Rhodium-Biphephos-Catalyzed Tandem Isomerization–Hydroformylation of Oleonitrile

 ,
,  and
and
Abstract
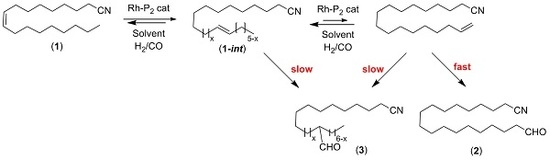
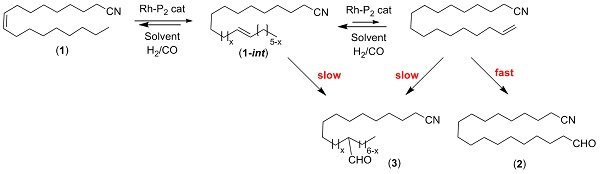
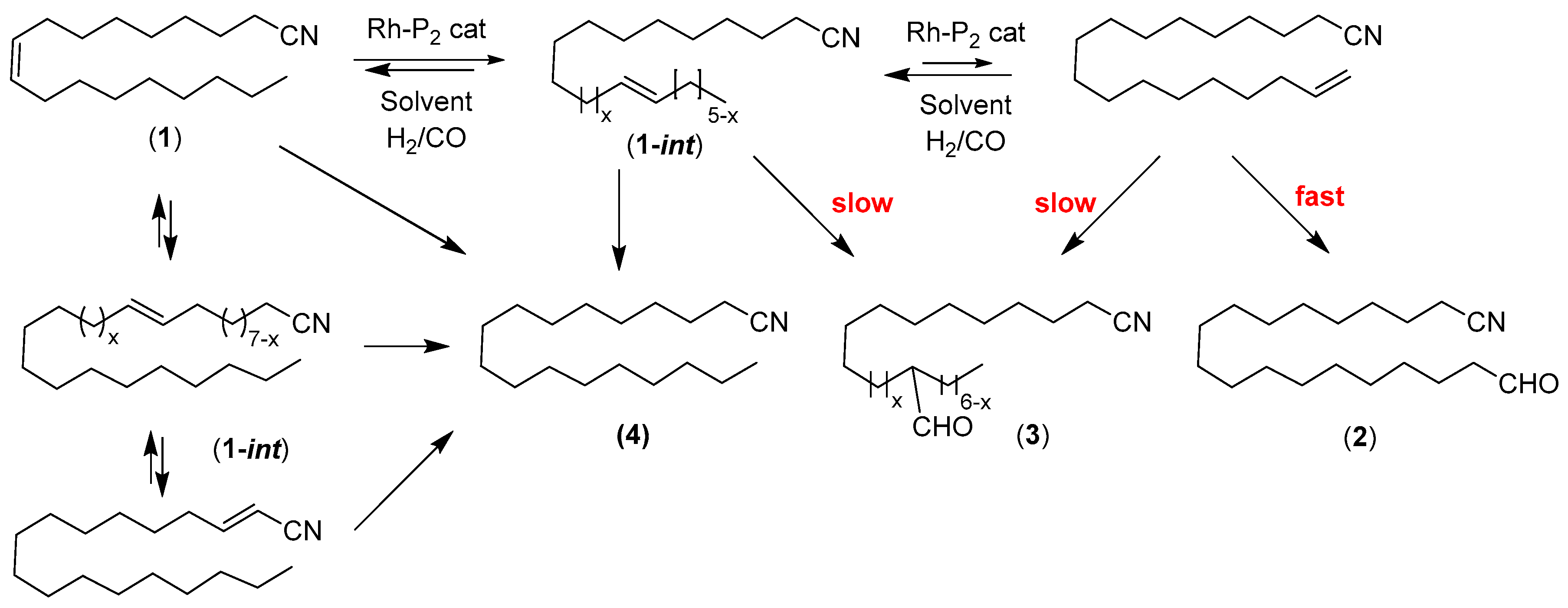
:
1. Introduction
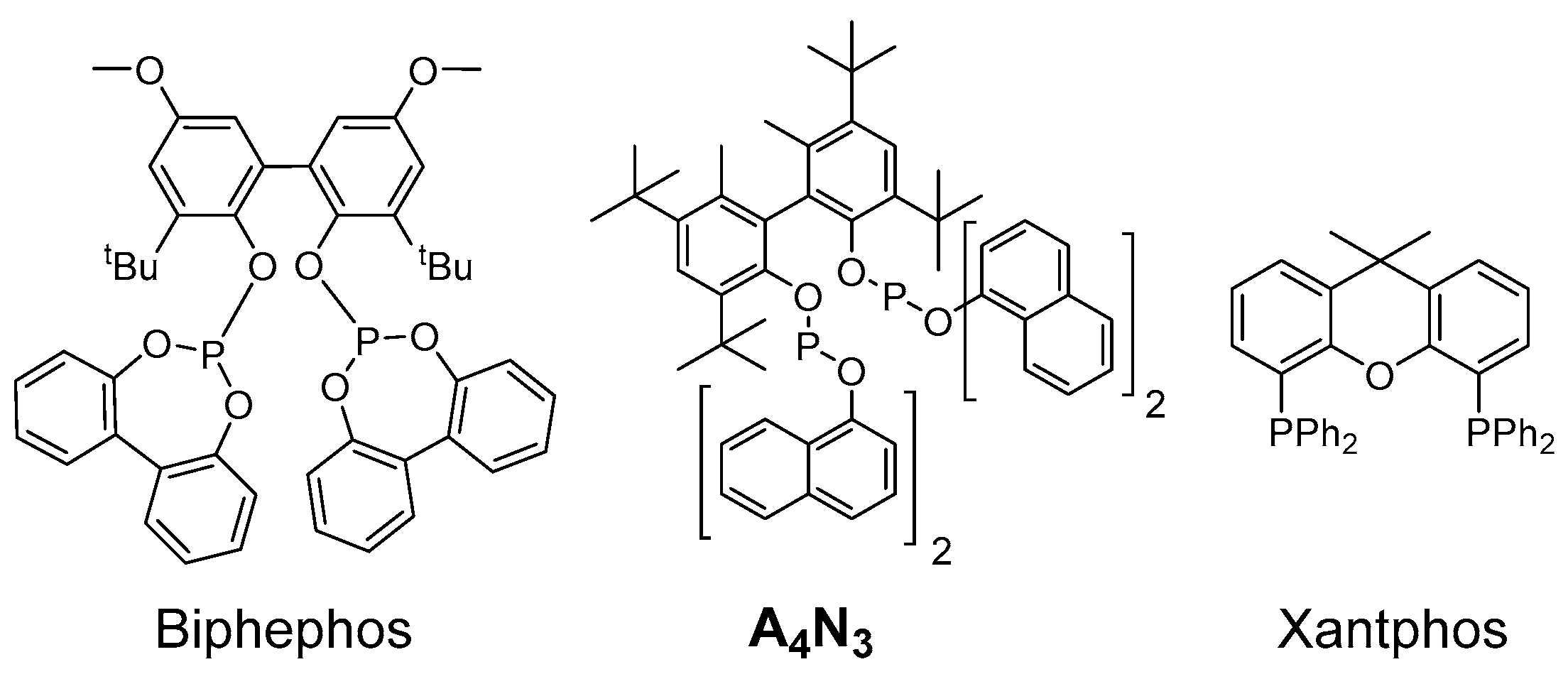
2. Results and Discussion
3. Materials and Methods
3.1. General Considerations
3.2. General Procedure for Hydroformylation Reactions with Oleonitrile
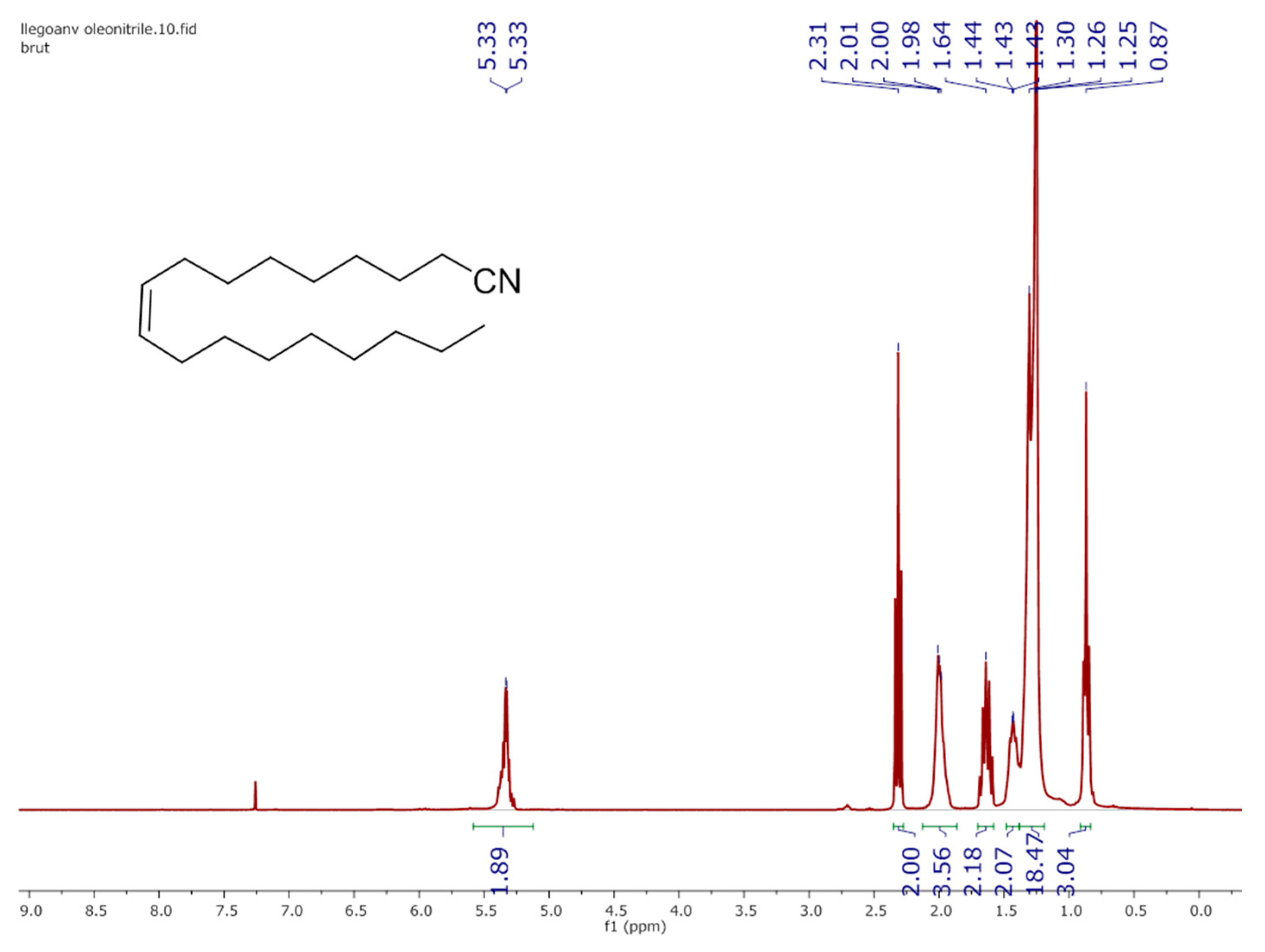
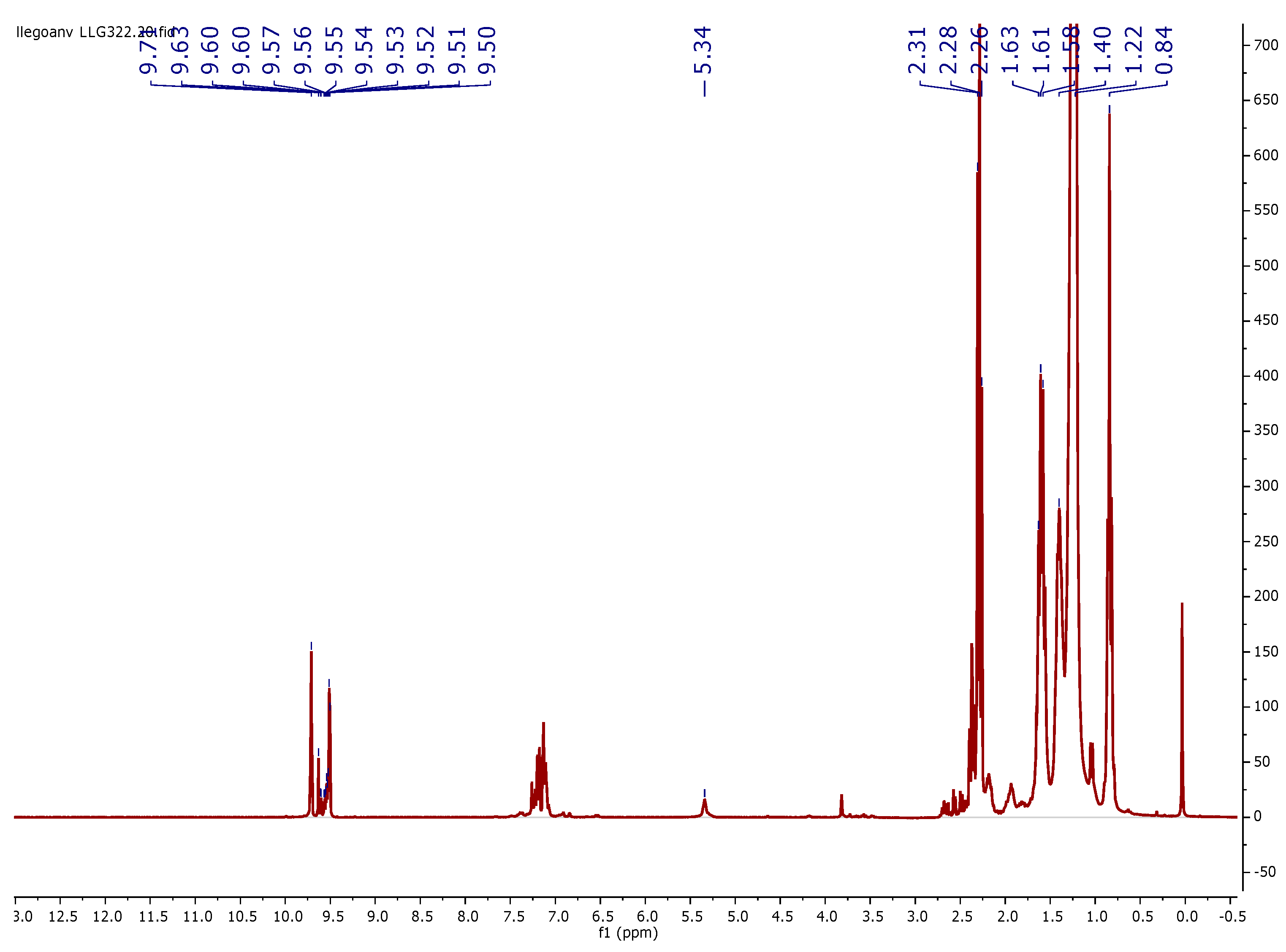
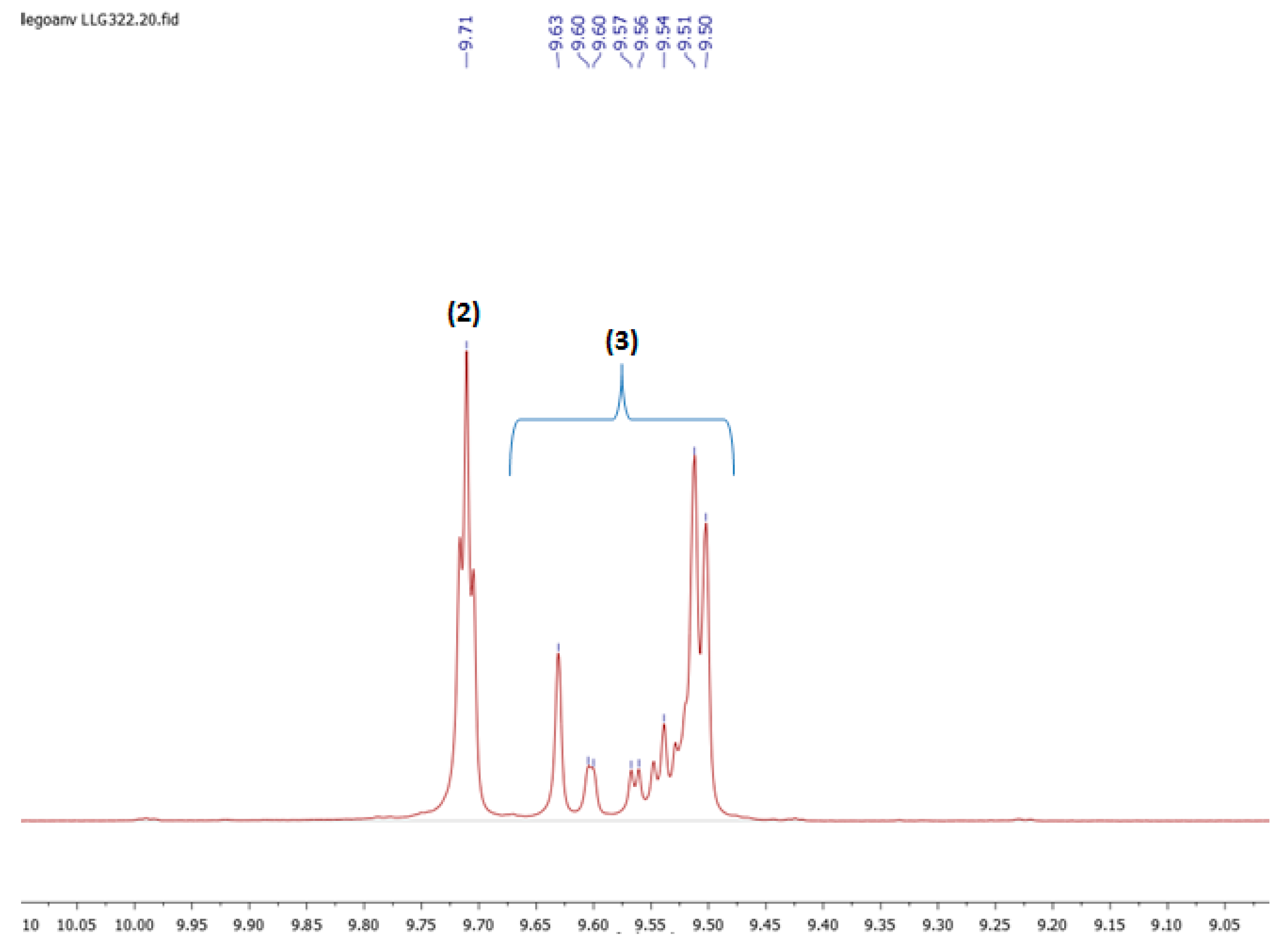
3.3. 1H NMR Characterization
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A




References
- Roelen, O. To Chemische Verwertungsgesellschaft Oberhausen m.b.H. German Patent DE 849548, 1938/1952; US Patent 2327066, 1943. Chem. Abstr. 1944, 38, 3631. [Google Scholar]
- Casey, C.P.; Paulsen, E.L.; Beuttenmueller, E.W.; Proft, B.R.; Petrovich, L.M.; Matter, B.A.; Powell, D.R. Electron withdrawing substituents on equatorial and apical phosphines have opposite effects on the regioselectivity of rhodium catalyzed hydroformylation. J. Am. Chem. Soc. 1997, 119, 11817–11825. [Google Scholar] [CrossRef]
