
Supported Catalysts for CO2 Methanation: A Review


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
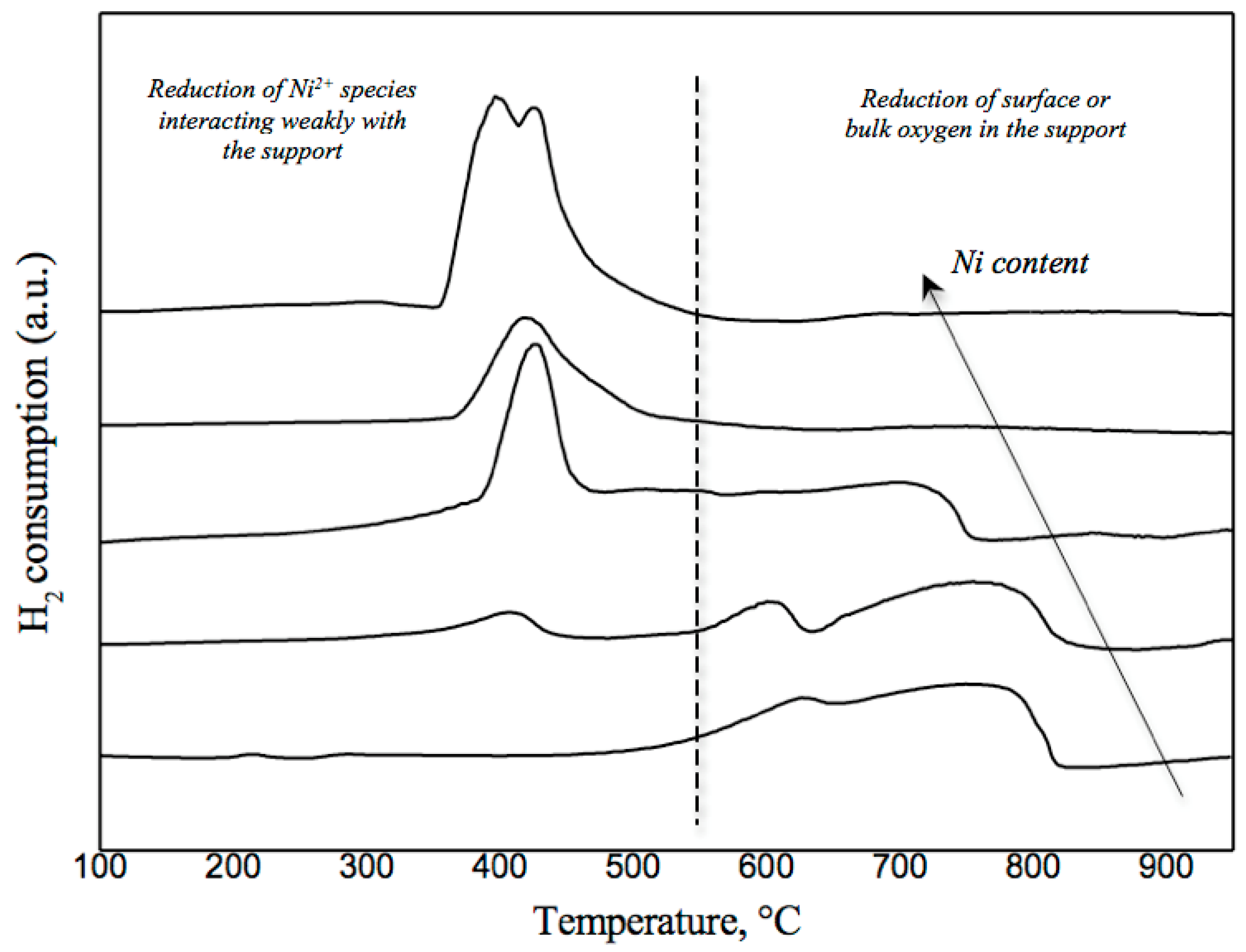{kind=link}
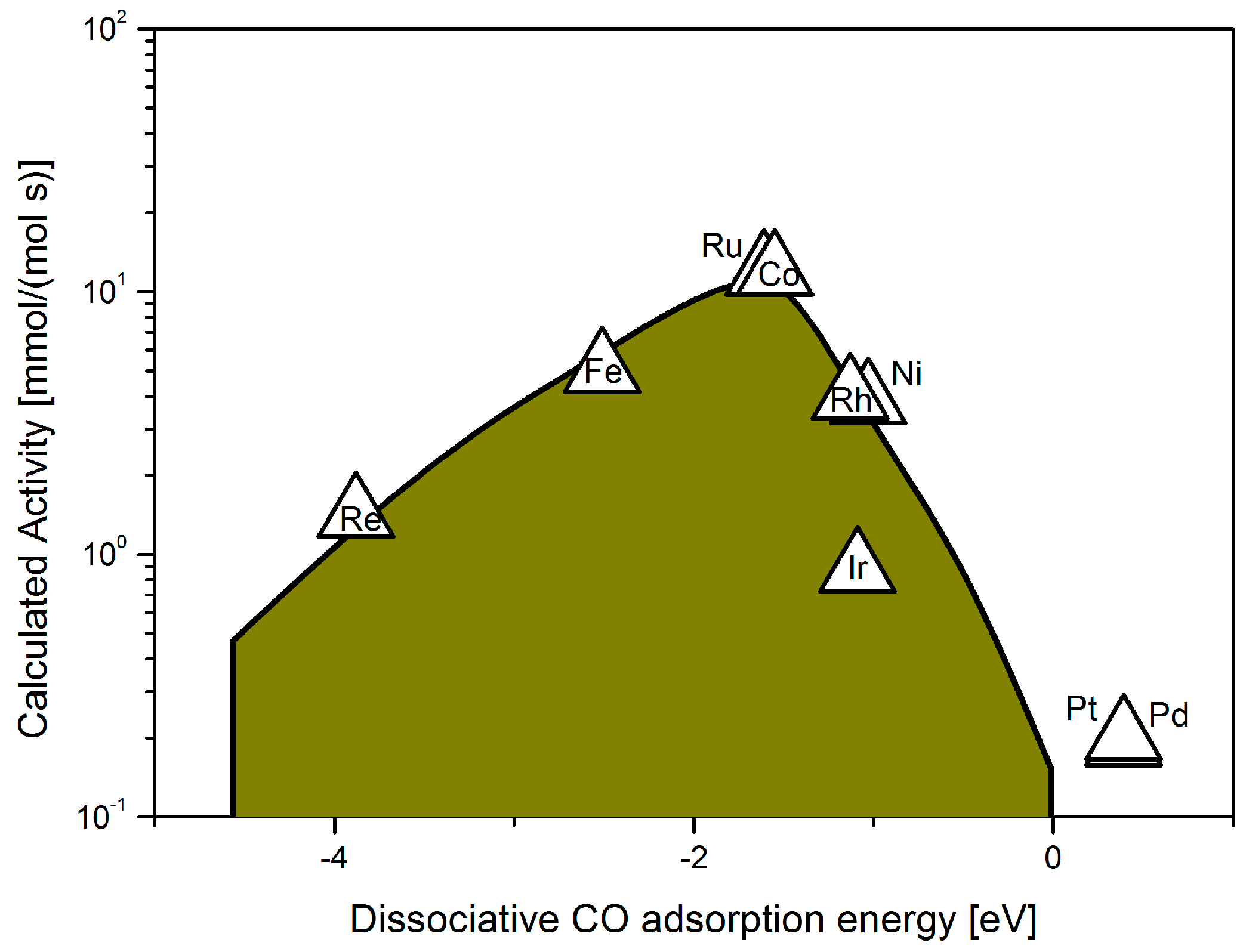{kind=link}
1. Introduction
2. Catalytic Materials for CO2 Methanation Reaction
2.1. Noble Metal Catalysts
2.1.1. Rhodium
2.1.2. Ruthenium
2.1.3. Palladium
2.2. Nickel-Based Catalysts
2.2.1. Alumina-Supported Nickel
2.2.2. Silica-Supported Nickel
2.2.3. Zirconia- and Ceria-Supported Catalysts
2.2.4. Hydrotalcite-Supported Nickel
2.2.5. Other Supported-Nickel Catalysts
2.2.6. Nickel–Iron-Based Catalysts
3. Industrial Applications and Concluding Remarks
Author Contributions
Conflicts of Interest
References
- Iglesias, G.M.; de Vries, C.; Claeys, M.; Schaub, G. Chemical energy storage in gaseous hydrocarbons via iron Fischer–Tropsch synthesis from H2/CO2-Kinetics, selectivity and process considerations. Catal. Today 2015, 242, 184–192. [Google Scholar] [CrossRef]
- Janke, C.; Duyar, M.S.; Hoskins, M.; Farrauto, R. Catalytic and adsorption studies for the hydrogenation of CO2 to methane. Appl. Catal. B Environ. 2014, 152–153, 184–191. [Google Scholar] [CrossRef]
- Koschany, F.; Schelereth, D.; Hinrichsen, O. On the kinetics of the methanation of carbon dioxide on coprecipitated NiAl(O)x. Appl. Catal. B Environ. 2016, 181, 504–516. [Google Scholar] [CrossRef]
- Ashok, J.; Ang, M.L.; Kawi, S. Enhanced activity of CO2 methanation over Ni/CeO2–ZrO2 catalysts: Influence of preparation method. Catal. Today 2017, 281, 304–311. [Google Scholar] [CrossRef]
- Tian, D.; Liu, Z.; Li, D.; Shi, H.; Pan, W.; Cheng, Y. Bimetallic Ni-Fe total-methanation catalyst for the production of substitute natural gas under high pressure. Fuel 2013, 104, 224–229. [Google Scholar] [CrossRef]
