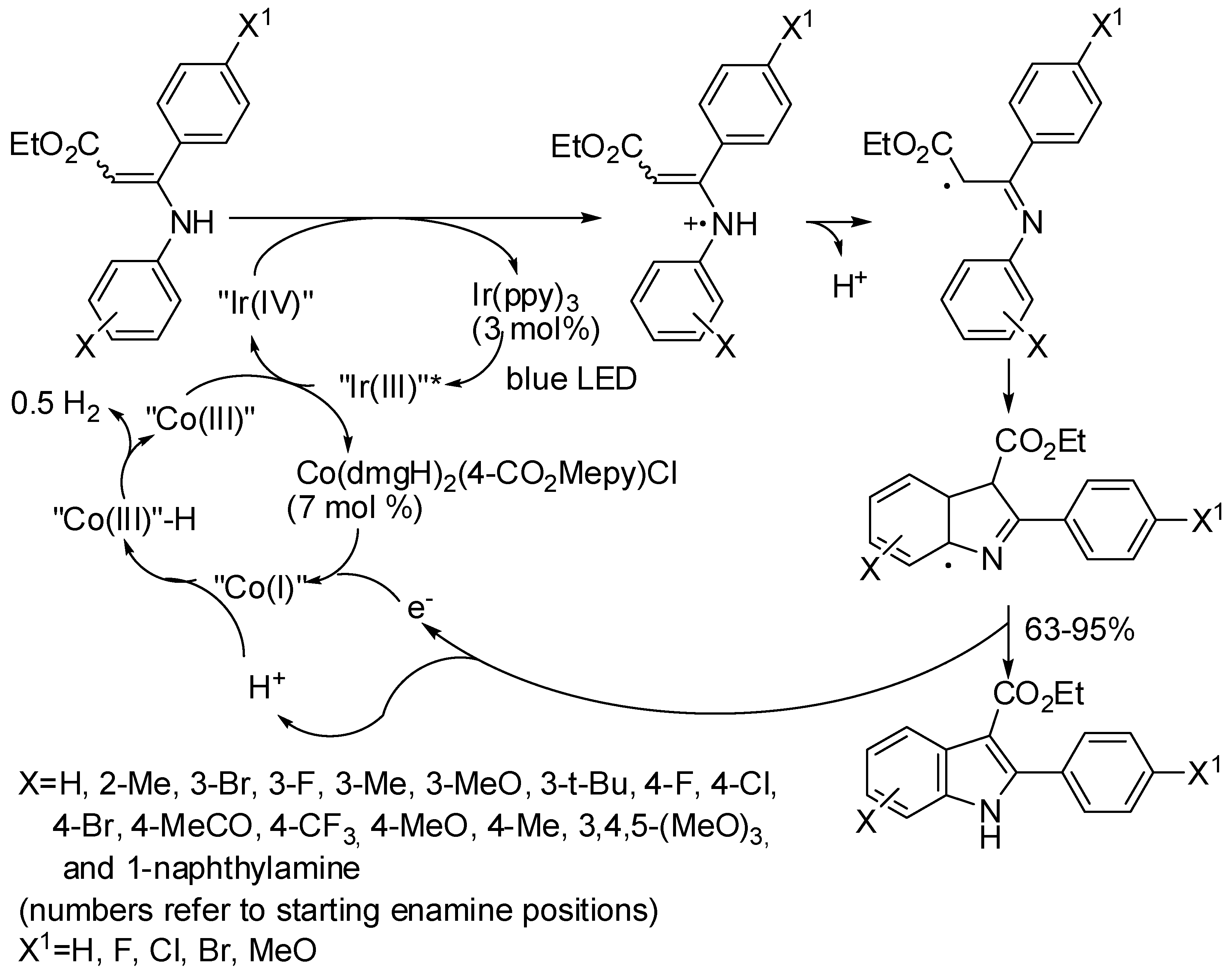
2. Cobalt
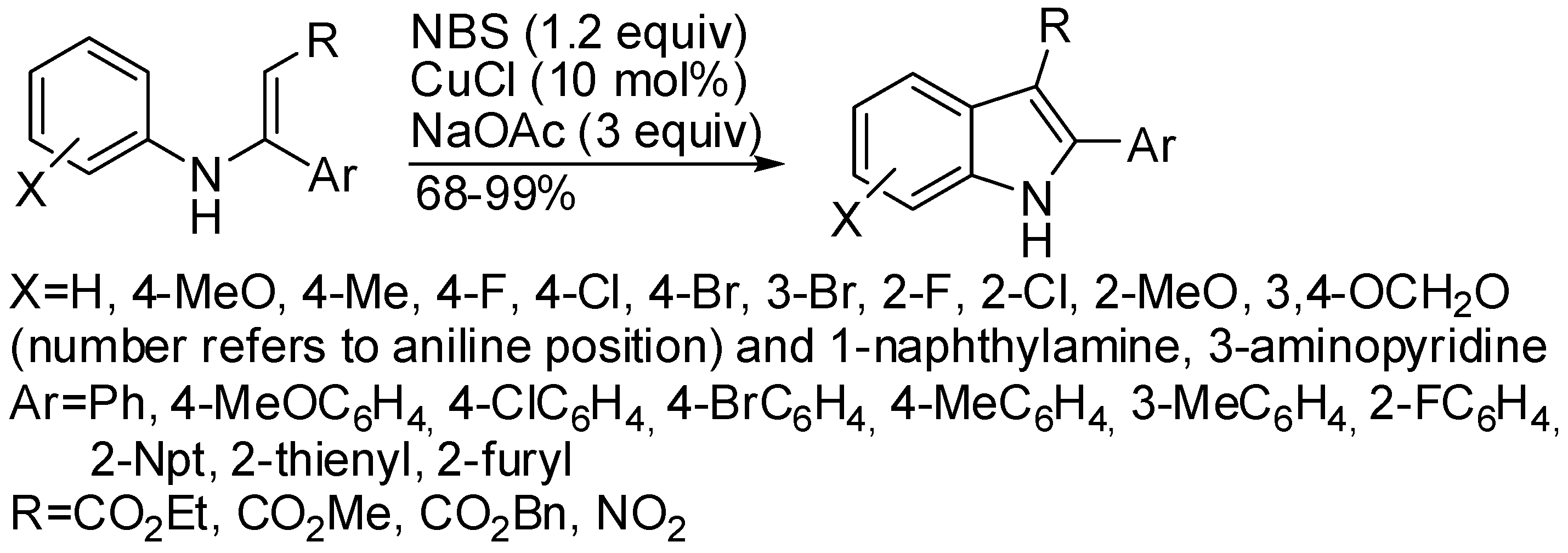
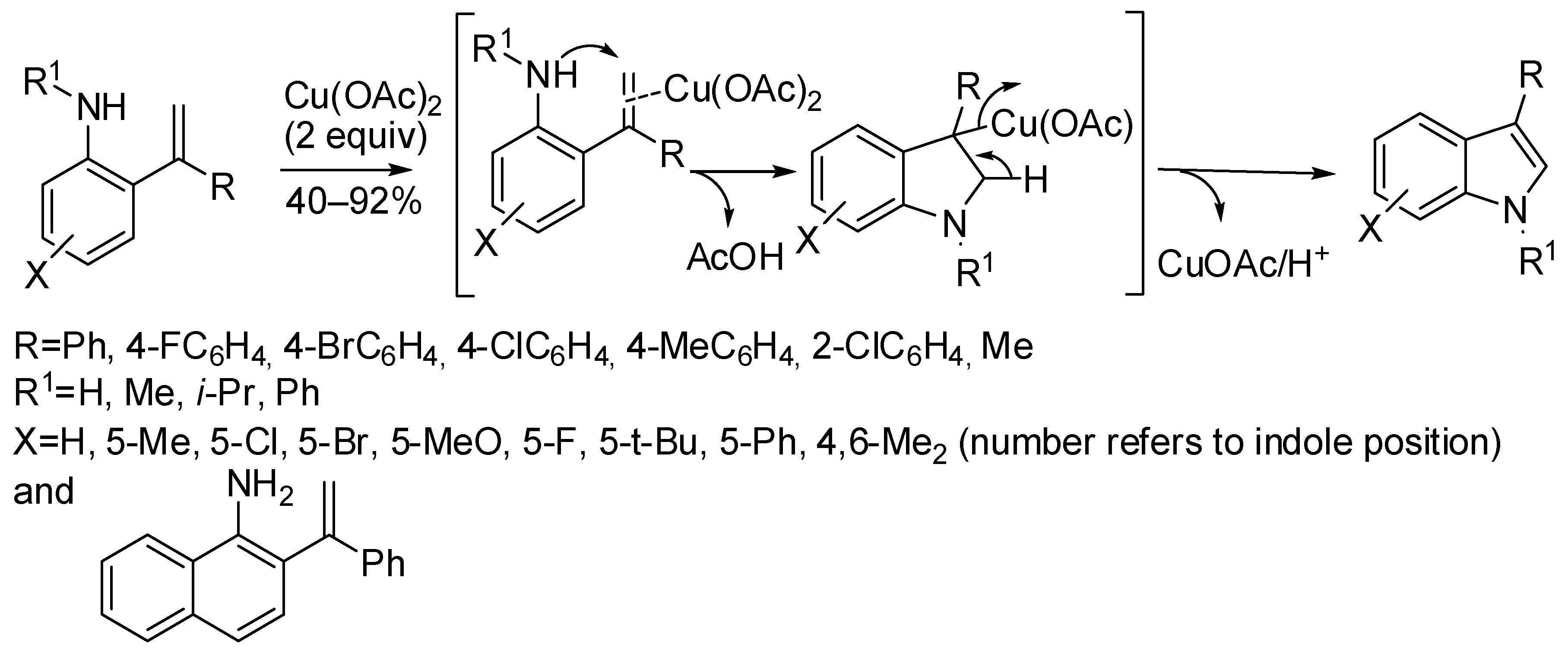
3. Copper
- in order to ensure the correct and exclusive regiochemistry in the deprotonation step, at least one C=C double bond has to be in conjugation to the α-center of the α-aminonitrile;
- bromoarenes are unsuitable for this reaction, unlike palladium used as catalyst (see later in the text);
- 1,10-phenanthroline and 1,2-diaminocyclohexane are not effective as complexing agents of copper.
- N-(3-bromophenyl)-β-enamino ester led to a mixture of 6-bromo- and 4-bromoindole in 29% and 61% yield, respectively, but N-(3-pyridinyl) or 3,4-(methylenedioxyphenyl)-β-enamino ester afforded exclusively the 4-aza- and the 5,6-(methylenedioxy)indole;
- 3-(2-furyl)-3-(4-methoxyphenylamino)acrylate gave the expected indole in 96% yield, but 3-(2-furyl)-3-(phenylamino)acrylate was unreactive;
- reduction of 3-nitroindoles with Zn/TMSCl afforded 3-aminoindoles, while reduction with H2/Pd/C in ethanol gave 3-(N,N-diethylamino)indoles in moderate to high yields.