- Klein, H.; Jackstell, R.; Wiese, K.-D.; Borgmann, C.; Beller, M. Highly selective catalyst systems for the hydroformylation of internal olefins to linear aldehydes. Angew. Chem. Int. Ed. 2001, 40, 3408–3411. [Google Scholar] [CrossRef]
- Van Rooy, A.; Kamer, P.C.J.; Van Leeuwen, P.W.N.M.; Goubitz, K.; Fraanje, J.; Veldman, N.; Spek, A.L. Bulky diphosphite modified rhodium catalyst; hydroformylation and characterization. Organometallics 1996, 15, 835–847. [Google Scholar] [CrossRef]
- Ohls, E.; Stein, M. The thermochemistry of long chain olefin isomers during hydroformylation. New J. Chem. 2017, 41, 7347–7355. [Google Scholar] [CrossRef]
- Roesle, P.; Durr, C.J.; Moller, H.M.; Cavallo, L.; Caporaso, L.; Mecking, S. Mechanistic features of isomerizing alkoxycarbonylation of methyl oleate. J. Am. Chem. Soc. 2012, 134, 17696–17703. [Google Scholar] [CrossRef] [PubMed]
- Vilches-Herrera, M.; Domke, L.; Börner, A. Isomerization-hydroformylation tandem reactions. ACS Catal. 2014, 4, 1706–1724. [Google Scholar] [CrossRef]
- Goldbach, V.; Roesle, P.; Mecking, S. Catalytic isomerizing ω-functionalization of fatty acids. ACS Catal. 2015, 5, 5951–5972. [Google Scholar] [CrossRef]
- Billig, E.; Abatjoglou, A.G.; Bryant, D.R.; Union Carbide Corporation. European Patent 213639. Chem. Abstr. 1987, 107, 7392. [Google Scholar]
