Acidity-Reactivity Relationships in Catalytic Esterification over Ammonium Sulfate-Derived Sulfated Zirconia

 ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Results and Discussion
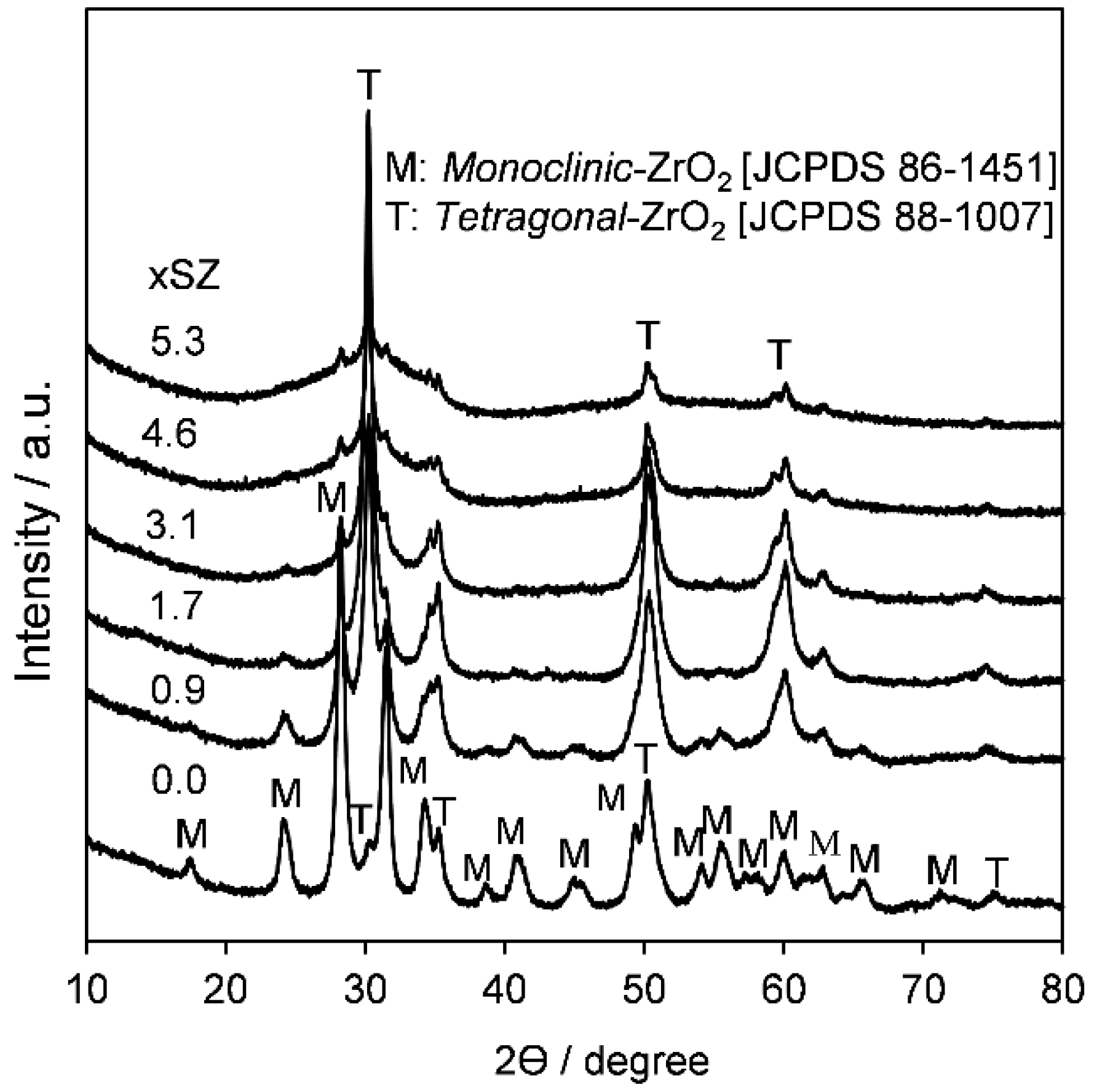
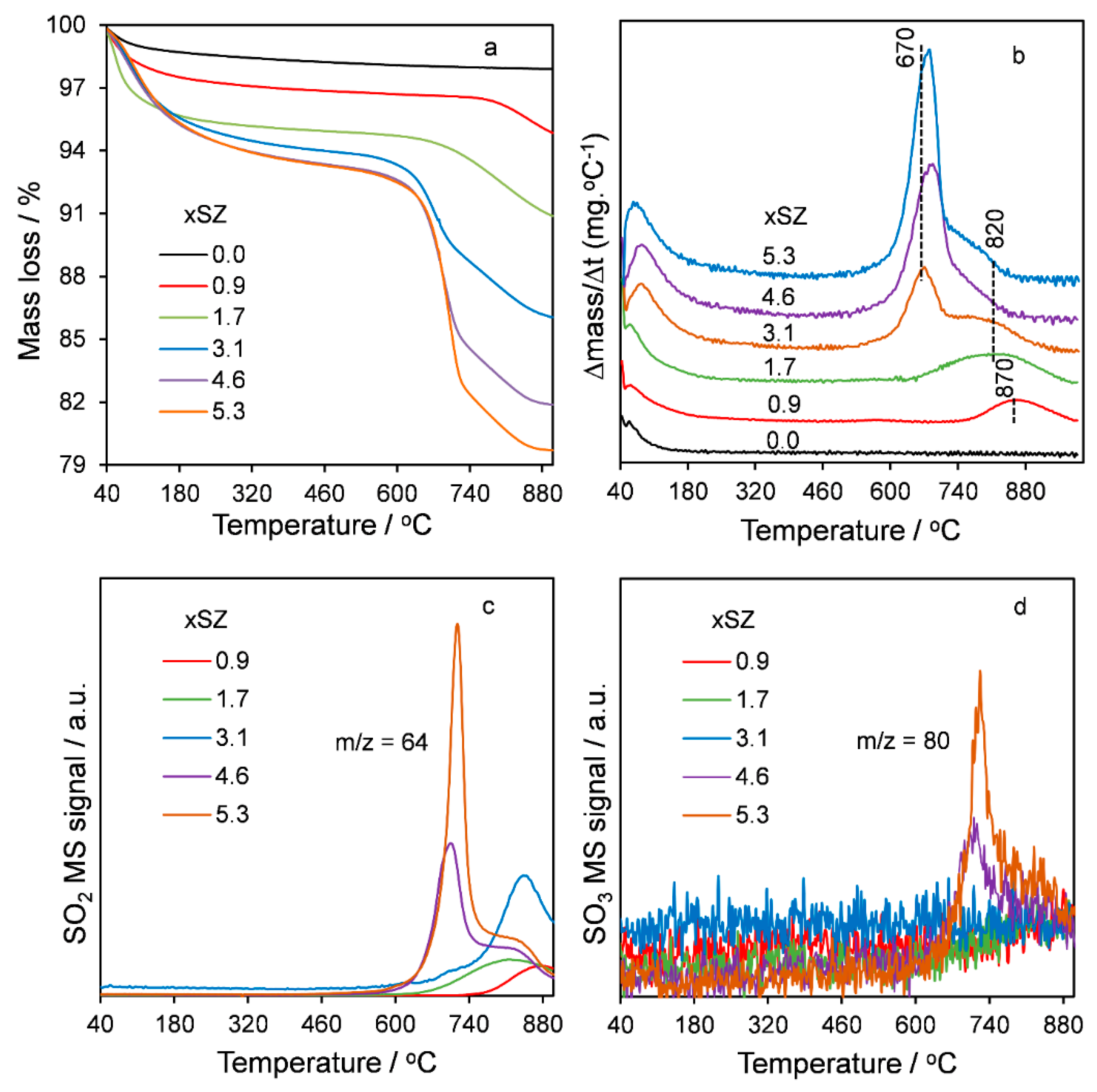
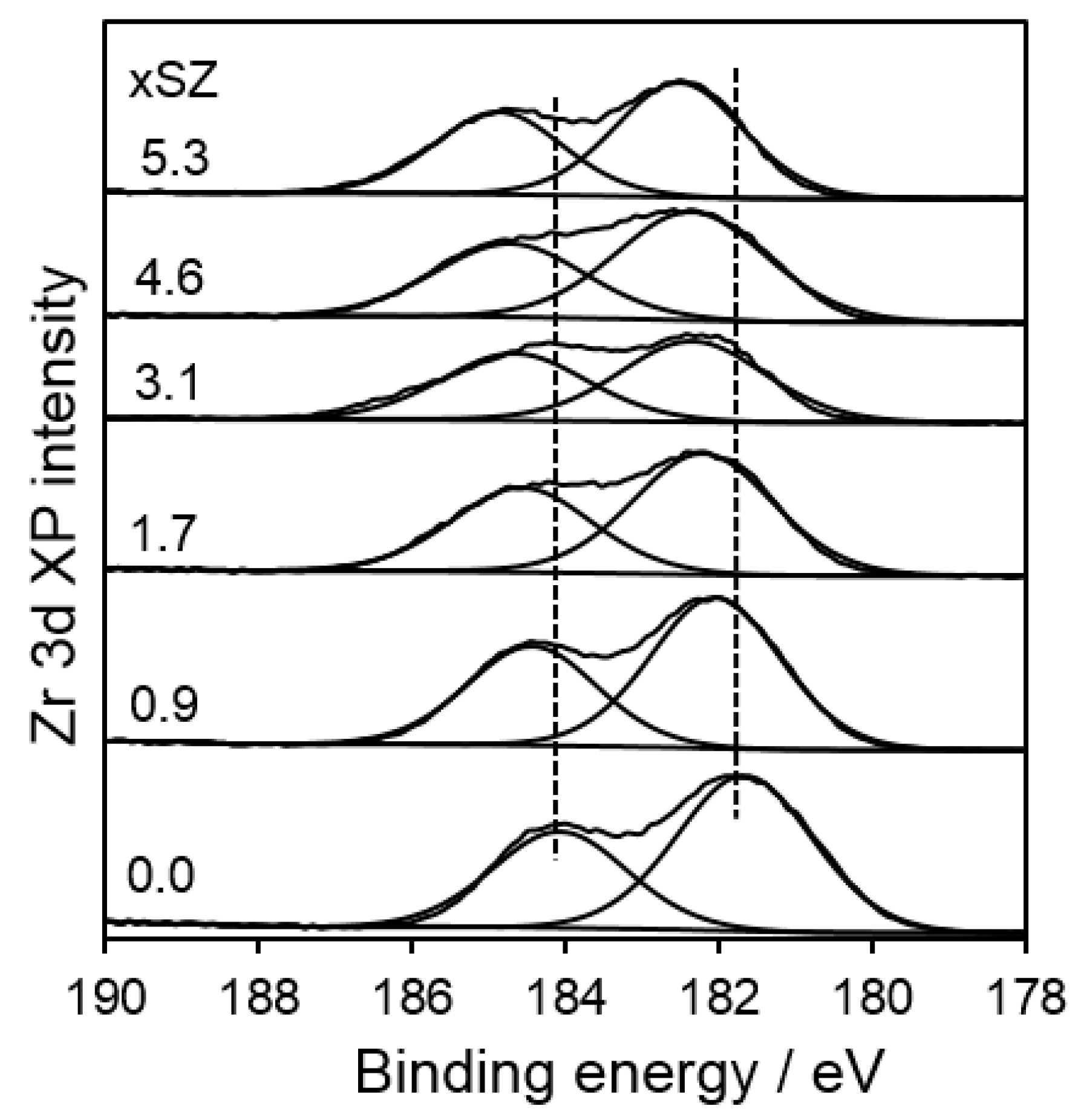
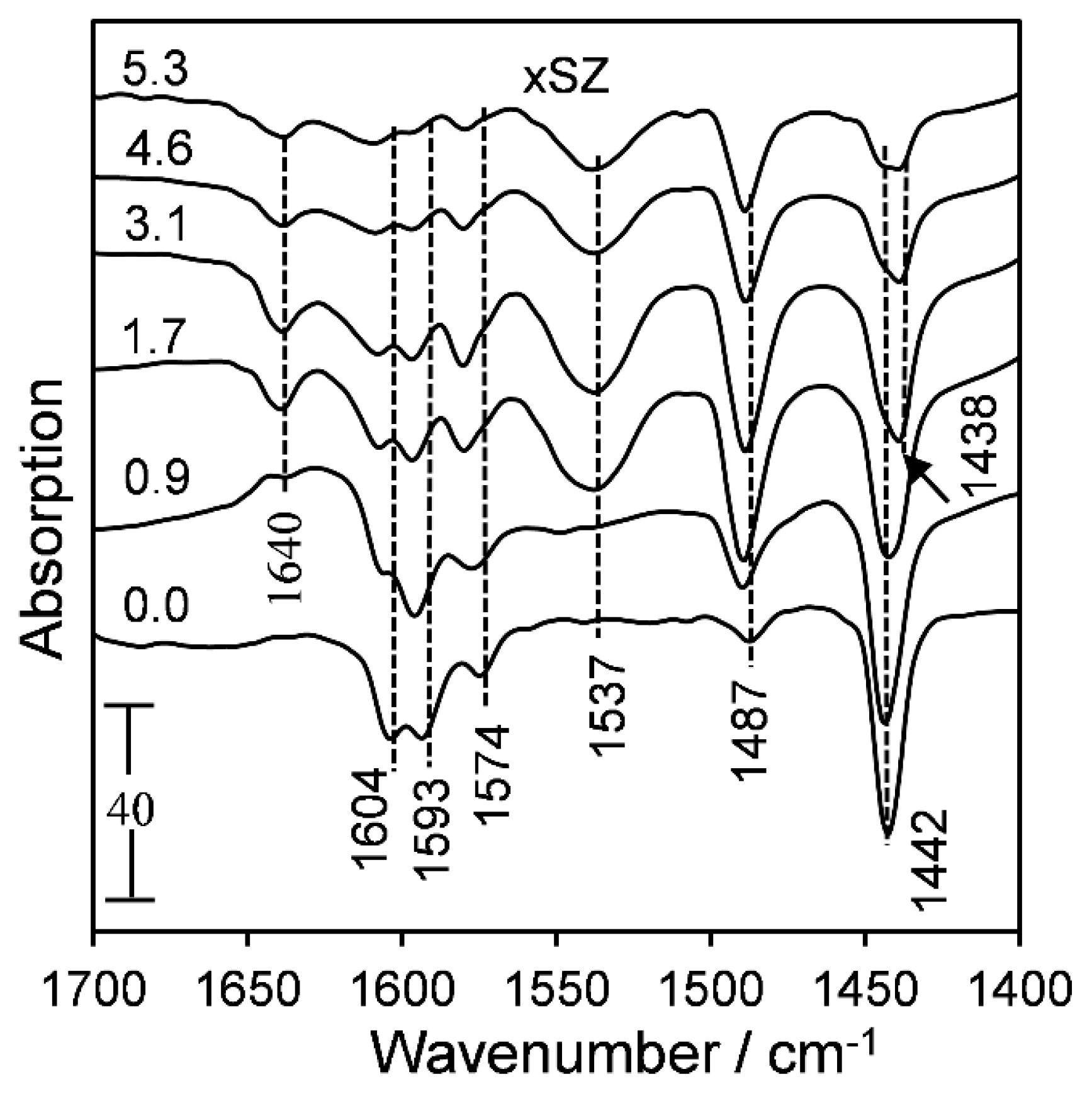
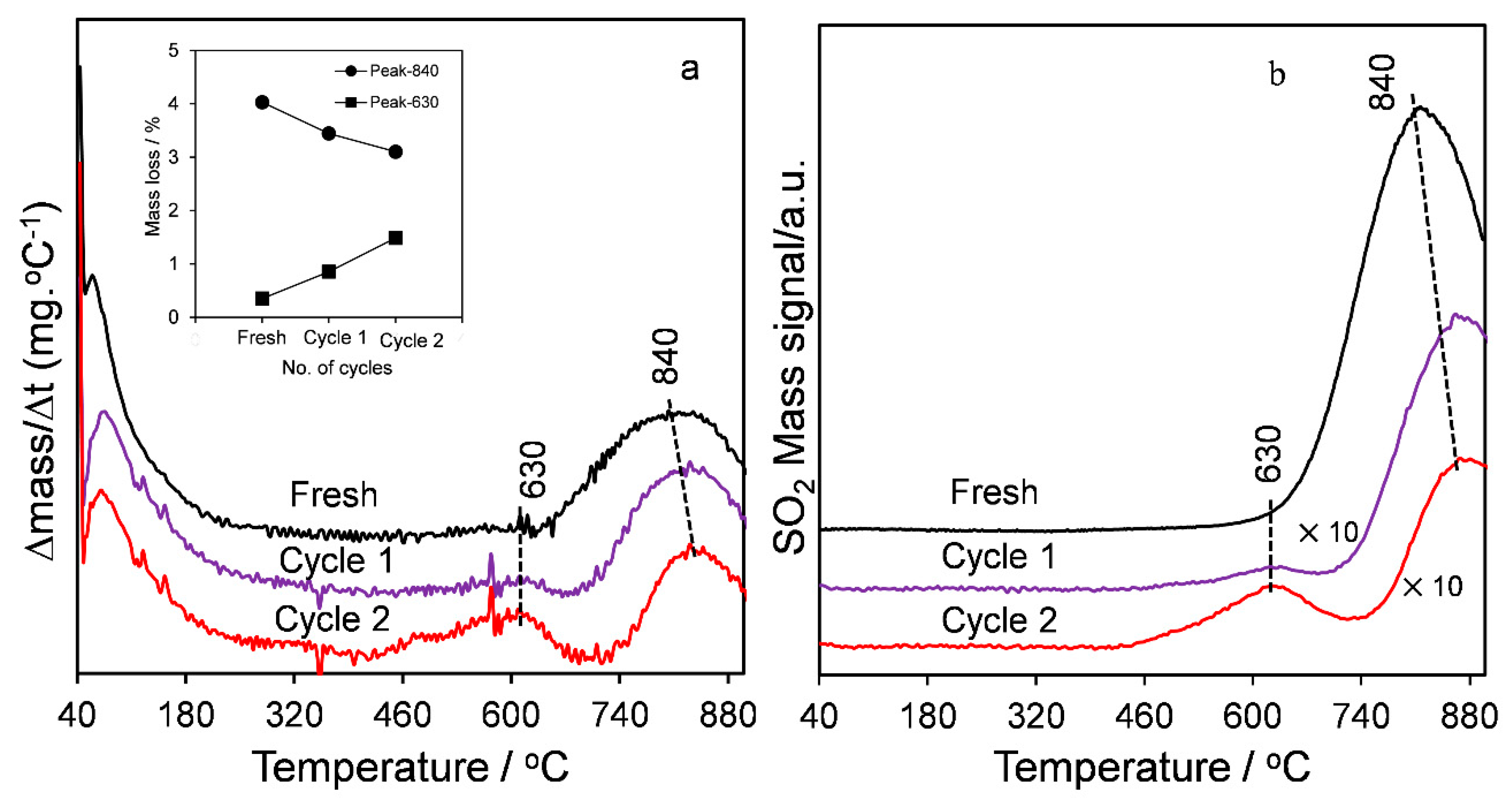
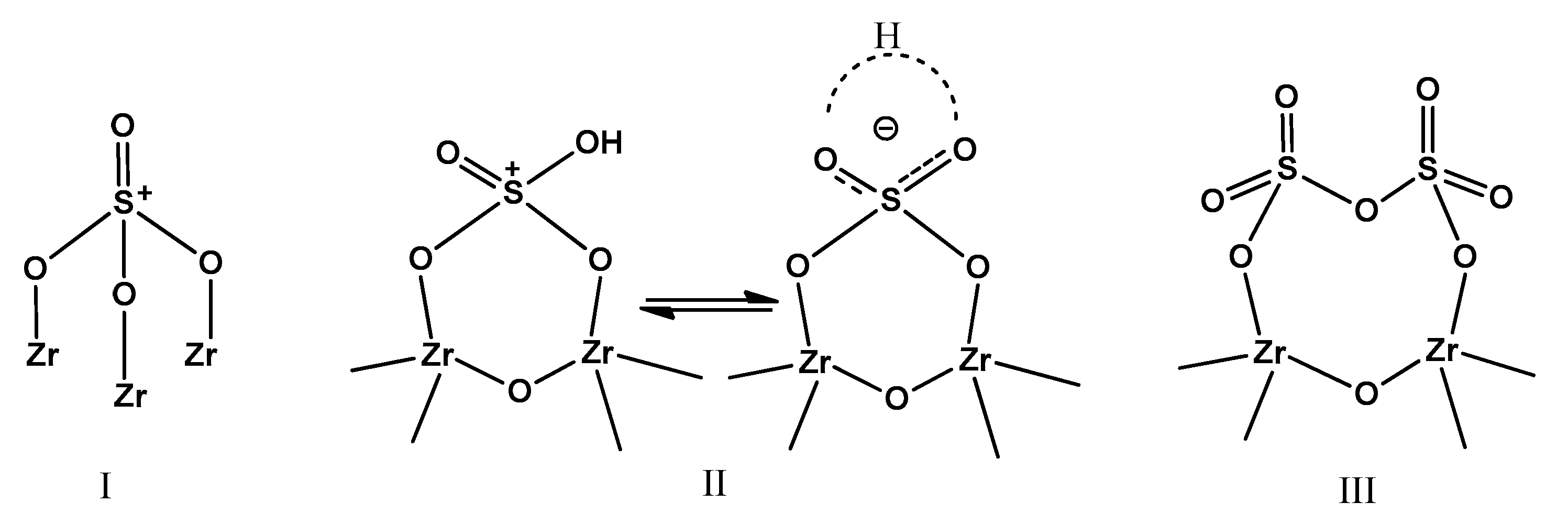
2.1. Catalyst Characterization
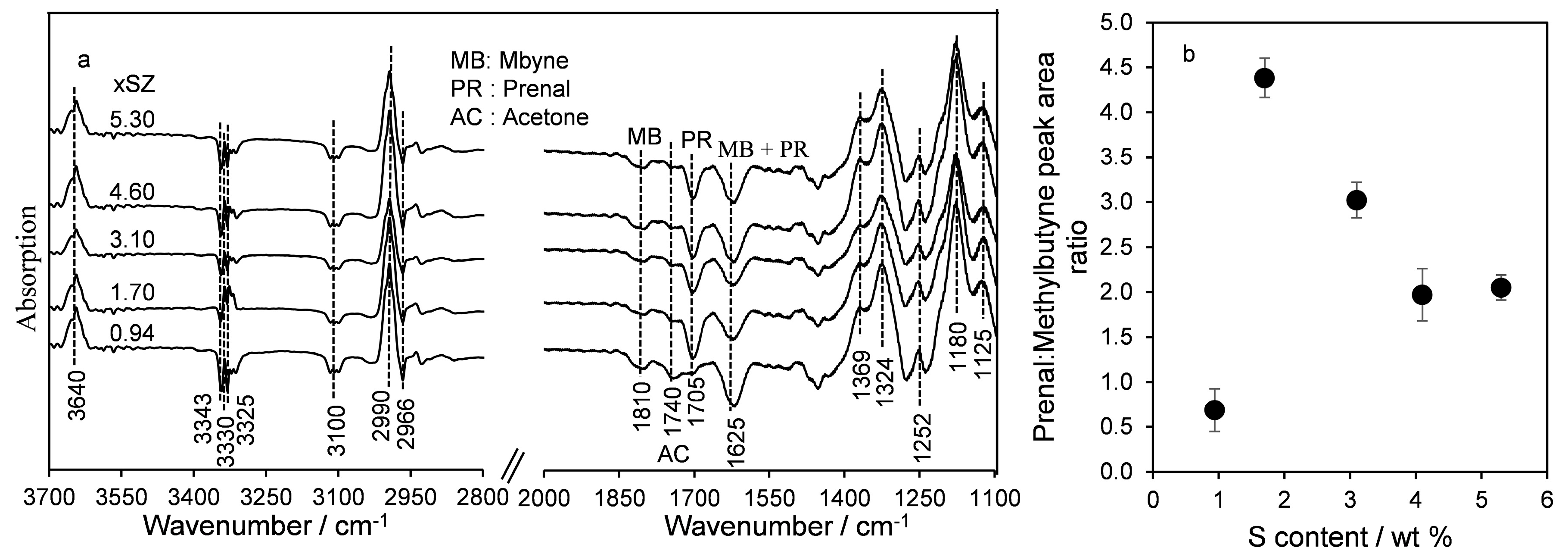
2.2. Surface Acidity
Methylbutynol Decomposition
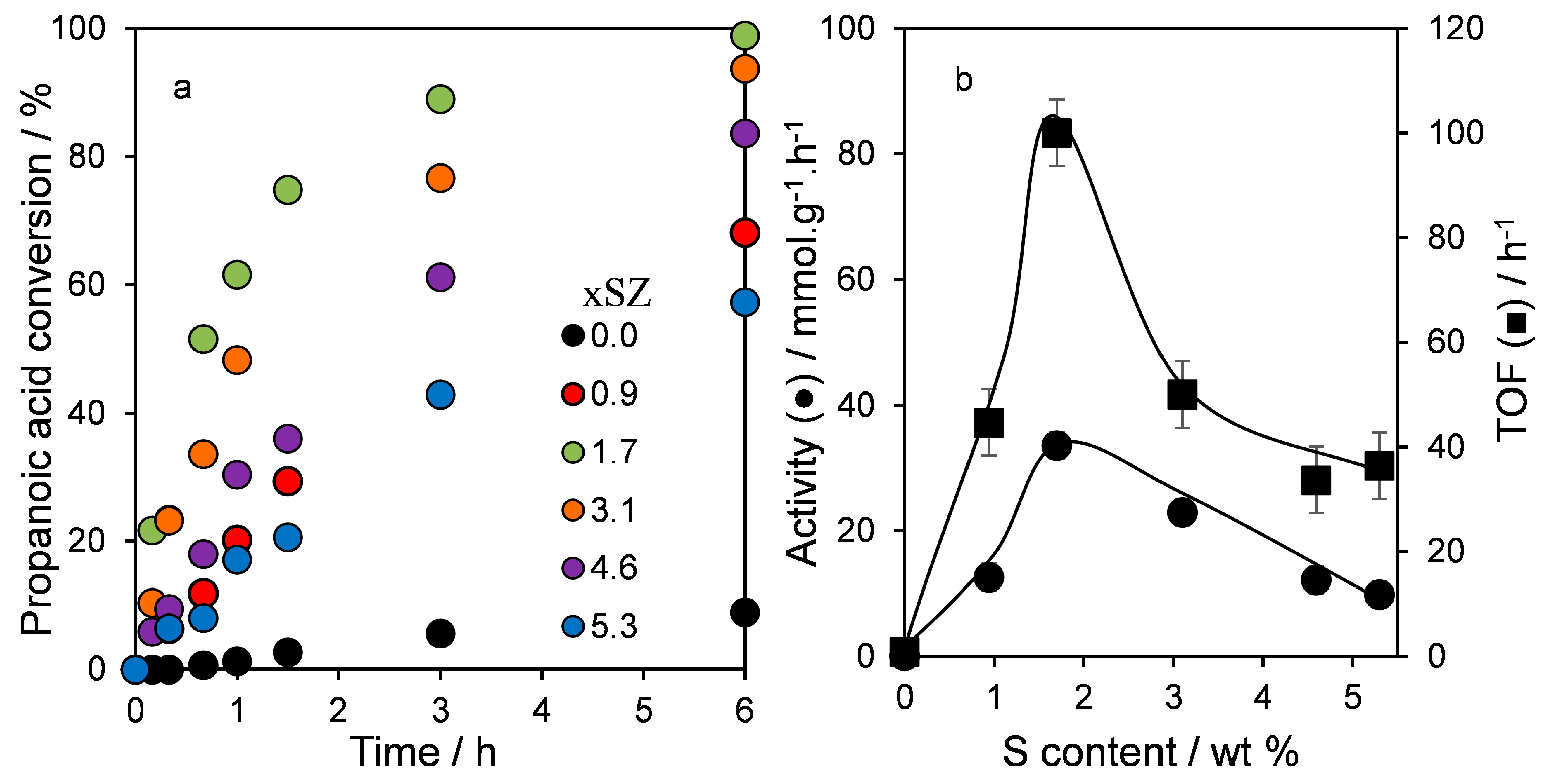
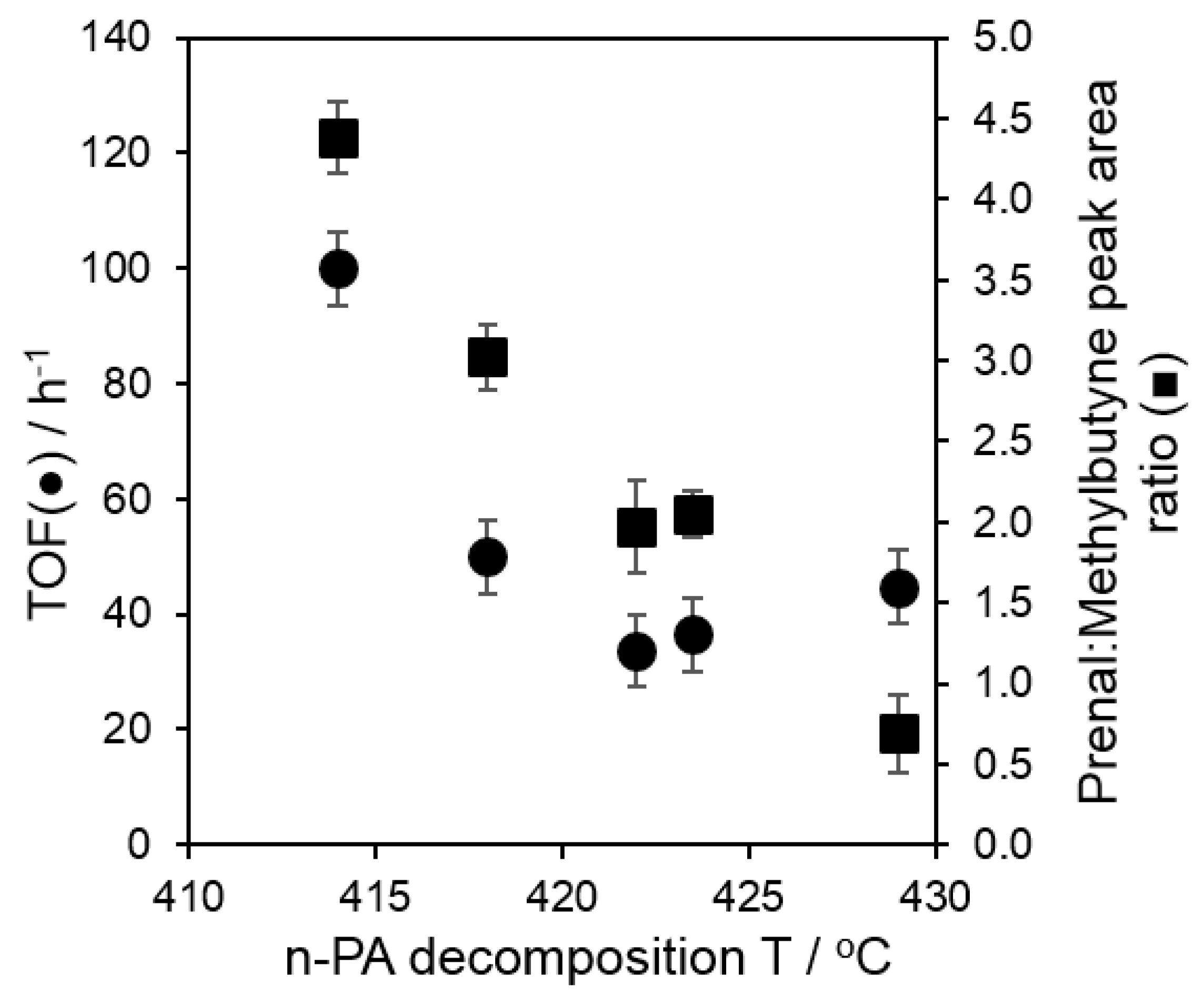
2.3. Catalytic Esterification of Propanoic Acid
3. Experimental
3.1. Catalyst Synthesis
3.2. Catalyst Characterization
3.3. Catalytic Esterification
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Osatiashtiani, A.; Lee, A.F.; Brown, D.R.; Melero, J.A.; Morales, G.; Wilson, K. Bifunctional SO4/ZrO2 catalysts for 5-hydroxymethylfufural (5-HMF) production from glucose. Catal. Sci. Technol. 2014, 4, 333–342. [Google Scholar] [CrossRef]
- Jung, K.T.; Bell, A.T. Effects of catalyst phase structure on the elementary processes involved in the synthesis of dimethyl carbonate from methanol and carbon dioxide over zirconia. Top. Catal. 2002, 20, 97–105. [Google Scholar] [CrossRef]
- Suwannakarn, K.; Lotero, E.; Goodwin, J.G.; Lu, C. Stability of sulfated zirconia and the nature of the catalytically active species in the transesterification of triglycerides. J. Catal. 2008, 255, 279–286. [Google Scholar] [CrossRef]