4. Gold
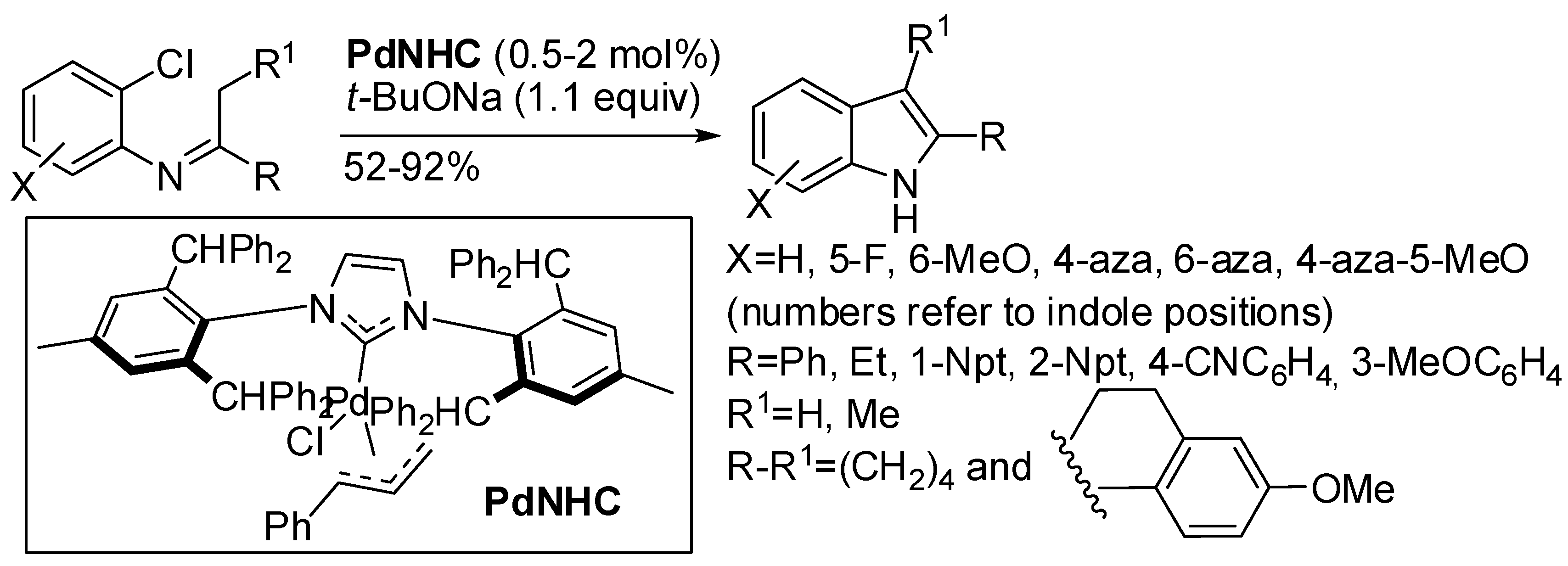
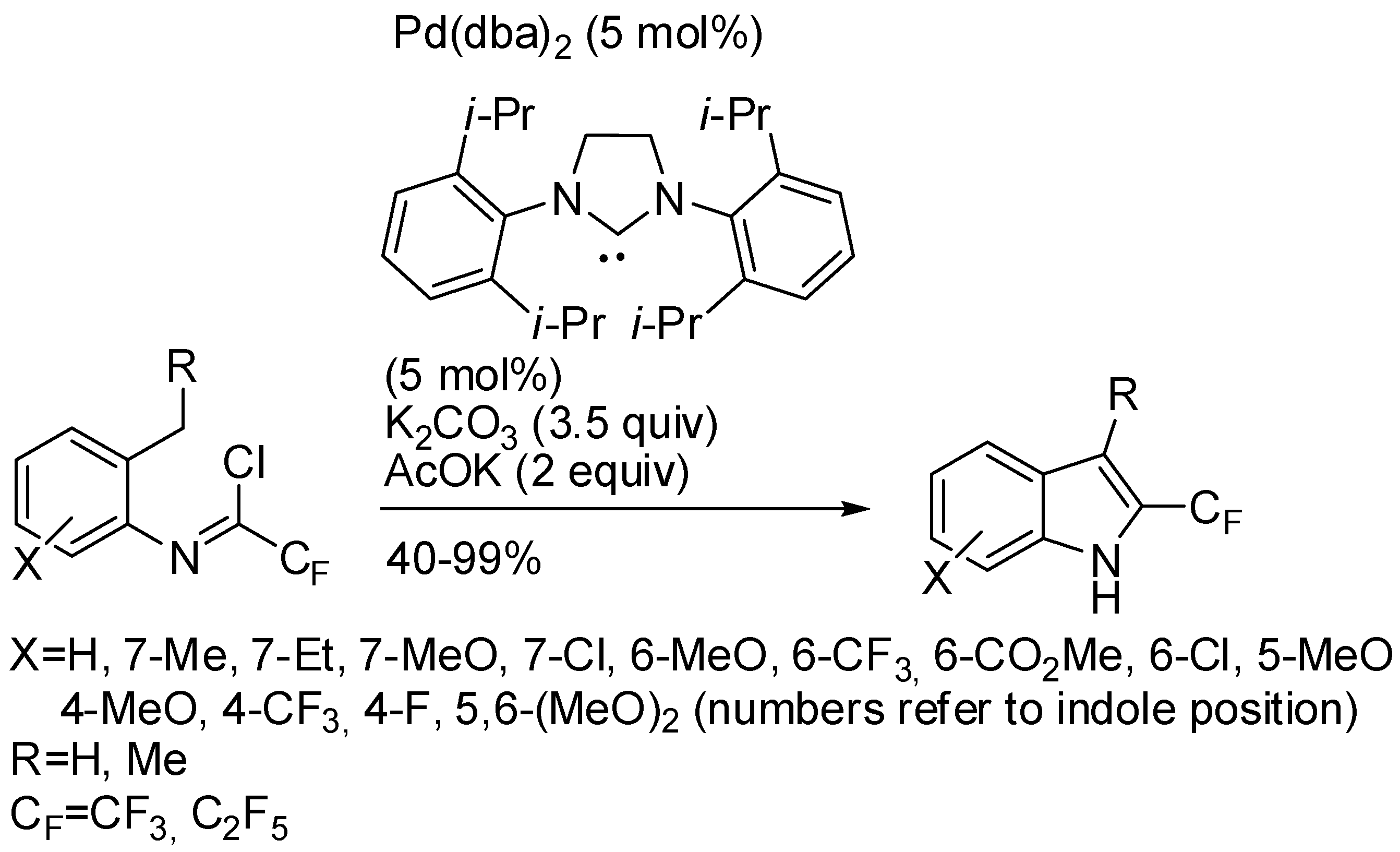
5. Palladium
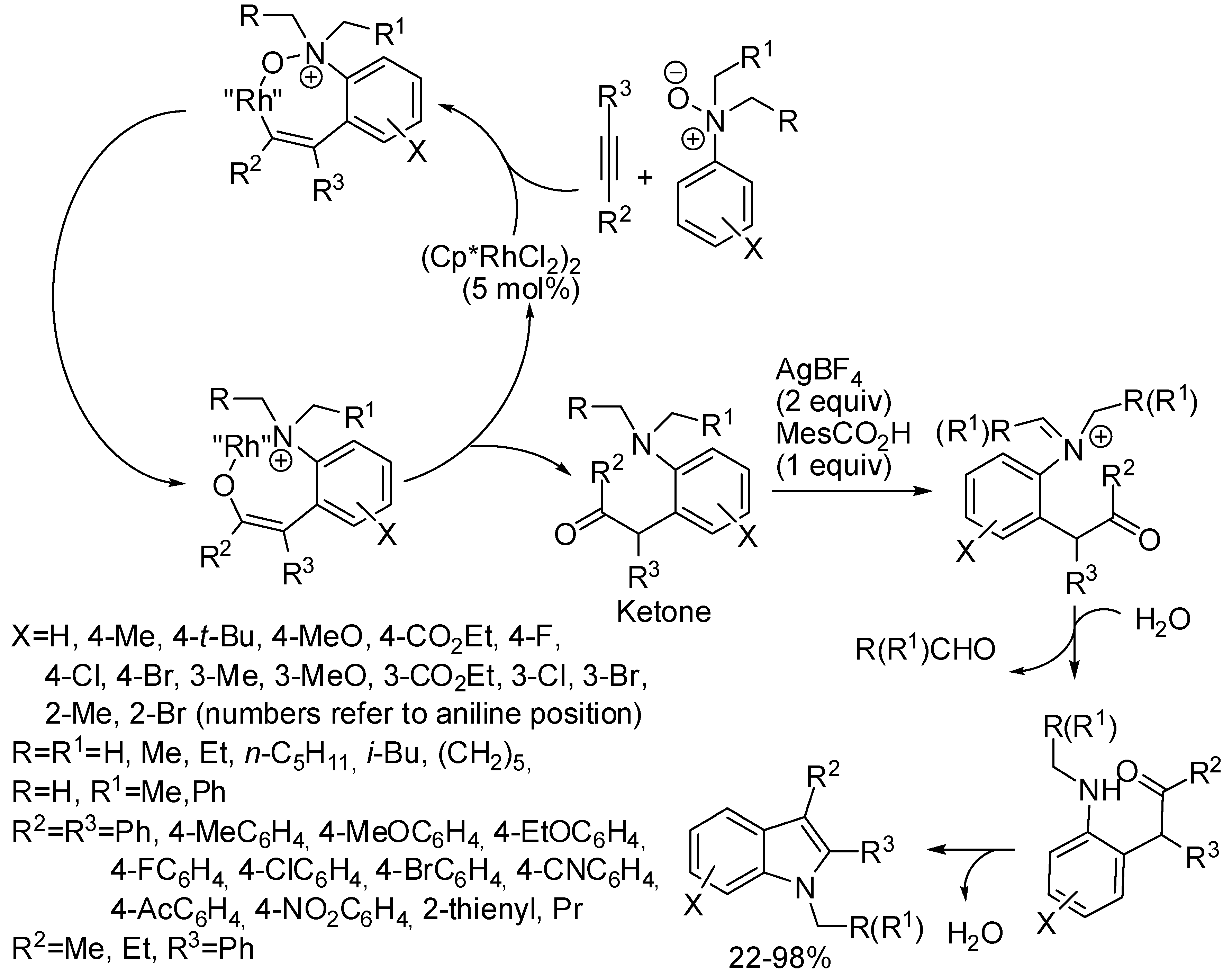
6. Rhodium
- N-oxides with two different alkyl groups were not selectively cleaved (N-ethyl vs. N-methyl 1.6:1 and N-benzyl vs. N-methyl 4.6:1);
- cyclization on meta-substituted aniline N-oxides always occurred at the less hindered position (6-substituted indoles) except for m-OMe derivatives which gave a nearly 1:1 mixture of 4- and 6-isomers;
- ortho-bromoaniline N-oxide gave partial debromination (33% and 5% yields of 7-bromo e 7-unsubstituted indole respectively);
- ortho methylaniline N-oxide gave 49% and 24% yields of indole and uncyclized ketone, respectively;
- from aryl alkyl-disubstituted alkynes the expected isomer with the aryl group in the 2-position was exclusively recovered;
- octyne gave indole with difficulty (22% yield, together with 40% of ketone);
- N-oxides of naphthylamines, strong electron-withdrawing 4-substituted anilines, N-methyldiphenylamine, and 1-ethyl-1,2,3,4-tetrahydroquinoline did not react;
- a gram scale reaction was performed recovering 1.28 g of product (85% yields).
- Both reactions gave only 6-substituted indoles from meta-substituted anilines and Wang [97] showed that 2,5-substituted substrates were unreactive, thus demonstrating that regioselectivity was driven by steric factors.
- Both research groups performed also some experiments to support the mechanism and envisaged the same mechanism as depicted in Scheme 52.
- On the other hand, Wang was able to selectively remove the C-3 ester group in 82% yield, and then the pyrimidyl group in 80% (64% overall) [97].
- α-Diazomalonates did not cyclize under both conditions, but Lin and Yao [96] recovered alkylated anilines from dimethyl 2-diazomalonic esters, methyl 2-diazo-2-(phenylsulfonyl)acetate and ethyl 2-diazo-2-(diethoxyphosphoryl)acetate in 30–47% yields.
- It should be noted that Lin and Yao reported that the diazo derivative of Meldrum’s acid was completely unreactive [96], while Wang obtained indole from 2-diazo-5,5-dimethylcyclohexane-1,3-dione in 67% yield [97], thus demonstrating that is the second ester group which prevent the cyclization to indole.
- Lin and Yao [96] obtained 1.16 g (82% yields) by scaling up their reaction.
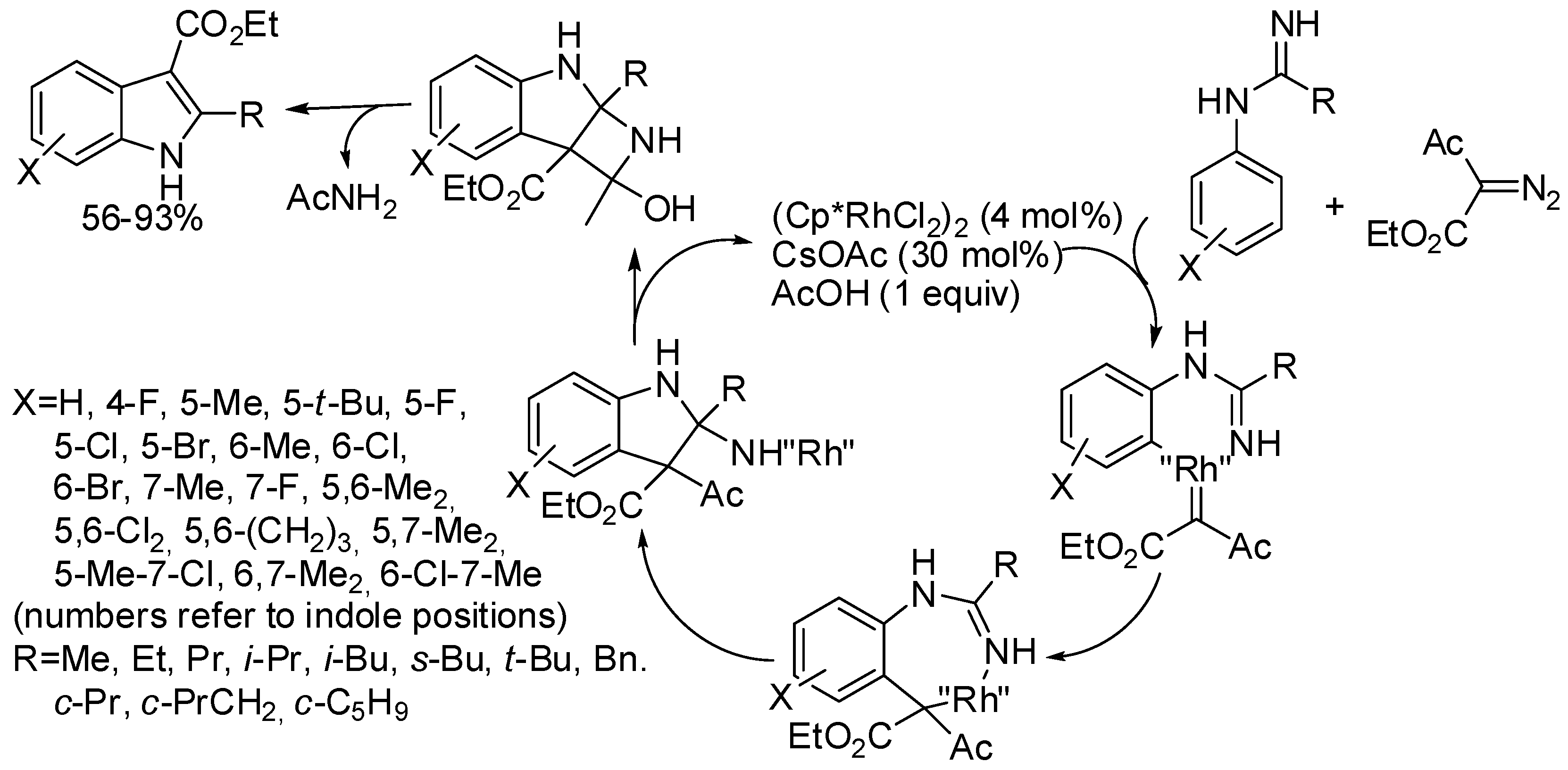
- Lin and Yao [96] further utilized the pyrimidyl group as directing group for C-7 functionalization and obtained many 7-substituted indoles in 44–87% yields.