- Billig, E.; Abatjoglou, A.G.; Bryant, D.R.; Union Carbide Corporation. European Patent 214622. Chem. Abstr. 1987, 107, 25126. [Google Scholar]
- Billig, E.; Abatjoglou, A.G.; Bryant, D.R.; Union Carbide Corporation. US Patent 4769498. Chem. Abstr. 1989, 111, 117287. [Google Scholar]
- Yan, Y.; Zhang, X.; Zhang, X. A tetraphosphorus ligand for highly regioselective isomerization-hydroformylation of internal olefins. J. Am. Chem. Soc. 2006, 128, 16058–16061. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Chie, Y.; Guan, Z.; Zhang, X. Highly regioselective isomerization—Hydroformylation of internal olefins to linear aldehyde using Rh complexes with tetraphosphorus ligands. Org. Lett. 2008, 10, 3469–3472. [Google Scholar] [CrossRef] [PubMed]
- Sémeril, D.; Matt, D.; Toupet, L. Highly regioselective hydroformylation with hemispherical chelators. Chem. Eur. J. 2008, 14, 7144–7155. [Google Scholar] [CrossRef] [PubMed]
- Behr, A.; Obst, D.; Schulte, C.; Schosser, T. Highly selective tandem isomerization-hydroformylation reaction of trans-4-octene to n-nonanal with rhodium-Biphephos catalysis. J. Mol. Catal. A Chem. 2003, 206, 179–184. [Google Scholar] [CrossRef]