- Osatiashtiani, A.; Durndell, L.J.; Manayil, J.C.; Lee, A.F.; Wilson, K. Influence of alkyl chain length on sulfated zirconia catalysed batch and continuous esterification of carboxylic acids by light alcohols. Green Chem. 2016, 18, 5529–5535. [Google Scholar] [CrossRef]
- Yadav, G.; Sengupta, S. Friedel-crafts alkylation of diphenyl oxide with benzyl chloride over sulphated zirconia. Org. Proc. Res. Dev. 2002, 6, 256–262. [Google Scholar] [CrossRef]
- Tabora, J.E.; Davis, R.J. On the superacidity of sulfated zirconia catalysts for low-temperature isomerization of butane. J. Am. Chem. Soc. 1996, 118, 12240–12241. [Google Scholar] [CrossRef]
- Ecormier, M.A.; Wilson, K.; Lee, A.F. Structure-reactivity correlations in sulphated-zirconia catalysts for the isomerisation of α-pinene. J. Catal. 2003, 215, 57–65. [Google Scholar] [CrossRef]
- Ahmed, I.; Khan, N.A.; Mishra, D.K.; Lee, J.S.; Hwang, J.-S.; Jhung, S.H. Liquid-phase dehydration of sorbitol to isosorbide using sulfated titania as a solid acid catalyst. Chem. Eng. Sci. 2013, 93, 91–95. [Google Scholar] [CrossRef]
- Yadav, G.D.; Nair, J.J. Sulfated zirconia and its modified versions as promising catalysts for industrial processes. Microporous Mesoporous Mater. 1999, 33, 1–48. [Google Scholar] [CrossRef]
- Jin, T.; Yamaguchi, T.; Tanabe, K. Mechanism of acidity generation on sulfur-promoted metal oxides. J. Phys. Chem. 1986, 90, 4794–4796. [Google Scholar] [CrossRef]
- Babou, F.; Coudurier, G.; Vedrine, J.C. Acidic properties of sulfated zirconia: An infrared spectroscopic study. J. Catal. 1995, 152, 341–349. [Google Scholar] [CrossRef]
- Chen, F.; Coudurier, G.; Joly, J.-F.; Vedrine, J. Superacid and catalytic properties of sulfated zirconia. J. Catal. 1993, 143, 616–626. [Google Scholar] [CrossRef]