- Lin and Yao [96] successfully performed the reaction of α-diazo-β-aldehyde esters.
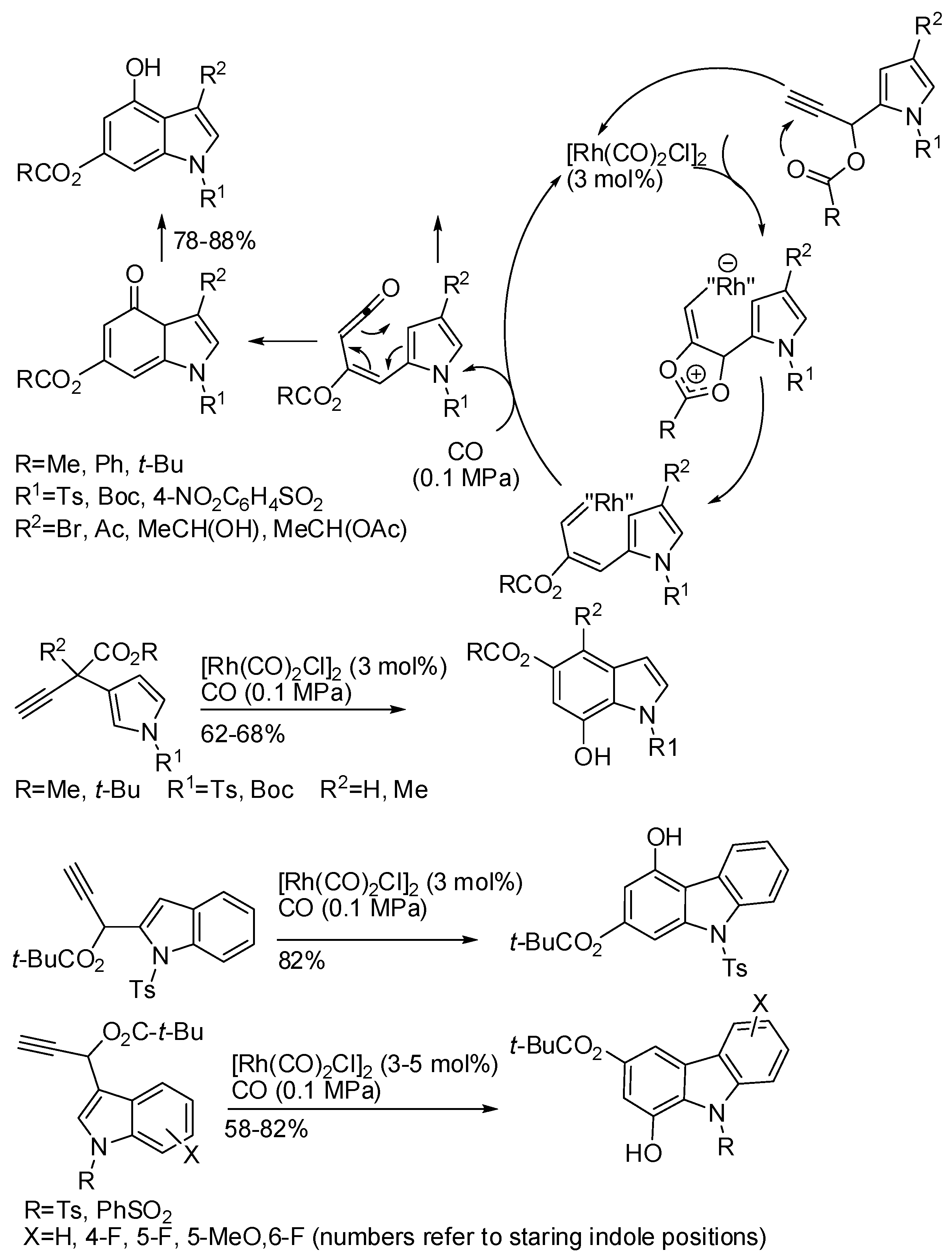
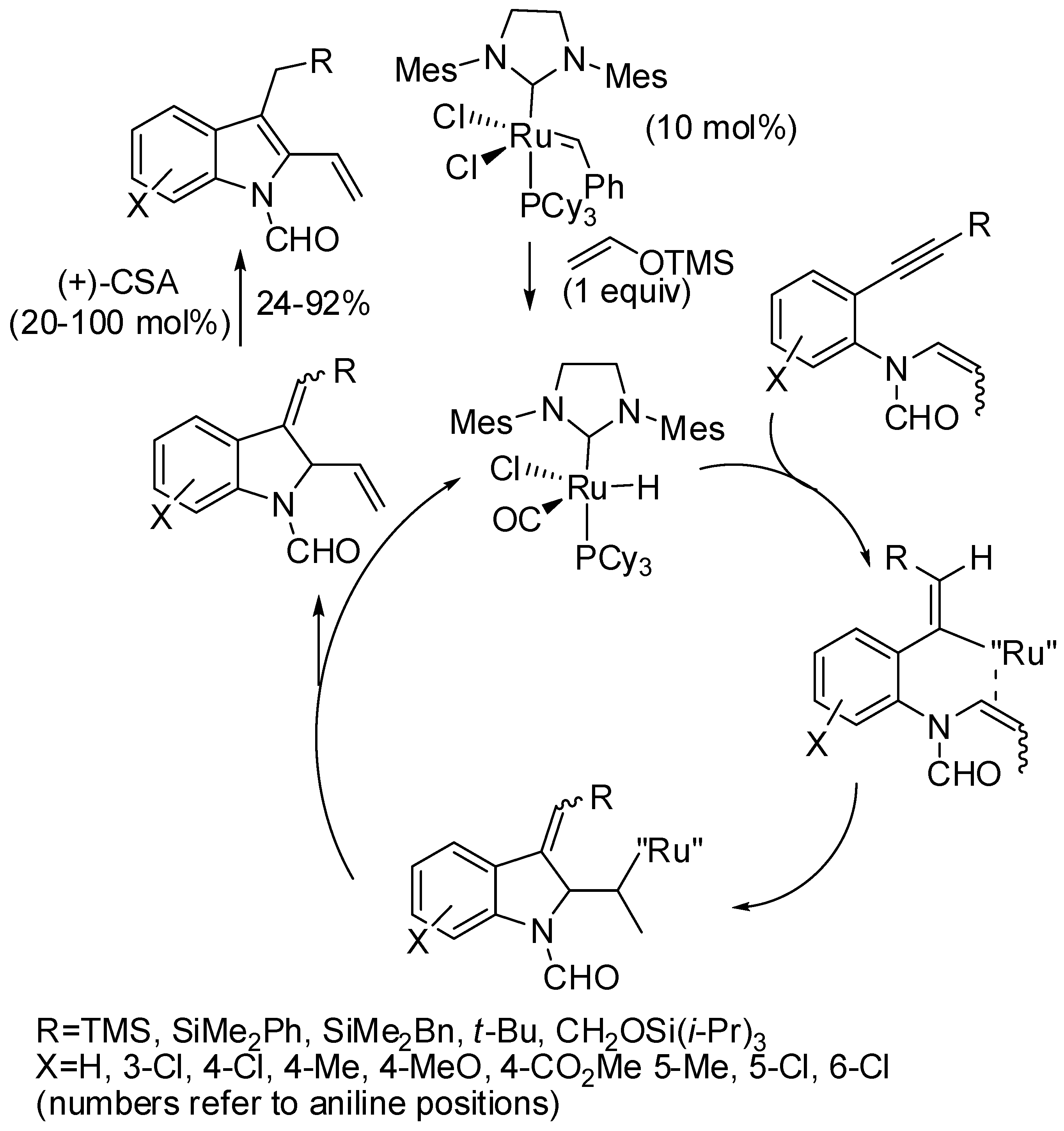
7. Ruthenium
- meta-halogeno substituted imidamides exclusively led to 6-substituted indoles except 3-fluoro derivative which exclusively afforded 4-fluoroindole and once more no explanation was given;
- methyl, benzyl, tert-butyl 2-diazo-3-oxobutanoate and α-diazoacetylacetone also gave indoles in 46–83% yields;
- the proposed mechanism was the same. However, under ruthenium catalysis, some benzimidamides were allowed to react and the C–H activation occurred exclusively at the N-aryl ring.