- Behr, A.; Obst, D.; Schulte, C. Kinetik der isomerisierenden Hydroformylierung von trans-4-Octen. Chem. Ing. Tech. 2004, 76, 904–910. [Google Scholar] [CrossRef]
- Vanbésien, T.; Monflier, E.; Hapiot, F. Hydroformylation of vegetable oils: More than 50 years of technical innovation, successful research, and development. Eur. J. Lipid Sci. Technol. 2016, 118, 26–35. [Google Scholar] [CrossRef]
- Muilwijk, K.F.; Kamer, P.C.J.; Van Leeuwen, P.W.N.M. A bulky phosphite-modified rhodium catalyst for the hydroformylation of unsaturated fatty acid esters. J. Am. Oil Chem. Soc. 1997, 74, 223–228. [Google Scholar] [CrossRef]
- Kandanarachchi, P.; Guo, A.; Petrovic, Z. The hydroformylation of vegetable oils and model compounds by ligand modified rhodium catalysis. J. Mol. Catal. A Chem. 2002, 184, 65–71. [Google Scholar] [CrossRef]
- Benetskiy, E.; Lühr, S.; Vilches-Herrera, M.; Selent, D.; Jiao, H.; Domke, L.; Dyballa, K.; Franke, R.; Börner, A. Rhodium-catalyzed non-isomerizing hydroformylation of methyl oleate applying lactame-based phosphoramidite ligands. ACS Catal. 2014, 4, 2130–2136. [Google Scholar] [CrossRef]
- Frankel, E.N.; Metlin, S.; Rohwedder, W.K.; Wender, I. Hydroformylation of fatty unsaturated esters. J. Am. Oil Chem. Soc. 1969, 46, 133–138. [Google Scholar] [CrossRef]