- Arata, K.; Hino, M. Preparation of superacids by metal oxides and their catalytic action. Mater. Chem. Phys. 1990, 26, 213–237. [Google Scholar] [CrossRef]
- Clearfield, A.; Serrette, G.; Khazi-Syed, A. Nature of hydrous zirconia and sulfated hydrous zirconia. Catal. Today 1994, 20, 295–312. [Google Scholar] [CrossRef]
- Venkatesh, K.R.; Hu, J.; Dogan, C.; Tierney, J.W.; Wender, I. Sulfated metal oxides and related solid acids: Comparison of protonic acid strengths. Energy Fuels 1995, 9, 888–893. [Google Scholar] [CrossRef]
- Bensitel, M.; Saur, O.; Lavalley, J.-C.; Morrow, B. An infrared study of sulfated zirconia. Mater. Chem. Phys. 1988, 19, 147–156. [Google Scholar] [CrossRef]
- Morterra, C.; Cerrato, G.; Bolis, V. Lewis and brønsted acidity at the surface of sulfate-doped ZrO2 catalysts. Catal. Today 1993, 17, 505–515. [Google Scholar] [CrossRef]
- Stichert, W.; Schüth, F. Synthesis of catalytically active high surface area monoclinic sulfated zirconia. J. Catal. 1998, 174, 242–245. [Google Scholar] [CrossRef]
- Stichert, W.; Schüth, F.; Kuba, S.; Knözinger, H. Monoclinic and tetragonal high surface area sulfated zirconias in butane isomerization: Co adsorption and catalytic results. J. Catal. 2001, 198, 277–285. [Google Scholar] [CrossRef]
- Son, J.R.; Gwon, T.D.; Kim, S.B. Characterization of zirconium sulfate supported on zirconia and activity for acid catalysis. Bull. Korean Chem. Soc. 2001, 22, 1309–1315. [Google Scholar]
- Reddy, B.M.; Patil, M.K. Organic syntheses and transformations catalyzed by sulfated zirconia. Chem. Rev. 2009, 109, 2185–2208. [Google Scholar] [CrossRef] [PubMed]