8. Miscellaneous
9. Conclusions
Funding
Conflicts of Interest
Abbreviations
| acac | Acetylacetonate |
| ACVA | cis-4,4′-azobis(4-cyanovaleric acid) |
| BHT | di-tert-Butylhydroxytoluene |
| Bn | Benzyl (phenylmethyl) |
| Boc | tert-Butoxycarbonyl |
| COD | 1,5-Cyclooctadiene |
| Cp | Cyclopentadienyl anion |
| Cp* | Pentamethylcyclopentadienyl anion |
| Cy | Cyclohexyl |
| dba | Dibenzylideneacetone |
| DBU | 1,5-diazabiciclo(5.4.0)undec-7-ene |
| DMPS | Dimethylphenylsilyl |
| dppe | 1,2-Bis(diphenylphosphino)ethane |
| dppf | 1,1′-Bis(diphenylphosphino)ferrocene |
| dppp | 1,3-Bis(diphenylphosphino)propane |
| dr | Diastereomeric ratio |
| H8-BINAP | 2,2′-Bis(diphenylphospino)-5,5′,6,6′,7,7′,8,8′-octahydro-1,1′-binaphthyl |
| Hex | Hexyl |
| hfacac | Hexafluoroacetylacetonate |
| HMDS | Hexamethyldisylazide |
| JohnPhos | (2-Biphenyl)di-tert-butylphosphine |
| Mes | 2,4,6-Trimethylphenyl (mesityl) |
| Ms | Methanesulfonyl (mesyl) |
| MS | Molecular sieves |
| MW | Microwave |
| NHC | N-heterocyclic carbene |
| Npt | Naphthyl |
| Oct | Octyl |
| Piv | Pivaloyl (2,2-dimethylpropanoyl) |
| PMP | 4-methoxyphenyl |
| ppy | Tris[2-phenylpyridinato-C2,N] |
| PS | Polystyrene nanoparticles |
| SET | Single electron transfer |
| TBAB | Tetrabutylammonium bromide |
| TBDPS | tert-Butyldiphenylsilyl |
| TBS | tert-Butyldimethylsilyl |
| TES | Triethylsilyl |
| Tf | trifluoromethanesulfonyl |
| TIPS | Triisopropylsilyl |
| TMS | Trimethylsilyl |
| Ts | 4-Methylbenzenesulfonyl (tosyl) |
| t-BuXPhos | 2-Di-tert-butylphosphino-2′,4′,6′-triisopropylbiphenyl |
| XPhos | 2-Dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl |
References
- Baeyer, A.; Emmerling, A. Synthese des Indols. Chem. Ber. 1869, 2, 679–682. [Google Scholar] [CrossRef]
- Gribble, G.W. (Ed.) Heterocyclic Scaffolds II: Reactions and Applications of Indoles. In Topics in Heterocyclic Chemistry; Springer: Berlin/Heidelberg, Germany, 2010; Volume 26, ISBN 978-3-642-15733-2. [Google Scholar]
- Gribble, G.W. Indole Ring Synthesis: From Natural Products to Drug Discovery; John Wiley & Sons: Chichester, UK, 2016; ISBN 978-0-470-51218-0. [Google Scholar]
- Leitch, J.A.; Bhonoah, Y.; Frost, C.G. Beyond C2 and C3: Transition-Metal-Catalyzed C–H Functionalization of Indole. ACS Catal. 2017, 7, 5618–5627. [Google Scholar] [CrossRef]
- Agasti, S.; Dey, A.; Maiti, D. Palladium-catalyzed benzofuran and indole synthesis by multiple C–H functionalizations. Chem. Commun. 2017, 53, 6544–6556. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Li, J.; Liu, Y.; Wu, C.; Wang, J.; Lan, J.; Liu, K.-Y. Progress in the Synthesis of 2,3-Disubstituted Indole by Cyclization. Chin. J. Org. Chem. 2016, 36, 2619–2633. [Google Scholar] [CrossRef]
- Guo, T.; Huang, F.; Yu, L.; Yu, Z. Indole synthesis through transition metal-catalyzed C–H activation. Tetrahedron Lett. 2015, 56, 296–302. [Google Scholar] [CrossRef]
- Inman, M.; Moody, C.J. Indole synthesis—Something old, something new. Chem. Sci. 2013, 4, 29–41. [Google Scholar] [CrossRef]
- Yoshikai, N.; Wei, Y. Synthesis of Pyrroles, Indoles, and Carbazoles through Transition-Metal-Catalyzed C–H Functionalization. Asian J. Org. Chem. 2013, 2, 466–478. [Google Scholar] [CrossRef]
- Shi, Z.; Glorius, F. Efficient and versatile synthesis of indoles from enamines and imines by cross-dehydrogenative coupling. Angew. Chem. Int. Ed. 2012, 51, 9220–9222. [Google Scholar] [CrossRef] [PubMed]
- Platon, M.; Amardeil, R.; Djakovitch, L.; Hierso, J.-C. Progress in palladium-based catalytic systems for the sustainable synthesis of annulated heterocycles: A focus on indole backbones. Chem. Soc. Rev. 2012, 41, 3929–3968. [Google Scholar] [CrossRef] [PubMed]
- Taber, D.F.; Tirunahari, P.K. Indole synthesis: A review and proposed classification. Tetrahedron 2011, 67, 7195–7210. [Google Scholar] [CrossRef] [PubMed]
- Cacchi, S.; Fabrizi, G. Update 1 of: Synthesis and Functionalization of Indoles through Palladium-Catalyzed Reactions. Chem. Rev. 2011, 111, PR215–PR283. [Google Scholar] [CrossRef] [PubMed]
- Youn, S.W.; Ko, T.Y. Metal-Catalyzed Synthesis of Substituted Indoles. Asian J. Org. Chem. 2018, 7, 1467–1487. [Google Scholar] [CrossRef]
- Satoh, T.; Kawamura, Y.; Miura, M.; Nomura, M. Palladium-Catalyzed Regioselective Mono- and Diarylation Reactions of 2-Phenylphenols and Naphthols with Aryl Halides. Angew. Chem. Int. Ed. Engl. 1997, 36, 1740–1742. [Google Scholar] [CrossRef]
- Palucki, M.; Buchwald, S.L. Palladium-Catalyzed α-Arylation of Ketones. J. Am. Chem. Soc. 1997, 119, 11108–11109. [Google Scholar] [CrossRef]
- Hamann, B.C.; Hartwig, J.F. Palladium-Catalyzed Direct α-Arylation of Ketones. Rate Acceleration by Sterically Hindered Chelating Ligands and Reductive Elimination from a Transition Metal Enolate Complex. J. Am. Chem. Soc. 1997, 119, 12382–12383. [Google Scholar] [CrossRef]
- Liu, B.; Song, C.; Sun, C.; Zhou, S.; Zhu, J. Rhodium(III)-Catalyzed Indole Synthesis Using N–N Bond as an Internal Oxidant. J. Am. Chem. Soc. 2013, 135, 16625–16631. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Moselage, M.; Gonzàlez, M.J.; Ackermann, L. Selective Synthesis of Indoles by Cobalt(III)-Catalyzed C–H/N–O Functionalization with Nitrones. ACS Catal. 2016, 6, 2705–2709. [Google Scholar] [CrossRef]
- Zhou, Z.; Liu, G.; Chen, Y.; Lu, X. Rhodium(III)-Catalyzed Redox-Neutral C–H Annulation of Arylnitrones and Alkynes for the Synthesis of Indole Derivatives. Adv. Synth. Catal. 2015, 357, 2944–2950. [Google Scholar] [CrossRef]
- Dateer, R.B.; Chang, S. Selective Cyclization of Arylnitrones to Indolines under External Oxidant-Free Conditions: Dual Role of Rh(III) Catalyst in the C–H Activation and Oxygen Atom Transfer. J. Am. Chem. Soc. 2015, 137, 4908–4911. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Wang, H.; Li, X.; Xin, X.; Wang, C.; Wan, B. Rhodium-Catalyzed C–H Annulation of Nitrones with Alkynes: A Regiospecific Route to Unsymmetrical 2,3-Diaryl-Substituted Indole. Angew. Chem. Int. Ed. 2015, 54, 10613–10617. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Vàsquez-Céspedes, S.; Gensch, T.; Glorius, F. Control over Organometallic Intermediate Enables Cp*Co(III) Catalyzed Switchable Cyclization to Quinolines and Indoles. ACS Catal. 2016, 6, 2352–2356. [Google Scholar] [CrossRef]
- Zhang, Z.-Z.; Liu, B.; Xu, J.-W.; Yan, S.-Y.; Shi, B.-F. Indole Synthesis via Cobalt(III)-Catalyzed Oxidative Coupling of N-Arylureas and Internal Alkynes. Org. Lett. 2016, 18, 1776–1779. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Jiao, N. Cationic Cobalt(III) Catalyzed Indole Synthesis: The Regioselective Intermolecular Cyclization of N-Nitrosoanilines and Alkynes. Angew. Chem. Int. Ed. 2016, 55, 4035–4039. [Google Scholar] [CrossRef] [PubMed]
- Lerchen, A.; Vàsquez-Céspedes, S.; Glorius, F. Cobalt(III)-Catalyzed Redox-Neutral Synthesis of Unprotected Indoles Featuring an N–N Bond Cleavage. Angew. Chem. Int. Ed. 2016, 55, 3208–3211. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Wang, J.; Wang, L.; Chen, K.; Song, C.; Zhu, J. Co(III)-Catalyzed, Internal and Terminal Alkyne-Compatible Synthesis of Indoles. Org. Lett. 2016, 18, 3806–3809. [Google Scholar] [CrossRef] [PubMed]
- Ghorai, J.; Reddy, A.C.S.; Anbarasan, P. Cobalt(III)-Catalyzed Intramolecular Cross-Dehydrogenative C−H/X−H Coupling: Efficient Synthesis of Indoles and Benzofurans. Chem. Eur. J. 2016, 22, 16042–16046. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.-Y.; Li, J.-H.; Wang, S.-Y.; Ji, S.-J. Cobalt(II)-catalyzed bis-isocyanide insertion reactions with sulfonyl azides via nitrene radicals: Chemoselective synthesis of sulfonylamidyl amide and 3-imine indole derivatives. Chem. Commun. 2017, 53, 11173–11176. [Google Scholar] [CrossRef] [PubMed]
- Bachon, A.-K.; Opatz, T. Synthesis of 1,2-Disubstituted Indoles from α-Aminonitriles and 2-Halobenzyl Halides. J. Org. Chem. 2016, 81, 1858–1869. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zhang, B.; Zhang, X.; Fan, X. Synthesis of 3-Cyano-1H-indoles and Their 2′-Deoxyribonucleoside Derivatives through One-Pot Cascade Reactions. J. Org. Chem. 2016, 81, 9530–9538. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-G.; Li, Z.-H.; Xie, J.-W.; Liu, P.; Zhang, J.; Dai, B. Copper-catalyzed synthesis of 2,3-disubstituted indoles from ortho-haloanilines and β-keto esters/β-diketone. Tetrahedron 2016, 72, 653–657. [Google Scholar] [CrossRef]
- Liu, J.; Wei, W.; Zhao, T.; Liu, X.Y.; Wu, J.; Yu, W.Q.; Chang, J.B. Iodine/Copper Iodide-Mediated C–H Functionalization: Synthesis of Imidazo[1,2-a]pyridines and Indoles from N-Aryl Enamines. J. Org. Chem. 2016, 81, 9326–9336. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, C.E.P.; Silva, P.J. Computational exploration of the reaction mechanism of the Cu+-catalysed synthesis of indoles from N-aryl enaminones. R. Soc. Open Sci. 2016, 3, 150582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, F.-Z.; Zhao, S.-H.; Chen, H.; Yu, S.-W.; Xu, X.-Y.; Yuan, W.-C.; Zhang, X.-M. Facile Synthesis of 2,3-Disubstituted Indoles by NBS/CuCl Mediated Oxidative Cyclization of N-Aryl Enamines. ChemistrySelect 2017, 2, 1409–1412. [Google Scholar] [CrossRef]