- Behr, A.; Obst, D.; Westfechtel, A. Isomerizing hydroformylation of fatty acid esters: Formation of ω-aldehydes. Eur. J. Lipid Sci. Technol. 2005, 107, 213–219. [Google Scholar] [CrossRef]
- Pandey, S.; Chikkali, S.H. Highly regioselective isomerizing hydroformylation of long-chain internal olefins catalyzed by a rhodium bis(phosphite) complex. ChemCatChem 2015, 7, 3468–3471. [Google Scholar] [CrossRef]
- Pandey, S.; Shinde, D.R.; Chikkali, S.H. Isomerizing hydroformylation of cashew nut shell liquid. ChemCatChem 2017, 9, 3997–4004. [Google Scholar] [CrossRef]
- Yuki, Y.; Takahashi, K.; Tanaka, Y.; Nozaki, K. Tandem isomerization/hydroformylation/hydrogenation of internal alkenes to n-alcohols using Rh/Ru dual- or ternary-catalyst systems. J. Am. Chem. Soc. 2013, 135, 17393–17400. [Google Scholar] [CrossRef] [PubMed]
- Gaide, T.; Bianga, J.; Schlipköter, K.; Behr, A.; Vorholt, A.J. Linear selective isomerization/hydroformylation of unsaturated fatty acid methyl esters: A bimetallic approach. ACS Catal. 2017, 7, 4163–4171. [Google Scholar] [CrossRef]