- Walrafen, G.; Irish, D.; Young, T. Raman spectral studies of molten potassium bisulfate and vibrational frequencies of S2O7 groups. J. Chem. Phys. 1962, 37, 662–670. [Google Scholar] [CrossRef]
- Moulder, J.F.; Stickle, W.F.; Sobol, P.E.; Bomben, K.D. Handbook of X-ray Photoelectron Spectroscopy: A Reference Book of Standard Spectra for Identification and Interpretation of Xps Data, 3rd ed.; Physical Electronics Eden Prairie: Chanhassen, MN, USA, 1992; p. 109. [Google Scholar]
- Chen, S.; Yin, Y.; Wang, D.; Liu, Y.; Wang, X. Structures, growth modes and spectroscopic properties of small zirconia clusters. J. Cryst. Growth 2005, 282, 498–505. [Google Scholar] [CrossRef]
- Qian, Z.; Shi, J. Characterization of pure and doped zirconia nanoparticles with infrared transmission spectroscopy. Nanostruct. Mater. 1998, 10, 235–244. [Google Scholar] [CrossRef]
- Mishra, M.K.; Tyagi, B.; Jasra, R.V. Synthesis and characterization of nano-crystalline sulfated zirconia by sol-gel method. J. Mol. Catal. A-Chem. 2004, 223, 61–65. [Google Scholar] [CrossRef]
- Deshmane, V.G.; Adewuyi, Y.G. Mesoporous nanocrystalline sulfated zirconia synthesis and its application for ffa esterification in oils. Appl. Catal. A-Gen. 2013, 462, 196–206. [Google Scholar] [CrossRef]
- Köck, E.-M.; Kogler, M.; Bielz, T.; Klötzer, B.; Penner, S. In situ FT-IR spectroscopic study of CO2 and CO adsorption on Y2O3, ZrO2, and yttria-stabilized ZrO2. J. Phys. Chem. C 2013, 117, 17666–17673. [Google Scholar] [CrossRef] [PubMed]
- Dobson, K.D.; McQuillan, A.J. An infrared spectroscopic study of carbonate adsorption to zirconium dioxide sol-gel films from aqueous solutions. Langmuir 1997, 13, 3392–3396. [Google Scholar] [CrossRef]