- Lopez, S.E.; Gallagher, R.; Gilliland, R.J.; Ghiviriga, I.; Dolbier, W.R. Jr. Synthesis of 3-phenylsulfonyl-2-trifluoromethyl-1H-indoles: A copper catalyzed cyclization approach. J. Fluor. Chem. 2017, 193, 118–125. [Google Scholar] [CrossRef]
- Yamamoto, C.; Takamatsu, K.; Hirano, K.; Miura, M. A Divergent Approach to Indoles and Oxazoles from Enamides by Directing-Group-Controlled Cu-Catalyzed Intramolecular C−H Amination and Alkoxylation. J. Org. Chem. 2017, 82, 9112–9118. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Yu, J.-T.; Sun, S.; Zheng, Q.; Cheng, J. Copper-mediated intramolecular aza-Wacker-type cyclization of 2-alkenylanilines toward 3-aryl indoles. Tetrahedron Lett. 2017, 58, 445–448. [Google Scholar] [CrossRef]
- Yu, J.; Zhang-Negrerie, D.; Du, Y. Cu(OAc)2-Mediated Cascade Annulation of Diarylalkyne Sulfonamides through Dual C–N Bond Formation: Synthesis of 5,10-Dihydroindolo[3,2-b]indoles. Org. Lett. 2016, 18, 3322–3325. [Google Scholar] [CrossRef] [PubMed]
- Ho, H.E.; Oniwa, K.; Yamamoto, Y.; Jin, T. N-Methyl Transfer Induced Copper-Mediated Oxidative Diamination of Alkynes. Org. Lett. 2016, 18, 2487–2490. [Google Scholar] [CrossRef] [PubMed]
- Villuri, B.K.; Kotipalli, T.; Kavala, V.; Ichake, S.S.; Bandi, V.; Kuo, C.-W.; Yao, C.-F. Synthesis of spiro isoindolinone-indolines and 1,2-disubstituted indoles from 2-iodobenzamide derivatives. RSC Adv. 2016, 6, 74845–74858. [Google Scholar] [CrossRef]
- Huang, C.-H.; Kuo, C.-W.; Konala, A.; Yang, T.H.; Lin, L.; Chen, Y.-W.; Kavala, V.; Yao, C.F. Synthesis of 3-arylindole derivatives from nitroalkane precursors. RSC Adv. 2016, 6, 96049–96056. [Google Scholar] [CrossRef]
- Li, Y.; Wang, R.; Wang, T.; Cheng, X.-F.; Zhou, X.; Fei, F.; Wang, X.-S. A Copper-Catalyzed Aerobic [1,3]-Nitrogen Shift through Nitrogen-Radical 4-exo-trig Cyclization. Angew. Chem. Int. Ed. 2017, 56, 15436–15440. [Google Scholar] [CrossRef] [PubMed]
- Debrouwer, W.; Heugebaert, T.S.A.; Roman, B.I.; Stevens, C.V. Homogeneous Gold-Catalyzed Cyclization Reactions of Alkynes with N- and S-Nucleophiles. Adv. Synth. Catal. 2015, 357, 2975–3006. [Google Scholar] [CrossRef]
- Liang, S.; Hammond, L.; Xu, B.; Hammond, G.B. Commercial Supported Gold Nanoparticles Catalyzed Alkyne Hydroamination and Indole Synthesis. Adv. Synth. Catal. 2016, 358, 3313–3318. [Google Scholar] [CrossRef]
- Qu, C.; Zhang, S.; Du, H.; Zhu, C. Cascade photoredox/gold catalysis: Access to multisubstituted indoles via aminoarylation of alkynes. Chem. Commun. 2016, 52, 14400–14403. [Google Scholar] [CrossRef] [PubMed]
- Michalska, M.; Grela, K. Simple and Mild Synthesis of Indoles via Hydroamination Reaction Catalysed by NHC-Gold Complexes: Looking for Optimized Condition. Synlett 2016, 27, 599–603. [Google Scholar] [CrossRef]
- Reiner, B.R.; Bezpalko, M.W.; Foxman, B.M.; Wade, C.R. Lewis Acid Catalysis with Cationic Dinuclear Gold(II,II) and Gold(III,III) Phosphorus Ylide Complexes. Organometallics 2016, 35, 2830–2835. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, X.; Zhang, V.; Cui, X.; Ma, M. Gold-Catalyzed Tandem Reactions of 2-Alkynyl Arylazides and Carboxylic Acids for the Synthesis of 1H-Indol-3-yl Esters. Chin. J. Org. Chem. 2015, 35, 1469–1474. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, X.; Fan, H.; Lyu, C.; Li, P.; Zhang, H.; Rao, W. Gold-catalyzed formation of indole derivatives from 2-alkynyl arylazides and oxygen-containing heterocycles. RSC Adv. 2016, 6, 56319–56322. [Google Scholar] [CrossRef]
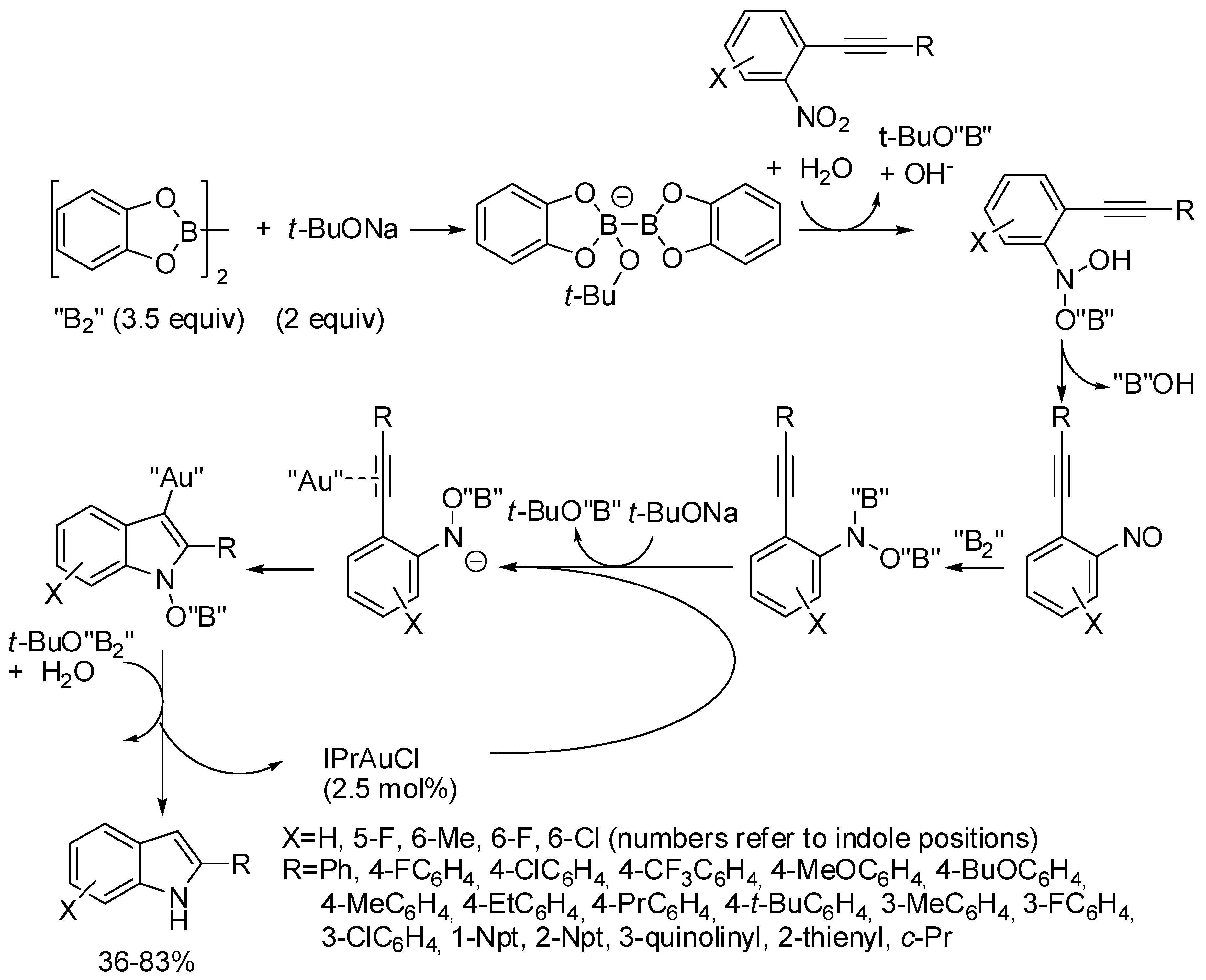
- Fu, W.; Yang, K.; Chen, J.; Song, Q. Gold(I)-catalyzed synthesis of 2-substituted indoles from 2-alkynylnitroarenes with diboron as reductant. Org. Biomol. Chem. 2017, 15, 8354–8360. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Huang, L.; Xie, J.; Rudolph, M.; Rominger, F.; Hashmi, A.S.K. Gold-Catalyzed C–H Annulation of Anthranils with Alkynes: A Facile, Flexible, and Atom-Economical Synthesis of Unprotected 7-Acylindoles. Angew. Chem. Int. Ed. 2016, 55, 794–797. [Google Scholar] [CrossRef] [PubMed]
- Marelli, E.; Corpet, M.; Minenkov, Y.; Neyyappadath, R.M.; Bismuto, A.; Buccolini, G.; Curcio, M.; Cavallo, L.; Nolan, S.P. Catalytic α-arylation of imines leading to N-unprotected indoles and azaindoles. ACS Catal. 2016, 6, 2930. [Google Scholar] [CrossRef] [PubMed]
- Solé, D.; Pérez-Janer, F.; Zulaica, E.; Guastavino, J.F.; Fernández, I. Pd-Catalyzed α-Arylation of Sulfones in a Three-Component Synthesis of 3-[2-(Phenyl/methylsulfonyl)ethyl]indoles. ACS Catal. 2016, 6, 1691–1700. [Google Scholar] [CrossRef] [Green Version]
- Solé, D.; Pérez-Janer, F.; Mancuso, R. Pd0-Catalyzed Intramolecular α-Arylation of Sulfones: Domino Reactions in the Synthesis of Functionalized Tetrahydroisoquinolines. Chem. Eur. J. 2015, 21, 4580–4584. [Google Scholar] [CrossRef] [PubMed]
- Gharpure, S.J.; Anuradha, D. Synthesis of Benzo[1,4]heterocycles using Palladium Catalyzed Heck Reaction to Vinylogous Carbonates/Carbamates: Unexpected Formation of Indoles via Carbopalladation Intercepted byNucleopalladation. Org. Lett. 2017, 19, 6136–6139. [Google Scholar] [CrossRef] [PubMed]
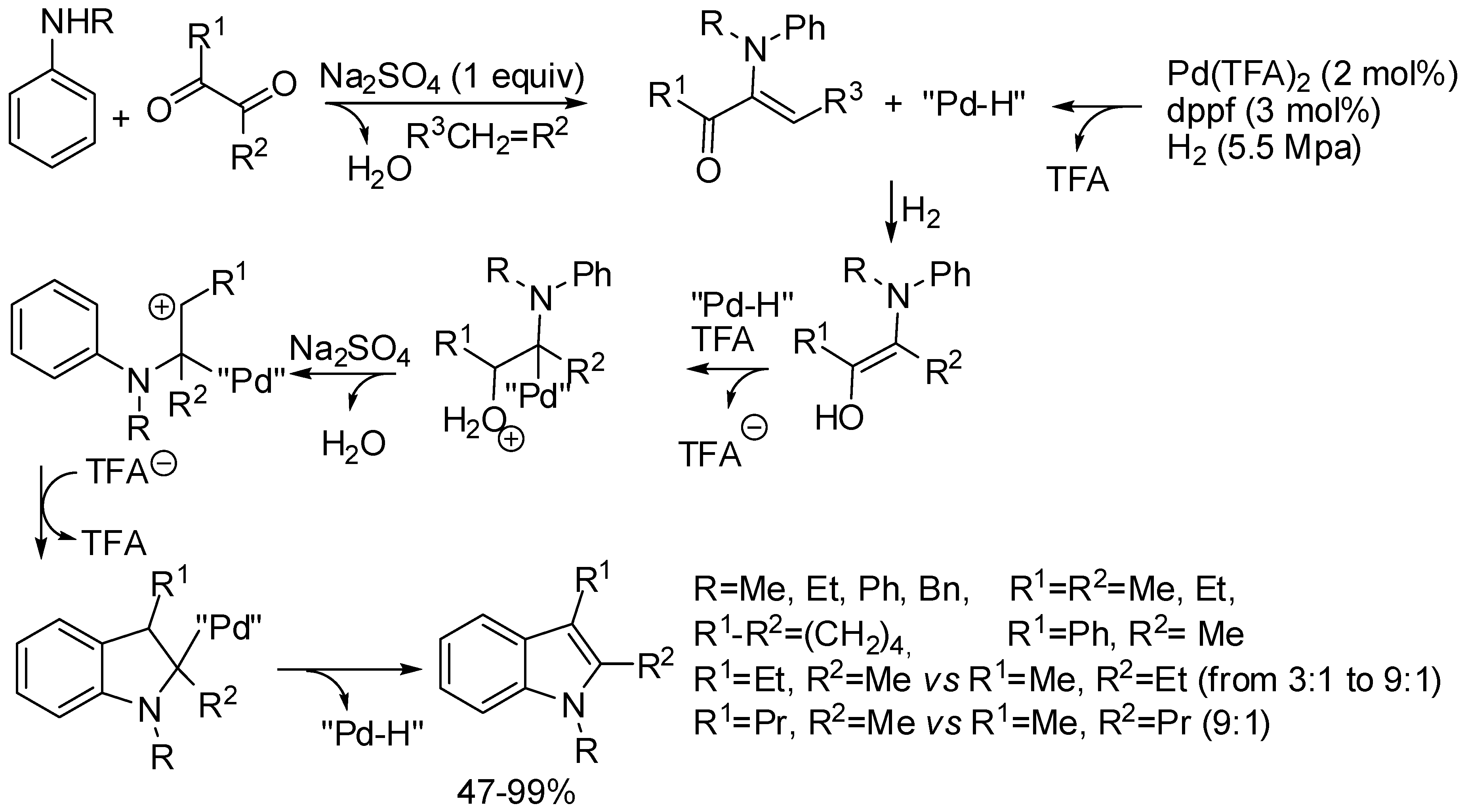
- Benitez-Medina, G.E.; Amézquita-Valencia, M.; Cabrera, A.; Sharma, P. Synthesis of 2,3-Disubstituted Indoles from α-Diketones and N-Substituted Anilines: One-Pot Pd-Catalyzed Reductive Amination. ChemCatChem 2017, 9, 1450–1460. [Google Scholar] [CrossRef]
- Yu, S.; Qi, L.; Hu, K.; Gong, J.; Cheng, T.; Wang, Q.; Chen, J.; Wu, H. The Development of a Palladium-Catalyzed Tandem Addition/Cyclization for the Construction of Indole Skeletons. J. Org. Chem. 2017, 82, 3631–3638. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Hu, K.; Gong, J.; Qi, L.; Zhu, J.; Zhang, Y.; Cheng, T.; Chen, J. Palladium-catalyzed tandem addition/cyclization in aqueous medium: Synthesis of 2-arylindoles. Org. Biomol. Chem. 2017, 15, 4300–4307. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M.; Akiyama, T.; Sasou, H.; Katsumata, H.; Manabe, K. One-Pot Synthesis of Substituted Benzo[b]furans and Indoles from Dichlorophenols/Dichloroanilines Using a Palladium–Dihydroxyterphenylphosphine Catalyst. J. Org. Chem. 2016, 81, 5450–5463. [Google Scholar] [CrossRef] [PubMed]
- Chuang, K.V.; Kieffer, M.E.; Reisman, S.E. A Mild and General Larock Indolization Protocol for the Preparation of Unnatural Tryptophans. Org. Lett. 2016, 18, 4750–4753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, C.B.; Bharti, R.; Kumara, S.; Das, P. Supported palladium nanoparticles-catalyzed decarboxylative coupling approaches to aryl alkynes, indoles and pyrrolines synthesis. RSC Adv. 2016, 6, 71117–71121. [Google Scholar] [CrossRef]
- Bruneau, A.; Gustafson, K.P.J.; Yuan, N.; Tai, C.-W.; Persson, I.; Zou, X.; Bäckvall, J.-E. Synthesis of Benzofurans and Indoles from Terminal Alkynes and Iodoaromatics Catalyzed by Recyclable Palladium Nanoparticles Immobilized on Siliceous Mesocellular Foam. Chem. Eur. J. 2017, 23, 12886–12891. [Google Scholar] [CrossRef] [PubMed]
- Panyam, P.K.R.; Gandhi, T. Palladium(II)/N-Heterocyclic Carbene-Catalyzed Regioselective Heteroannulation of Tertiary Propargyl Alcohols and o-Haloanilines to form 2-Alkenylindoles. Adv. Synth. Catal. 2017, 359, 1144–1151. [Google Scholar] [CrossRef]
- Shi, D.; Guan, Z.; Yao, X.; Shi, W.; Chen, H. One-Pot Synthesis of Indoles from Alkynes and Anilines through Palladium-Catalyzed Double C–H Activation using O2 as the oxidant. ChemistrySelect 2016, 1, 119–121. [Google Scholar] [CrossRef]
- Youn, S.W.; Ko, T.Y.; Jang, Y.H. Palladium-Catalyzed Regioselective Synthesis of 3-Arylindoles from N-Ts-Anilines and Styrenes. Angew. Chem. Int. Ed. 2017, 56, 6636–6640. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Zhao, F.; Du, Y.; Zhao, L.; Chen, L.; Wang, J.; Liu, H. Highly selective intramolecular addition of C–N and S-N bonds to alkynes catalyzed by palladium: A practical access to two distinct functional indoles. RSC Adv. 2016, 6, 70682–70690. [Google Scholar] [CrossRef]
- Ikeda, A.; Omote, M.; Kusumoto, K.; Komori, M.; Tarui, A.; Sato, K.; Ando, A. A dramatic enhancing effect of InBr3 towards the oxidative Sonogashira cross-coupling reaction of 2-ethynylanilines. Org. Biomol. Chem. 2016, 14, 2127–2133. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Han, X.; Lu, X. Enantioselective Synthesis of Tetrahydropyrano[3,4-b]indoles: Palladium(II)-Catalyzed Aminopalladation/1,4-Addition Sequence. Angew. Chem. Int. Ed. 2017, 56, 14698–14701. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Han, X.; Lu, X. Atom-Economic Synthesis of Pentaleno[2,1-b]indoles via Tandem Cyclization of Alkynones Initiated by Aminopalladation. J. Org. Chem. 2017, 82, 1977–1985. [Google Scholar] [CrossRef] [PubMed]