- Furst, M.R.L.; Korkmaz, V.; Gaide, T.; Seidensticker, T.; Behr, A.; Vorholt, A.J. Tandem reductive hydroformylation of castor oil derived substrates and catalyst recycling by selective product crystallisation. ChemCatChem 2017, 9, 4319–4323. [Google Scholar] [CrossRef]
- Ternel, J.; Couturier, J.-L.; Dubois, J.-L.; Carpentier, J.-F. Rhodium-catalyzed tandem isomerization/hydroformylation of the bio-sourced 10-Undecenenitrile: Selective and productive catalysts for production of polyamide-12 precursor. Adv. Synth. Catal. 2013, 355, 3191–3204. [Google Scholar] [CrossRef]
- Ternel, J.; Couturier, J.-L.; Dubois, J.-L.; Carpentier, J.-F. Rhodium versus Iridium catalysts in the controlled tandem hydroformylation-isomerization of functionalized unsaturated fatty substrates. ChemCatChem 2015, 7, 513–520. [Google Scholar] [CrossRef]
- Le Goanvic, L.; Couturier, J.-L.; Dubois, J.-L.; Carpentier, J.-F. Ruthenium-catalyzed hydroformylation of the functional unsaturated fatty nitrile 10-undecenitrile. J. Mol. Catal. A Chem. 2016, 417, 116–121. [Google Scholar] [CrossRef]
- Van der Veen, L.A.; Kamer, P.C.J.; Van Leeuwen, P.W.N.M. Hydroformylation of internal olefins to linear aldehydes with novel rhodium catalysts. Angew. Chem. Int. Ed. 1999, 38, 336–338. [Google Scholar] [CrossRef]