- Carniti, P.; Gervasini, A.; Biella, S.; Auroux, A. Intrinsic and effective acidity study of niobic acid and niobium phosphate by a multitechnique approach. Chem. Mater. 2005, 17, 6128–6136. [Google Scholar] [CrossRef]
- Hess, A.; Kemnitz, E. Surface acidity and catalytic behavior of modified zirconium and titanium dioxides. Appl. Catal. A-Gen. 1997, 149, 373–389. [Google Scholar] [CrossRef]
- Zaki, M.I.; Hasan, M.A.; Al-Sagheer, F.A.; Pasupulety, L. In situ ftir spectra of pyridine adsorbed on SiO2-Al2O3, TiO2, ZrO2 and CeO2: General considerations for the identification of acid sites on surfaces of finely divided metal oxides. Colloids Surf. A Physicochem. Eng. Asp. 2001, 190, 261–274. [Google Scholar] [CrossRef]
- Platon, A.; Thomson, W.J. Quantitative lewis/brönsted ratios using drifts. Ind. Eng. Chem. Res. 2003, 42, 5988–5992. [Google Scholar] [CrossRef]
- Waqif, M.; Bachelier, J.; Saur, O.; Lavalley, J.-C. Acidic properties and stability of sulfate-promoted metal oxides. J. Mol. Catat. 1992, 72, 127–138. [Google Scholar] [CrossRef]
- Matsuhashi, H.; Motoi, H.; Arata, K. Determination of acid strength of solid superacids by temperature programmed desorption using pyridine. Catal. Lett. 1994, 26, 325–328. [Google Scholar] [CrossRef]
- Farneth, W.; Gorte, R. Methods for characterizing zeolite acidity. Chem. Rev. 1995, 95, 615–635. [Google Scholar] [CrossRef]
- Bourikas, K.; Vakros, J.; Kordulis, C.; Lycourghiotis, A. Potentiometric mass titrations: Experimental and theoretical establishment of a new technique for determining the point of zero charge (PZC) of metal (HYDR) oxides. J. Phys. Chem. B 2003, 107, 9441–9451. [Google Scholar] [CrossRef]