- Minami, M.; Kanayama, T.; Tanaka, R.; Okamoto, N.; Sueda, T.; Yanada, R. Regioselective Arylative Ring-Closing Reaction of 2-Alkynylphenyl Derivatives: Formation of Arylated Benzoxazin-2-ones, Benzoxazin-2-amines and 2,3-Disubstituted Indoles. Eur. J. Org. Chem. 2016, 5990–6000. [Google Scholar] [CrossRef]
- Li, J.; Li, C.; Ouyang, L.; Li, C.; Wu, W.; Jiang, H. N-Heterocyclic carbene palladium-catalyzed cascade annulation/alkynylation of 2-alkynylanilines with terminal alkynes. Org. Biomol. Chem. 2017, 15, 7898–7908. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, P.; Lyu, C.; Yong, W.; Li, J.; Zhu, X.; Rao, W. Synthesis of 1H-indole-3-sulfonates via palladium catalyzed tandem reactions of 2-alkynyl arylazides with sulfonic acid. Org. Biomol. Chem. 2017, 15, 6080–6083. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Zhang, Z.; Zhou, Y.; Li, S.; Zhang, Y.; Wang, J. Palladium-Catalyzed Synthesis of Indoles and Isoquinolines with in situ Generated Phosphinimine. J. Org. Chem. 2017, 82, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Battistuzzi, G.; Cacchi, S.; Fabrizi, G. The Aminopalladation/Reductive Elimination Domino Reaction in the Construction of Functionalized Indole Rings. Eur. J. Org. Chem. 2002, 2671–2681. [Google Scholar] [CrossRef]
- Glotz, G.; Gutmann, B.; Hanselmann, P.; Kulesza, A.; Roberge, D.; Kappe, C.O. Continuous flow synthesis of indoles by Pd-catalyzed deoxygenation of 2-nitrostilbenes with carbon monoxide. RSC Adv. 2017, 7, 10469–10478. [Google Scholar] [CrossRef] [Green Version]
- Ansari, N.H.; Dacko, C.A.; Akhmedov, N.G.; Söderberg, B.C.G. Double Palladium Catalyzed Reductive Cyclizations. Synthesis of 2,2′-, 2,3′-, and 3,3′-Bi-1H-indoles, Indolo[3,2-b]indoles, and Indolo[2,3-b]indoles. J. Org. Chem. 2016, 81, 9337–9349. [Google Scholar] [CrossRef] [PubMed]
- Clagg, K.; Hou, H.; Weinstein, A.B.; Russell, D.; Stahl, S.S.; Koenig, S.G. Synthesis of Indole-2-carboxylate Derivatives via Palladium-Catalyzed Aerobic Amination of Aryl C–H Bonds. Org. Lett. 2016, 18, 3586–3589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.; Chen, W.-W.; Xu, B.; Xu, M.-H. Access to Indole-Fused Polyheterocycles via Pd-Catalyzed Base-Free Intramolecular Cross Dehydrogenative Coupling. J. Org. Chem. 2016, 81, 11501–11507. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Simaan, M.; Marek, I.; Wei, Y.; Shi, M. Palladium-catalyzed oxidative cyclization of aniline-tethered alkylidenecyclopropanes with O2: A facile protocol to selectively synthesize 2- and 3-vinylindoles. Chem. Commun. 2017, 53, 216–219. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhou, X.; Yu, B.; Huang, H. Palladium-Catalyzed Dehydrogenative Difunctionalization of Aminoalkenes with Aminals as Oxidants and Electrophiles. Org. Lett. 2017, 19, 4600–4603. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Ali, M.A.; Jana, S.; Samanta, S.; Hajra, A. Palladium-catalyzed synthesis of indole fused coumarins via cross-dehydrogenative coupling. Tetrahedron Lett. 2017, 58, 313–316. [Google Scholar] [CrossRef]
- Della Ca’, N.; Fontana, M.; Motti, E.; Catellani, M. Pd/norbornene: A winning combination for selective aromatic functionalization via C–H bond activation. Acc. Chem. Res. 2016, 49, 1389–1400. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Shi, Z.; Sperger, T.; Yasukawa, Y.; Kingston, C.; Schoenebeck, F.; Lautens, M. Remote C–H alkylation and C–C bond cleavage enabled by an in situ generated palladacycle. Nat. Chem. 2017, 9, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Pedroni, J.; Cramer, N. 2-(Trifluoromethyl)indoles via Pd(0)-Catalyzed C(sp3)-H Functionalization of Trifluoroacetimidoyl Chlorides. Org. Lett. 2016, 18, 1932–1935. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, Y.; Mita, T.; Sato, Y. Palladium-Catalyzed Intramolecular Arylative Carboxylation of Allenes with CO2 for the Construction of 3-Substituted Indole-2-carboxylic Acids. Org. Lett. 2017, 19, 2710–2713. [Google Scholar] [CrossRef] [PubMed]
- Bzeih, T.; Lamaa, D.; Frison, G.; Hachem, A.; Jaber, N.; Bignon, J.; Retailleau, P.; Alami, M.; Hamze, A. Csp2−Csp2 and Csp2−N Bond Formation in a One-Pot Reaction between N-Tosylhydrazones and Bromonitrobenzenes: An Unexpected Cyclization to Substituted Indole Derivatives. Org. Lett. 2017, 19, 6700–6703. [Google Scholar] [CrossRef] [PubMed]
- McCabe, S.R.; Wipf, P. Eight-Step Enantioselective Total Synthesis of (−)-Cycloclavine. Angew. Chem. Int. Ed. 2017, 56, 324–327. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wipf, P. Indole synthesis by palladium-catalyzed tandem allylic isomerization—Furan Diels–Alder reaction. Org. Biomol. Chem. 2017, 15, 7093–7096. [Google Scholar] [CrossRef] [PubMed]
- Mizukami, A.; Ise, Y.; Kimachi, T.; Inamoto, K. Rhodium-Catalyzed Cyclization of 2-Ethynylanilines in the Presence of Isocyanates: Approach toward Indole-3-carboxamides. Org. Lett. 2016, 18, 748–751. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Tong, X.; Liu, G. Rhodium(III)-Catalyzed Cascade Cyclization/Electrophilic Amidation for the Synthesis of 3-Amidoindoles and 3-Amidofurans. Org. Lett. 2016, 18, 2058–2061. [Google Scholar] [CrossRef] [PubMed]
- Hermann, G.N.; Junga, C.L.; Bolm, C. Mechanochemical indole synthesis by rhodium-catalysed oxidative coupling of acetanilides and alkynes under solventless conditions in a ball mill. Green Chem. 2017, 19, 2520–2523. [Google Scholar] [CrossRef]
- Li, B.; Xu, H.; Wang, H.; Wang, B. Rhodium-Catalyzed Annulation of Tertiary Aniline N-Oxides to N-Alkylindoles: Regioselective C–H Activation, Oxygen-Atom Transfer, and N-Dealkylative Cyclization. ACS Catal. 2016, 6, 3856–3862. [Google Scholar] [CrossRef]
- Zhou, S.; Wang, J.; Zhang, F.; Song, C.; Zhu, J. A Versatile, Traceless C–H Activation-Based Approach for the Synthesis of Heterocycles. Org. Lett. 2016, 18, 2427–2730. [Google Scholar] [CrossRef] [PubMed]
- .Fan, Z.; Lu, H.; Li, W.; Genga, K.; Zhang, A. Rhodium-catalyzed redox-neutral coupling of phenidones with alkynes. Org. Biomol. Chem. 2017, 15, 5701–5708. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Gao, S.; Xu, J.; Wu, X.; Lin, A.; Yao, H. Multiple Roles of the Pyrimidyl Group in the Rhodium-Catalyzed Regioselective Synthesis and Functionalization of Indole-3-carboxylic Acid Esters. Adv. Synth. Catal. 2016, 358, 188. [Google Scholar] [CrossRef]
- Yu, K.; Liang, Y.; Li, B.; Wang, B. Rhodium(III)-Catalyzed Synthesis of Indole Derivatives From Pyrimidyl-Substituted Anilines and Diazo Compounds. Adv. Synth. Catal. 2016, 358, 661–666. [Google Scholar] [CrossRef]
- Allu, S.; Ravi, M.; Swamy, K.C.K. Rhodium(III)-Catalysed Carbenoid C(sp2)–H Functionalisation of Aniline Substrates with α-Diazo Esters: Formation of Oxindoles and Characterisation/Utility of an Intermediate-Like Rhodacycle. Eur. J. Org. Chem. 2016, 5697–5705. [Google Scholar] [CrossRef]
- Huynh, U.; Uddin, M.; Wengryniuk, S.E.; McDonald, S.L.; Coltart, D.M. A simple and efficient approach to the N-amination of oxazolidinones using monochloroamine. Tetrahedron Lett. 2016, 57, 4799–4802. [Google Scholar] [CrossRef]
- Kaga, A.; Peng, X.; Hirao, H.; Chiba, S. Diastereo-Divergent Synthesis of Saturated Azaheterocycles Enabled by t-BuOK-Mediated Hydroamination of Alkenyl Hydrazones. Chem. Eur. J. 2015, 21, 19112–19118. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Chen, H.; Liu, P. Rhodium-Catalyzed Oxidative Annulation of Hydrazines with Alkynes Using a Nitrobenzene Oxidant. Org. Lett. 2014, 16, 6176. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Yu, K.; Li, B.; Xu, S.; Song, H.; Wang, B. Rh(III)-catalyzed synthesis of 1-aminoindole derivatives from 2-acetyl-1-arylhydrazines and diazo compounds in water. Chem. Commun. 2014, 50, 6130–6133. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Lin, X.; Wang, L.; Cui, X. Facile synthesis of 1-aminoindoles via Rh(III)-catalysed intramolecular three-component annulation. Org. Chem. Front. 2017, 4, 2179–2183. [Google Scholar] [CrossRef]
- Shi, P.; Wang, L.; Guo, S.; Chen, K.; Wang, J.; Zhu, J. A C–H Activation-Based Strategy for N-Amino Azaheterocycle Synthesis. Org. Lett. 2017, 19, 4359–4362. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Shi, J.; Wu, B.; Guo, Y.; Huang, J.; Yi, W. One-pot synthesis of 2,3-difunctionalized indoles via Rh(III)-catalyzed carbenoid insertion C–H activation/cyclization. Org. Biomol. Chem. 2017, 15, 8054–8058. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.; Yu, S.; Li, X. Rh(III)-Catalyzed Synthesis of N-Unprotected Indoles from Imidamides and Diazo Ketoesters via C–H Activation and C–C/C–N Bond Cleavage. Org. Lett. 2016, 18, 700–703. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Han, J.; Liu, Y.; Qin, M.; Zhang, X.; Chen, B. Synthesis of 2,3-Disubstituted NH Indoles via Rhodium(III)-Catalyzed C–H Activation of Arylnitrones and Coupling with Diazo Compounds. J. Org. Chem. 2017, 82, 11505–11511. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, M.; Chen, K.; Zha, S.; Song, C.; Zhu, J. C–H Activation-Based Traceless Synthesis via Electrophilic Removal of a Directing Group. Rhodium(III)-Catalyzed Entry into Indoles from N-Nitroso and α-Diazo-β-keto Compounds. Org. Lett. 2016, 18, 1178–1181. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Lin, S.; Yi, X.; Xi, C. Substrate-Controlled Transformation of Azobenzenes to Indazoles and Indoles via Rh(III)-Catalysis. J. Org. Chem. 2017, 82, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.; Han, S.H.; Mishra, N.K.; De, U.; Lee, J.; Kim, H.S.; Jung, Y.H.; Kim, I.S. Synthesis and Anticancer Evaluation of 2,3-Disubstituted Indoles Derived from Azobenzenes and Internal Olefins. Eur. J. Org. Chem. 2017, 6265–6273. [Google Scholar] [CrossRef]