- Van Leeuwen, P.W.N.M.; Kamer, P.C.J. Featuring Xantphos. Catal. Sci. Technol. 2018. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Solvent | [1]0/[Rh] (mol Ratio) | [L]/[Rh] (mol Ratio) | [1]0 (mol L−1) | Time (h) | Conversion 1 (%) 2 | 1 + 1-int (%) 3 | 2 + 3 (%) 3 | 4 (%) 3 | HF (%) 4 | 2/3 (l/b) 5 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | toluene | 200 | 10 | 0.1 | 96 | 98 | 2 | 65 | 33 | 66 | 21:79 |
| 2 | dioxane | 200 | 10 | 0.1 | 96 | 98 | 4 | 69 | 27 | 72 | 19:81 |
| 3 | toluene | 2000 | 10 | 1.0 | 71 | 21 | 76 | 15 | 9 | 62 | 25:75 |
| 4 | toluene | 200 | 2 | 0.1 | 112 | 99 | 1 | 89 | 9 | 90 | 11:89 |
| Entry | Temperature (°C) | p (bar) | Time (h) | Conversion 1 (%) 2 | 1 + 1-int (%) 3 | 2 + 3 (%) 3 | 4 (%) 3 | HF (%) 4 | 2/3 (l/b) 5 |
|---|---|---|---|---|---|---|---|---|---|
| 5 | 120 | 20 | 112 | 99 | 1 | 89 | 9 | 90 | 11:89 |
| 6 | 120 | 10 | 109 | 94 | 6 | 56 | 38 | 60 | 58:42 |
| 7 | 120 | 5 | 120 | 98 | 1 | 69 | 30 | 70 | 52:48 |
| 8 | 100 | 5 | 160 | 94 | 6 | 81 | 13 | 87 | 37:63 |
| 9 | 140 | 5 | 92 | 98 | 2 | 47 | 51 | 48 | 56:44 |
| Entry | Temperature (°C) | p (bar) | Time (h) | Conversion 1 (%) 2 | 1 + 1-int (%) 3 | 2 + 3 (%) 3 | 4 (%) 3 | HF (%) 4 | 2/3 (l/b) 5 |
|---|---|---|---|---|---|---|---|---|---|
| 10 | 120 | 10 | 112 | 98 | 2 | 77 | 21 | 79 | 46:54 |