- Kustov, L.; Kazansky, V.; Figueras, F.; Tichit, D. Investigation of the acidic properties of ZrO2 modified by SO2-4 anions. J. Catal. 1994, 150, 143–149. [Google Scholar] [CrossRef]
- Adeeva, V.; Dehaan, J.; Janchen, J.; Lei, G.; Schunemann, V.; Vandeven, L.; Sachtler, W.; Vansanten, R. Acid sites in sulfated and metal-promoted zirconium dioxide catalysts. J. Catal. 1995, 151, 364–372. [Google Scholar] [CrossRef]
- Penzien, J.; Abraham, A.; van Bokhoven, J.A.; Jentys, A.; Müller, T.E.; Sievers, C.; Lercher, J.A. Generation and characterization of well-defined Zn2+ lewis acid sites in ion exchanged zeolite bea. J. Phys. Chem. B 2004, 108, 4116–4126. [Google Scholar] [CrossRef]
- Audry, F.; Hoggan, P.; Saussey, J.; Lavalley, J.; Lauron-Pernot, H.; Le Govic, A. Infrared study and quantum calculations of the conversion of methylbutynol into hydroxymethylbutanone on zirconia. J. Catal. 1997, 168, 471–481. [Google Scholar] [CrossRef]
- Lauron-Pernot, H.; Luck, F.; Popa, J. Methylbutynol: A new and simple diagnostic tool for acidic and basic sites of solids. Appl. Catal. 1991, 78, 213–225. [Google Scholar] [CrossRef]
- Lauron-Pernot, H. Evaluation of surface acido-basic properties of inorganic-based solids by model catalytic alcohol reaction networks. Catal. Rev. 2006, 48, 315–361. [Google Scholar] [CrossRef]
- Mekhemer, G.A.; Zaki, M.I. In situ ftir spectroscopic assessment of methylbutynol catalytic conversion products in relation to the surface acid-base properties of systematically modified aluminas. Surf. Sci. 2016, 652, 269–277. [Google Scholar] [CrossRef]