- Rajagopal, B.; Chou, C.H.; Chung, C.C.; Lin, P.C. Synthesis of Substituted 3-Indolylimines and Indole-3-carboxaldehydes by Rhodium(II)-Catalyzed Annulation. Org. Lett. 2014, 16, 3752–3755. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Fu, J.; Yuan, H.; Gong, J.; Yang, Z. Synthesis of 2,3-Disubstituted Indoles and Benzofurans by the Tandem Reaction of Rhodium(II)-Catalyzed Intramolecular C–H Insertion and Oxygen-Mediated Oxidation. J. Org. Chem. 2016, 81, 10180–10182. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xie, H.; Fu, X.; Liu, J.-T.; Wang, H.-Y.; Xi, B.-M.; Liu, P.; Xu, X.; Tang, W. Rhodium(I)-Catalyzed Benzannulation of Heteroaryl Propargylic Esters: Synthesis of Indoles and Related Heterocycles. Chem. Eur. J. 2016, 22, 10410–10414. [Google Scholar] [CrossRef] [PubMed]
- Gabriele, B.; Mancuso, R.; Salerno, G.; Lupinacci, E.; Ruffolo, G.; Costa, M. Versatile Synthesis of Quinoline-3-Carboxylic Esters and Indol-2-Acetic Esters by Palladium-Catalyzed Carbonylation of 1-(2-Aminoaryl)-2-Yn-1-Ols. J. Org. Chem. 2008, 73, 4971–4977. [Google Scholar] [CrossRef] [PubMed]
- Gabriele, B.; Veltri, L.; Salerno, G.; Mancuso, R.; Costa, M. Multicomponent Cascade Reactions: A Novel and Expedient Approach to Functionalized Indoles by an Unprecedented Nucleophilic Addition-Heterocyclization-Oxidative Alkoxycarbonylation Sequence. Adv. Synth. Catal. 2010, 352, 3355–3363. [Google Scholar] [CrossRef]
- Gabriele, B.; Veltri, L.; Salerno, G.; Mancuso, R.; Costa, M. A General Synthesis of Indole-3-carboxylic Esters by Palladium-Catalyzed Direct Oxidative Carbonylation of 2-Alkynylaniline Derivatives. Eur. J. Org. Chem. 2012, 2549–2559. [Google Scholar] [CrossRef]
- Kanno, H.; Nakamura, K.; Noguchi, K.; Shibata, Y.; Tanaka, K. Rhodium-Catalyzed Cycloisomerization of 2-Silylethynyl Phenols and Anilines via 1,2-Silicon Migration. Org. Lett. 2016, 18, 1654–1657. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Mutoh, Y.; Saito, S. Ruthenium-Catalyzed Cycloisomerization of 2-Alkynylanilides: Synthesis of 3-Substituted Indoles by 1,2-Carbon Migration. J. Am. Chem. Soc. 2017, 139, 7749–7752. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, L.; Fenner, S. Ruthenium-Catalyzed C–H/N–O Bond Functionalization: Green Isoquinolone Syntheses in Water. Org. Lett. 2011, 13, 6548–6551. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Ma, J.; Wang, N.; Feng, H.; Xu, S.; Wang, B. Ruthenium-Catalyzed Oxidative C–H Bond Olefination of N-Methoxybenzamides Using an Oxidizing Directing Group. Org. Lett. 2012, 14, 736–739. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Ackermann, L. Dehydrative C–H/N–OH Functionalizations in H2O by Ruthenium(II) Catalysis: Subtle Effect of Carboxylate Ligands and Mechanistic Insight. J. Org. Chem. 2014, 79, 12070–12082. [Google Scholar] [CrossRef] [PubMed]
- Nakanowatari, S.; Ackermann, L. Ruthenium(II)-Catalyzed C–H Functionalizations with Allenes: Versatile Allenylations and Allylations. Chem. Eur. J. 2015, 21, 16246–16251. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Jiang, H.; Huang, Y. Ruthenium-Catalyzed Redox-Neutral C–H Activation via N–N Cleavage: Synthesis of N-Substituted Indoles. Org. Lett. 2014, 16, 5976–5979. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Wang, J.; Chen, P.; Chen, K.; Zhu, J. Ruthenium(II)-Catalyzed Traceless C−H Functionalization Using an N−N Bond as an Internal Oxidant. Chem. Eur. J. 2016, 22, 14508–14512. [Google Scholar] [CrossRef] [PubMed]
- Manna, M.K.; Bhunia, S.K.; Jana, R. Ruthenium(II)-catalyzed intermolecular synthesis of 2-arylindolines through C–H activation/oxidative cyclization cascade. Chem. Commun. 2017, 53, 6906–6909. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Qi, Z.; Wang, H.; Yang, X.; Li, X. Ruthenium(II)-Catalyzed C–H Activation of Imidamides and Divergent Couplings with Diazo Compounds: Substrate-Controlled Synthesis of Indoles and 3H-Indoles. Angew. Chem. Int. Ed. 2016, 55, 11877–11881. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.S. Ruthenium-catalysed synthesis of indoles from anilines and trialkanolamines in the presence of tin(II) chloride dehydrate. Chem. Commun. 1998, 995–996. [Google Scholar] [CrossRef]
- Cho, C.S.; Oh, B.H.; Kim, J.S.; Kim, T.-J.; Shim, S.C. Synthesis of quinolines via ruthenium-catalysed amine exchange reaction between anilines and trialkylamines. Chem. Commun. 2000, 1885–1886. [Google Scholar] [CrossRef]
- Cho, C.S.; Kim, J.S.; Oh, B.H.; Kim, T.-J.; Shim, S.C.; Yoon, N.S. Ruthenium-Catalyzed Synthesis of Quinolines from Anilines and Allylammonium Chlorides in an Aqueous Medium via Amine Exchange Reaction. Tetrahedron 2000, 56, 7747–7750. [Google Scholar] [CrossRef]
- Tursky, M.; Lorentz-Petersen, L.L.R.; Olsen, L.B.; Madsen, R. Iridium- and ruthenium-catalysed synthesis of 2,3-disubstituted indoles from anilines and vicinal diols. Org. Biomol. Chem. 2010, 8, 5576–5582. [Google Scholar] [CrossRef] [PubMed]
- Monrad, R.N.; Madsen, R. Ruthenium-catalysed synthesis of 2- and 3-substituted quinolines from anilines and 1,3-diols. Org. Biomol. Chem. 2011, 9, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Xie, F.; Wang, X.T.; Yan, F.; Wang, T.; Chen, M.; Ding, Y. Improved indole syntheses from anilines and vicinal diols by cooperative catalysis of ruthenium complex and acid. RSC Adv. 2013, 3, 6022–6029. [Google Scholar] [CrossRef]
- Lee, H.; Yi, C.S. Catalytic Synthesis of Substituted Indoles and Quinolines from the Dehydrative C–H Coupling of Arylamines with 1,2- and 1,3-Diols. Organometallics 2016, 35, 1973–1977. [Google Scholar] [CrossRef]
- Takamoto, K.; Ohno, S.; Hyogo, N.; Fujioka, H.; Arisawa, M. Ruthenium-Catalyzed 1,6-Aromatic Enamide−Silylalkyne Cycloisomerization: Approach to 2,3-Disubstituted Indoles. J. Org. Chem. 2017, 82, 8733–8742. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, A.; Yorimitsu, H.; Oshima, K. Synthesis of 2-Indolylphosphines by Palladium-Catalyzed Annulation of 1-Alkynylphosphine Sulfides with 2-Iodoanilines. Org. Lett. 2010, 12, 1476–1479. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Lu, G.; Zhang, P.; Zhang, L.; Tang, G.; Zhao, Y. A Cascade Phosphinoylation/Cyclization /Desulfonylation Process for the Synthesis of 3-Phosphinoylindoles. Org. Lett. 2016, 18, 1242–1245. [Google Scholar] [CrossRef] [PubMed]
- Hou, Z.-W.; Mao, Z.-Y.; Zhao, H.-B.; Melcamu, Y.Y.; Lu, X.; Song, J.; Xu, H.-C. Electrochemical C–H/N-H Functionalization for the Synthesis of Highly Functionalized (Aza)indoles. Angew. Chem. Int. Ed. 2016, 55, 9168–9172. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-J.; Meng, Q.-Y.; Lei, T.; Zhong, J.-J.; Liu, W.-Q.; Zhao, L.-M.; Li, Z.-J.; Chen, B.; Tung, C.-H.; Wu, L.-Z. An Oxidant-Free Strategy for Indole Synthesis via Intramolecular C–C Bond Construction under Visible Light Irradiation: Cross-Coupling Hydrogen Evolution Reaction. ACS Catal. 2016, 6, 4635. [Google Scholar] [CrossRef]
- Tang, G.-D.; Pan, C.-L.; Li, X. Iridium(III)- and rhodium(III)-catalyzed coupling of anilines with α-diazoesters via chelation-assisted C–H activation. Org. Chem. Front. 2016, 3, 87–90. [Google Scholar] [CrossRef]
- Patel, P.; Borah, G. Direct Access to Indoles by IrIII-Catalyzed C–H Functionalization of Acetanilides with Diazo Compounds. Eur. J. Org. Chem. 2017, 2272–2279. [Google Scholar] [CrossRef]
- Alt, I.T.; Plietker, B. Iron-Catalyzed Intramolecular C(sp(2))-H Amination. Angew. Chem. Int. Ed. 2016, 55, 1519–1522. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Leslie, B.E.; Driver, T.G. Dirhodium(II)-catalyzed intramolecular C–H amination of aryl azides. Angew. Chem. Int. Ed. 2008, 47, 5056–5059. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhao, Y.; Ma, L.; Ma, M.; Jiang, J.; Wan, X. Radical-carbene coupling reaction: Mn-catalyzed synthesis of indoles from aromatic amines and diazo compounds. Chem. Commun. 2017, 53, 5993–5996. [Google Scholar] [CrossRef] [PubMed]
- Ohtaka, A.; Kozono, M.; Takahashi, K.; Hamasaka, G.; Uozumi, Y.; Shinagawa, T.; Shimomura, O.; Nomura, R. Linear Polystyrene-stabilized Pt Nanoparticles Catalyzed Indole Synthesis in Water via Aerobic Alcohol Oxidation. Chem. Lett. 2016, 45, 758–760. [Google Scholar] [CrossRef]