| 11 | 120 | 20 | 48 | 94 | 5 | 74 | 21 | 78 | 42:58 |
| 12 | 140 | 10 | 110 | 92 | 7 | 49 | 44 | 53 | 46:54 |
| Entry | Catalytic System | Time (h) | Conversion 1 (%) 2 | 1 + 1-int (%) 3 | 2 + 3 (%) 3 | 4 (%) 3 | HF (%) 4 | 2/3 (l/b) 5 |
|---|---|---|---|---|---|---|---|---|
| 13 | Rh-Biphephos | 120 | 98 | 1 | 69 | 30 | 70 | 52:48 |
| 14 | Rh-A4N3 | 112 | 72 | 25 | 39 | 36 | 52 | 64:36 |
| 15 | Rh-Xantphos 6 | 96 | 22 | 78 | 15 | 7 | 68 | 11:89 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le Goanvic, L.; Ternel, J.; Couturier, J.-L.; Dubois, J.-L.; Carpentier, J.-F. Rhodium-Biphephos-Catalyzed Tandem Isomerization–Hydroformylation of Oleonitrile. Catalysts 2018, 8, 21. https://doi.org/10.3390/catal8010021
Le Goanvic L, Ternel J, Couturier J-L, Dubois J-L, Carpentier J-F. Rhodium-Biphephos-Catalyzed Tandem Isomerization–Hydroformylation of Oleonitrile. Catalysts. 2018; 8(1):21. https://doi.org/10.3390/catal8010021
Chicago/Turabian StyleLe Goanvic, Lucas, Jérémy Ternel, Jean-Luc Couturier, Jean-Luc Dubois, and Jean-François Carpentier. 2018. "Rhodium-Biphephos-Catalyzed Tandem Isomerization–Hydroformylation of Oleonitrile" Catalysts 8, no. 1: 21. https://doi.org/10.3390/catal8010021