- Abelló, S.; Berrueco, C.; Montané, D. High-loaded nickel–alumina catalyst for direct CO2 hydrogenation into synthetic natural gas (SNG). Fuel 2013, 113, 598–609. [Google Scholar] [CrossRef]
- Tao, M.; Xin, Z.; Meng, X.; Bian, Z.; Lv, Y. Highly dispersed nickel within mesochannels of SBA-15 for CO methanation with enhanced activity and excellent thermostability. Fuel 2017, 188, 267–276. [Google Scholar] [CrossRef]
- Schaaf, T.; Grünig, J.; Roman Schuster, M.; Rothenfluh, T.; Orth, A. Methanation of CO2—Storage of renewable energy in a gas distribution system. Energy Sustain. Soc. 2014, 2–14. [Google Scholar] [CrossRef]
- Sahebdelfar, S.; Ravanchi, M.T. Carbon dioxide utilization for methane production: A thermodynamic analysis. J. Pet. Sci. Eng. 2015, 134, 14–22. [Google Scholar] [CrossRef]
- Su, X.; Xu, J.; Liang, B.; Duan, H.; Hou, B.; Huang, Y. Catalytic carbon dioxide hydrogenation to methane: A review of recent studies. J. Energy Chem. 2016, 25, 553–565. [Google Scholar] [CrossRef]
- Ronsch, S.; Schneider, J.; Matthischke, S.; Schluter, M.; Gotz, M.; Lefebre, J.; Prabhakaran, P.; Bajohr, S. Review on methanation—From fundamentals to current projects. Fuel 2016, 166, 276–296. [Google Scholar] [CrossRef]
- Aziz, M.A.A.; Jalil, A.A.; Triwahyono, S.; Ahmad, A. CO2 methanation over heterogeneous catalysts: Recent progress and future prospects. Green Chem. 2015, 17, 2647–2663. [Google Scholar] [CrossRef]
- Puga, A.V. Light-Promoted Hydrogenation of Carbon dioxide—An overview. Top. Catal. 2016, 59, 1268–1278. [Google Scholar] [CrossRef]
- Wang, W.; Gong, J. Methanation of carbon dioxide: An overview. Front. Chem. Sci. Eng. 2011, 5, 2–10. [Google Scholar]
- Liu, J.X.; Hou, W.H. Study on Ru-based catalyst used in reductive reaction of CO2. Space Med. Med. Eng. 2004, 17, 457–460. [Google Scholar]