- Allegretti, P.A.; Huynh, K.; Ozumerzifon, T.J.; Ferreira, E.M. Lewis Acid Mediated Vinylogous Additions of Enol Nucleophiles into an α,β-Unsaturated Platinum Carbene. Org. Lett. 2016, 18, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Ilies, L.; Isomura, M.; Yamauchi, S.-I.; Nakamura, T.; Nakamura, E. Indole Synthesis via Cyclative Formation of 2,3-Dizincioindoles and Regioselective Electrophilic Trapping. J. Am. Chem. Soc. 2017, 139, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.; Zhou, B.; Wu, Z.; Ji, X.; Zhang, Y. Synthesis of Carbazoles from 2-Iodobiphenyls by Palladium-Catalyzed C–H Activation and Amination with Diaziridinone. Adv. Synth. Catal. 2018, 360, 887–892. [Google Scholar] [CrossRef]
- Kumar, S.; Singh, R.; Singh, K.N. Palladium Catalyzed C–C and C–N Bond Formation via ortho-C–H Activation and Decarboxylative Strategy: A Practical Approach towards N-Acylated Indoles. Adv. Synth. Catal. 2018, 360, 422–426. [Google Scholar] [CrossRef]
- Hidayat, Y.; Armunanto, R.; Pranowo, H.D. Investigation of rubidium(I) ion solvation in liquid ammonia using QMCF-MD simulation and NBO analysis of first solvation shell structure. J. Mol. Model. 2018, 24, 122. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Li, D.; Zeng, F. Palladium-Catalyzed Sequential Vinylic C−H Arylation/Amination of 2-Vinylanilines with Aryl boronic Acids: Access to 2-Arylindoles. J. Org. Chem. 2018, 83, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Peng, J.; Chen, X.; Mo, B.; Li, X.; Sun, P.; Chen, C. Copper-Catalyzed Synthesis of Multisubstituted Indoles through Tandem Ullmann-Type C−N Formation and Cross-dehydrogenative Coupling Reactions. J. Org. Chem. 2018, 83, 5288–5294. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Chen, Y.; Ma, Z.; Zou, L.; Li, J.; Liu, Y. One-Pot Synthesis of Indole-3-acetic Acid Derivatives through the Cascade Tsuji−Trost Reaction and Heck Coupling. J. Org. Chem. 2018, 83, 6805–6814. [Google Scholar] [CrossRef] [PubMed]
- Panyam, P.K.R.; Sreedharan, R.; Gandhi, T. Synthesis of topologically constrained naphthalimide appended palladium(II)-N-heterocyclic carbene complexes—Insights into additive controlled product selectivity. Org. Biomol. Chem. 2018, 16, 4357–4364. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, S.; Wang, B.; Li, B. The regioselective synthesis of 2-phosphinoylindoles via Rh(III)-catalyzed C–H activation. Org. Chem. Front. 2018, 5, 88–91. [Google Scholar] [CrossRef]
- Duan, S.; Cheng, B.; Duan, X.; Bao, B.; Li, Y.; Zhai, H. Synthesis of cis-5,5a,6,10b-Tetrahydroindeno[2,1-b]indoles through Palladium-Catalyzed Decarboxylative Coupling of Vinyl Benzoxazinanones with Arynes. Org. Lett. 2018, 20, 1417–1420. [Google Scholar] [CrossRef] [PubMed]
- Onishia, K.; Oikawa, K.; Yano, H.; Suzuki, T.; Obora, Y. N,N-Dimethylformamide-stabilized palladium nanoclusters as a catalyst for Larock indole synthesis. RSC Adv. 2018, 8, 11324–11329. [Google Scholar] [CrossRef]
- Arcadi, A.; Cacchi, S.; Fabrizi, G.; Ghirga, F.; Goggiamani, A.; Iazzetti, A.; Marinelli, F. Palladium-Catalyzed Cascade Approach to 12-(Aryl)indolo-[1,2-c]quinazolin-6(5H)-ones. Synthesis 2018, 50, 1133–1140. [Google Scholar] [CrossRef]
- Wei, F.; Shen, X.-Q.; Chu, J.-J.; Hu, B.-L.; Zhang, X.-G. Palladium-catalyzed intramolecular aerobic C–H amination of enamines for the synthesis of 2-trifluoromethylindoles. Tetrahedron 2018, 74, 720–725. [Google Scholar] [CrossRef]
- Cirujano, F.; Lopez-Maya, E.; Navarro, J.A.R.; De Vos, D.E. Pd(II)–Ni(II) Pyrazolate Framework as Active and Recyclable Catalyst for the Hydroamination of Terminal Alkynes. Top. Catal. 2018. [Google Scholar] [CrossRef]













































































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
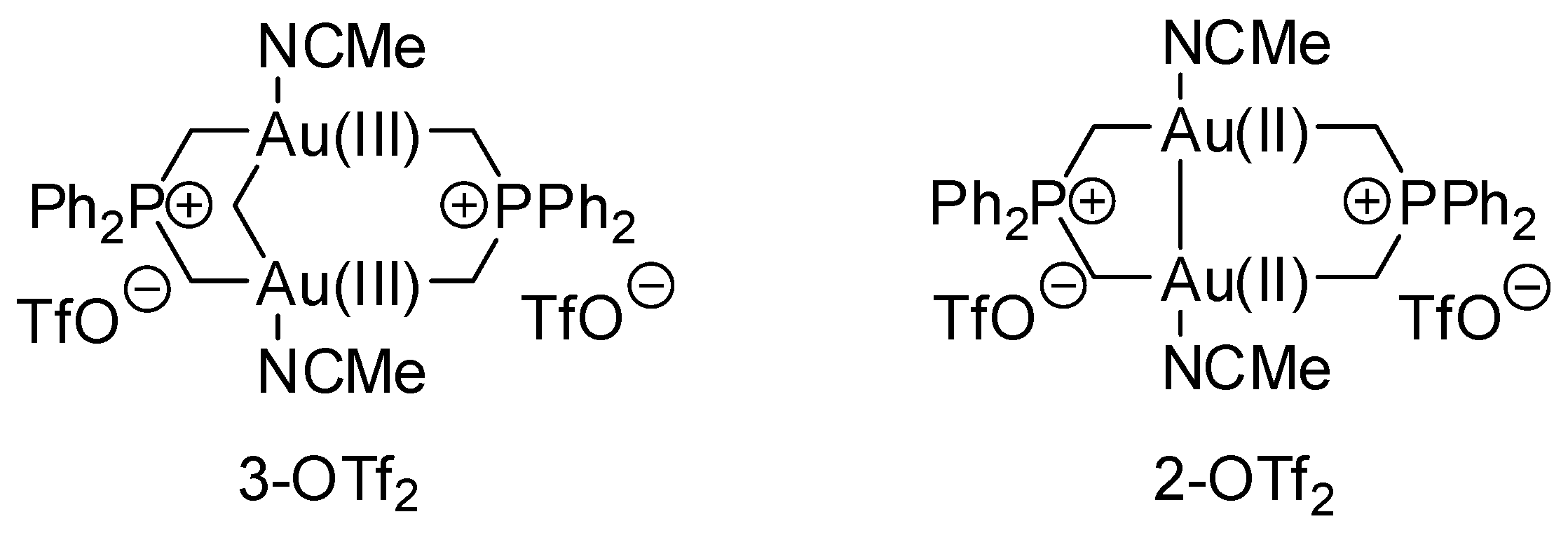{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2-Haloaniline | Alkyne | Reaction Conditions | Indole Yields (%) | Ref |
|---|---|---|---|---|
 X = 3-Cl, 4-Me, 4-F, 4-CF3, 4-Cl, 5-Cl, 5-MeO |  (2 equiv) R = C10H21, Ph(CH2)2, Ph, 4-MeOC6H4, |  (4 mol %) PdCl2(MeCN)2 (2 mol %) t-BuOLi (2.4 equiv) then H2O |  37–83 a | [60] |
 X = H, 3-MeCONH, 3-Cl, 4-MeO, 4-F, 4-NO2, 4-CN, 4-(4,4,5,5-Me4-dioxaborolan-2-yl, 5-Cl, 6-Cl, 6-Me, 6-Br, 4,5,6-F3, 6-aza R = H, Me, Ph, Ac |  | Pd[(PBu)3]2 (5 mol %) Cy2NMe (2.5 equiv) |  52–88 b | [61] |
 X = H, 4-Me, 4-Cl, |  R = Ph, Me, 4-ClC6H4 | Pd@PS (3 mol %) DBU (3 equiv) |  65–76 c | [62] |
 X = H, 5-Cl, 4-CN, 6-aza |  R = Ph, 4-MeC6H4, 4-FC6H4,n-C5H11, OH(CH2)3, cyclohex-1-en-1-yl | Pd0-AmP-MCF (2.5 mol %) CuI (5 mol %) NEt3 (1.2 equiv) |  90–95 d | [63] |
 Hal = I, Br, Cl X = H, Me, MeO, F, CN, CO2Et, MeCO R = H, Me, Bu, MeCO (H in the product) CF3CO (H in the product), |  Ar = Ph, 4-MeC6H4, 4-MeOC6H4, 4-CNC6H4, 4-NO2C6H4, 2-Npt, 2-thienyl R1 = H, Me, (CH2)3 |  Pd(II)/NHC (4 mol %) K2CO3 (2 equiv) Bu4NBr (1 equiv) then AcOH |  52–81 e | [64] |
| Alkynylaniline | Reaction Conditions | Indole Yields (%) | Ref. |
|---|---|---|---|
 X = H, 4-F, 4-Cl, 4-CN, 4-CF3, 5-Br, 5-Me R = Bu, c-Pr, Cy, Ph, AcO(CH2)3, Cl(CH2)3 R1 = Me, Et, Ph, 4-MeC6H4, | Pd(PPh3)4 (10 mol %) |  85–99 a | [67] |
| PdCl2(MeCN)2 (10 mol %) |  36–92 a | ||
 X = H, 4-Br, 4-Cl, 4-MeCO, 4-CO2Et, 4-Me, 4-MeO, 6-MeCO b | Pd(OAc)2 (10 mol %) |  39–65 | [68] |
 R = H, Me R1 = Me, Et, i-Pr, t-Bu, Ph, Bn, Cy X = H, 4-F, 4-Cl, 4-Br, 4-OMe, 4-TBSOCH2, 4-Ph, 4-CF3, 4-Me, 3-Me | Pd(OAc)2 (5 mol %) 2,2′-bypyridine (10 mol %) or  (10 mol %) (10 mol %) |  72–92% c 93–96% ee (positive αD) | [69] |
 X = H, 4-Me, 4-F, 4-Cl, 4-CF3, 4-MeO, 4-CO2Me, 3-Me, 2-Me-4-CO2Me, 2,4-Me2 R = CO2Me, CN, CH2OH, Me | Pd(OAc)2 (5 mol %) 2,2′-bypyridine (10 mol %) AcOH |  70–83 d | [70] |
 X = H, Me, F R = Et, Me Ar = Ph, 4-MeC6H4, 4-F C6H4 | Pd(OAc)2 (10 mol %) Ar2IOTf |  62–92 e | [71] |
 X = H, 3-F, 4-MeO, 4-F, 4-Cl, 4-Br, 4-CF3, 4-CN, 4-CO2Me, 5-Me, 5-Cl, 5-Br, 4,6-Me2, 4,6-Cl2, 4-Cl-6-F R = Ph, 4-MeC6H4, 4-BuC6H4, 4-t-BuC6H4, 4-ClC6H4, 4-BrC6H4, 4-Me2NC6H4, 4-CNC6H4, 4-CF3C6H4, 4-CHOC6H4, 4-NO2C6H4, 3-FC6H4, 3-BrC6H4, 2-thienyl, cyclohex-1-en-1-yl |  (1 mol %) K2CO3 (1 equiv)  (R1 = Ph, 2-FC6H4, 2-ClC6H4, 3-ClC6H4, 3-MeC6H4, 4-MeC6H4, 4-EtC6H4, 4-PrC6H4, 4-BuC6H4, 4-(n-C5H11)C6H4, 4-CF3C6H4, 4-BrC6H4, 4-MeOC6H4, 2,4-Me2C6H4, 2,6-Me2C6H4, c-C5H9, Cy, CyCH2, n-Hex, Bn, Ph(CH2)3, Cl(CH2)3, CN(CH2)3, 3-thienyl, 2-thienyl, oct-2-yn-1-yl |  53–89 | [72] |
 X = H, 4-Cl, 4-CF3, 4-Me, 5-Cl, 4,6-Cl2 R = Ph, 4-MeC6H4, 4-ClC6H4, 4-CO2MeC6H4, 3-thienyl, Bu, n-C5H11, c-Pr, t-Bu, BnO(CH2)2 | Pd(OAc)2 (5 mol %) R1SO3H (3 equiv) (R1 = Me, Ph, 4-MeC6H4, 4-ClC6H4, 1-Npt) |  70–97 f | [73] |
| Arene | Reaction Conditions | Indole Yield (%) | Ref. |
|---|---|---|---|
 X = H, 4-Me, 4-Ph, 4-F, 4-CF3, 4-NMe2, 4-MeO, 4-Cl, 4-(5-pyrimidyl), 3-Me, 3-Ph, 3-F, 3- CF3, 3-MeO, 2-Me, 2-Ph and  | Pd(OAc)2 (10 mol %) 3 Å MS, O2 (0,1 MPa) |  36–95 a | [78] |
 X = H, 2-Me, 2-MeO, 3-Me b, 3-MeO b, 3-F b, 4-MeO, 4-F, 4-Me, 4-Cl, 3,5-Me2, 3,5-(MeO)2, and 1-Npt X1 = H, 6-Me, 6-F, 6-Cl, 7-MeO, 7-F, 8-Me, 6,8-Me2, 5,7-Me2, 6.7-(MeO)2, Y = O, NH | Pd(OAc)2 (15 mol %), air or Pd(OAc)2 (10 mol %), AgOAc (2 equiv) |  76–99 | [79] |
 R = Me, Ph, X = H, 4-Me, 4-F, 4-MeO, 2-MeO, 3,5-Me2 | Pd(OAc)2 (10 mol %), AgOAc (2 equiv) |  76–94 | |
 X = H, 4-Cl, R = Ph, 4-MeC6H4, 4-BrC6H4, 4-FC6H4, 4-CF3C6H4, 4-PhC6H4, 4-BnOC6H4, 4-t-BuC6H4, 2-MeOC6H4, 2-ClC6H4, 3-MeC6H4, 3-MeOC6H4, 2-thienyl, Bu R1 = Ts, Ac, H, 4-BrC6H4SO2, 4-NO2C6H4SO2 | Pd[P(4-CF3C6H4)3]2Cl2 (10 mol %) KHCO3 (1 equiv), O2 (0.1 MPa) |  33–74 c | [80] |
 X = H, 3-MeO, 4-Br, 4-Me, 5-Cl, 6-Me, 5,6-Me2 R1 = Ts, Ms, 2,4-(MeO)2C6H3 | Pd[P(PMP)3]2Cl2 (10 mol %) KHCO3 (1 equiv), O2 (0.1 MPa) |  39–58 | |
 X = H, 4-MeO, 4-Cl, 4-F, 5-CN, 6-Cl, 3,5-Me2, 3,5-Cl2, R = H, Me, OHCH2, AcOCH2, PivOCH2 R1 = Ts, Ms, 4-NO2C6H4SO2, canphorylSO2 | Pd(BINAP)Cl2 (5 mol %) AgOTf (10 mol %)  (2 equiv) d Y = NBn2, (4-ClC6H4CH2)2N, (4-CF3C6H4CH2)2N, NBu2, (2-BrC6H4CH2)2N, (2-ClC6H4CH2)2N, MeO R2 = Bn, 4-ClC6H4CH2, 4-CF3C6H4CH2, Bu, 2-BrC6H4CH2, 2-BrC6H4CH2, Pr, –(CH2)2O(CH2)2- (2 equiv) d Y = NBn2, (4-ClC6H4CH2)2N, (4-CF3C6H4CH2)2N, NBu2, (2-BrC6H4CH2)2N, (2-ClC6H4CH2)2N, MeO R2 = Bn, 4-ClC6H4CH2, 4-CF3C6H4CH2, Bu, 2-BrC6H4CH2, 2-BrC6H4CH2, Pr, –(CH2)2O(CH2)2- |  28–98 | [81] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mancuso, R.; Dalpozzo, R. Recent Progress in the Transition Metal Catalyzed Synthesis of Indoles. Catalysts 2018, 8, 458. https://doi.org/10.3390/catal8100458
Mancuso R, Dalpozzo R. Recent Progress in the Transition Metal Catalyzed Synthesis of Indoles. Catalysts. 2018; 8(10):458. https://doi.org/10.3390/catal8100458
Chicago/Turabian StyleMancuso, Raffaella, and Renato Dalpozzo. 2018. "Recent Progress in the Transition Metal Catalyzed Synthesis of Indoles" Catalysts 8, no. 10: 458. https://doi.org/10.3390/catal8100458