- Dacquin, J.-P.; Cross, H.E.; Brown, D.R.; Duren, T.; Williams, J.J.; Lee, A.F.; Wilson, K. Interdependent lateral interactions, hydrophobicity and acid strength and their influence on the catalytic activity of nanoporous sulfonic acid silicas. Green Chem. 2010, 12, 1383–1391. [Google Scholar] [CrossRef]
- Satyarthi, J.K.; Srinivas, D.; Ratnasamy, P. Influence of surface hydrophobicity on the esterification of fatty acids over solid catalysts. Energy Fuels 2010, 24, 2154–2161. [Google Scholar] [CrossRef]
- Lee, A.F.; Bennett, J.A.; Manayil, J.C.; Wilson, K. Heterogeneous catalysis for sustainable biodiesel production via esterification and transesterification. Chem. Soc. Rev. 2014, 43, 7887–7916. [Google Scholar] [CrossRef] [PubMed]
- Corma, A. Solid acid catalysts. Curr. Opin. Solid St. Mater. Sci. 1997, 2, 63–75. [Google Scholar] [CrossRef]
- Kiss, A.A.; Dimian, A.C.; Rothenberg, G. Solid acid catalysts for biodiesel production-towards sustainable energy. Adv. Synth. Catal. 2006, 348, 75–81. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of gases in multimolecular layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Peri, J.B.; Hannan, R.B. Surface hydroxyl groups on γ-alumina1. J. Phys. Chem. 1960, 64, 1526–1530. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
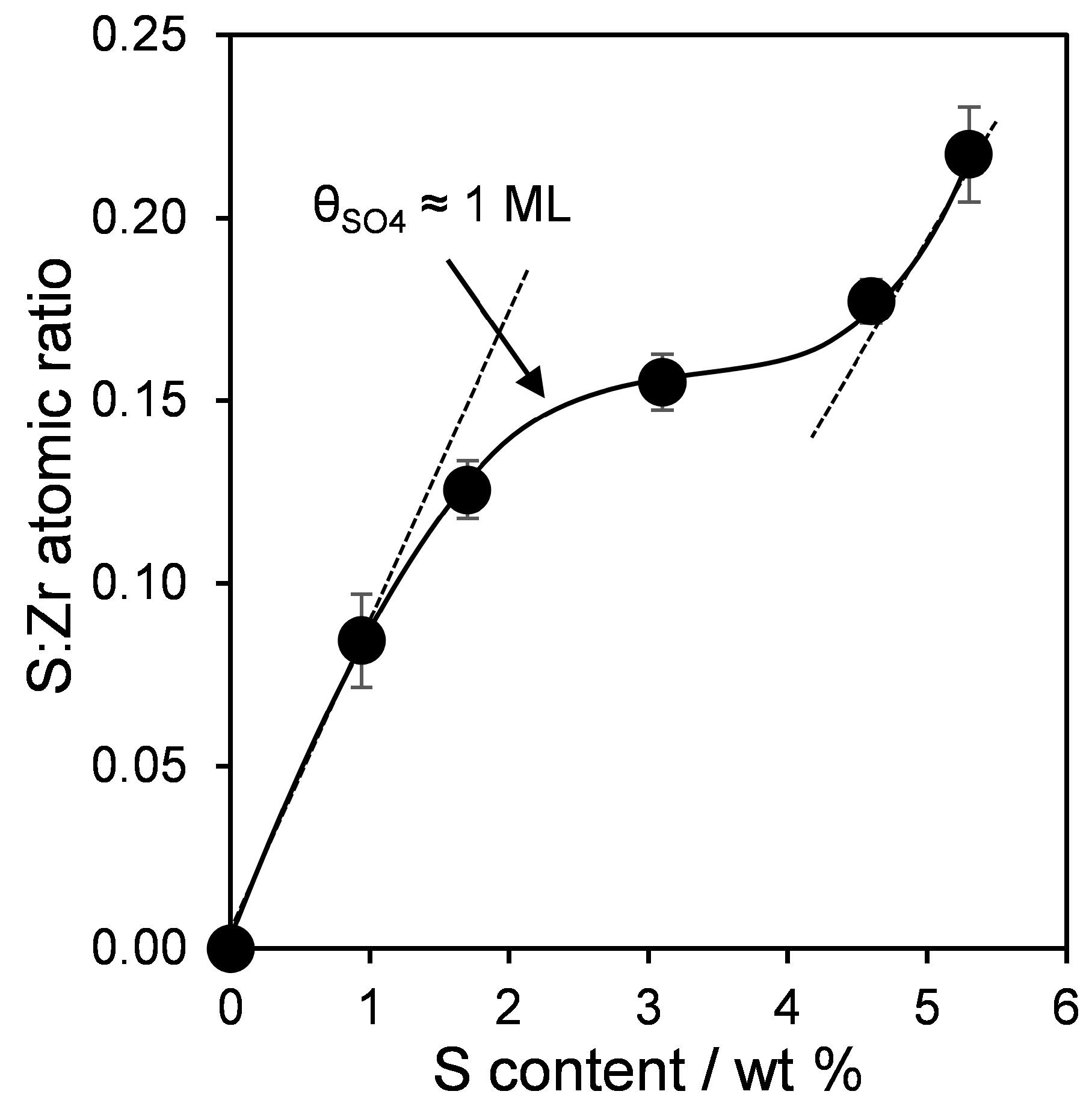
| Sample | Surface Area (m2·g−1) | Bulk S Loading a (wt %) | Surface Composition b (atom %) | S:Zr c Atomic Ratio | |
|---|---|---|---|---|---|
| S | Zr | ||||
| 0SZ | 40 | 0.0 | 0.0 | 30.9 | 0.00 |
| 1SZ | 100 | 0.9 | 2.5 | 29.7 | 0.08 (0.03) |
| 2SZ | 121 | 1.7 | 3.5 | 27.9 | 0.13 (0.05) |
| 3SZ | 90 | 3.1 | 4.0 | 26.1 | 0.16 (0.10) |
| 4SZ | 49 | 4.6 | 4.7 | 26.6 | 0.18 (0.14) |
| 5SZ | 34 | 5.3 | 5.6 | 25.8 | 0.22 (0.16) |
| S Content (wt %) | Mass Loss (±0.1) (%) | Evolved Species b | |
|---|---|---|---|
| 40–350 °C | 600–1000 °C a | ||
| 0.0 | 1.42 | 0.21 | H2O (80–350 °C) |
| 0.9 | 2.53 | 1.94 (0.97) | H2O, SO2 (840–870 °C) |
| 1.7 | 3.42 | 3.73 (1.86) | |
| 3.1 | 5.00 | 7.45 (3.73) | |
| 4.6 | 5.80 | 9.99 (4.99) | H2O, SO2, SO3 (670 °C) |
| 5.3 | 5.30 | 11.89 (5.95) | |
| S Loading (wt %) | Brønsted:Lewis Ratio a | pHzcp | Acid Loading b (µmol·g−1) | MBOH Conversion c (%) |
|---|---|---|---|---|
| 0.0 | - | 4.68 | 68 | 54 |
| 0.9 | 0.05 | 3.33 | 282 | 76 |
| 1.7 | 0.86 | 2.82 | 336 | 90 |
| 3.1 | 1.01 | 2.90 | 458 | 73 |
| 4.6 | 1.13 | 2.96 | 362 | 74 |
| 5.3 | 1.57 | 2.96 | 270 | 59 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rabee, A.I.M.; Mekhemer, G.A.H.; Osatiashtiani, A.; Isaacs, M.A.; Lee, A.F.; Wilson, K.; Zaki, M.I. Acidity-Reactivity Relationships in Catalytic Esterification over Ammonium Sulfate-Derived Sulfated Zirconia. Catalysts 2017, 7, 204. https://doi.org/10.3390/catal7070204
Rabee AIM, Mekhemer GAH, Osatiashtiani A, Isaacs MA, Lee AF, Wilson K, Zaki MI. Acidity-Reactivity Relationships in Catalytic Esterification over Ammonium Sulfate-Derived Sulfated Zirconia. Catalysts. 2017; 7(7):204. https://doi.org/10.3390/catal7070204
Chicago/Turabian StyleRabee, Abdallah I. M., Gamal A. H. Mekhemer, Amin Osatiashtiani, Mark A. Isaacs, Adam F. Lee, Karen Wilson, and Mohamed I. Zaki. 2017. "Acidity-Reactivity Relationships in Catalytic Esterification over Ammonium Sulfate-Derived Sulfated Zirconia" Catalysts 7, no. 7: 204. https://doi.org/10.3390/catal7070204