- Meng, Y.Y.; Shang, C.X. A study on CO2 methanization reduction technology. Space Med. Med. Eng. 1994, 7, 115–120. [Google Scholar]
- Zhou, K.H.; Wu, B.Z.; Ren, C.B. Comparative analysis of Sabatier CO2 reduction system for space station. Space Med. Med. Eng. 2011, 24, 384–390. [Google Scholar]
- Liu, C.; Cundari, T.R.; Wilson, A.K. CO2 reduction on transition metal (Fe, Co, Ni, and Cu) surface: In comparison with homogeneous catalysis. J. Phys. Chem. C. 2012, 116, 5681–5688. [Google Scholar] [CrossRef]
- Choe, S.J.; Park, D.H.; Huh, D.S. Adsorption and dissociation reaction of carbon dioxide on Pt(111) and Fe(111) surface: MO-study. Bull. Korean Chem. Soc. 2000, 21, 779–784. [Google Scholar]
- Solymosi, F. The bonding, structure and reaction of CO2 adsorbed on clean and promoted metal surface. J. Mol. Catal. 1991, 65, 337–358. [Google Scholar] [CrossRef]
- Fechete, I.; Vedrine, J.V. Nanoporous materials as new engineered catalysts for the synthesis of green fuels. Molecules 2015, 20, 5638–5666. [Google Scholar] [CrossRef] [PubMed]
- Italiano, G.; Espro, C.; Arena, F.; Frusteri, F.; Parmaliana, A. Catalytic features of Mg modified Ni/SiO2/Silica cloth systems in the decomposition of methane for making COx-Free Hydrogen. Catal. Lett. 2008, 124, 7–12. [Google Scholar] [CrossRef]
- Frontera, P.; Aloise, A.; Macario, A.; Crea, F.; Antonucci, P.L.; Giordano, G.; Nagy, J.B. Zeolite-supported Ni catalyst for methane reforming with carbon dioxide. Res. Chem. Intermed. 2011, 37, 267–279. [Google Scholar] [CrossRef]
- Mauriello, F.; Vinci, A.; Espro, C.; Gumina, B.; Musolino, M.G.; Pietropalo, R. Hydrogenolysis vs. aqueous phase reforming (APR) of glycerol promoted by a heterogeneous Pd/Fe catalyst. Catal. Sci. Technol. 2015, 5, 4466–4473. [Google Scholar] [CrossRef]
- Frontera, P.; Macario, A.; Aloise, A.; Antonucci, P.L.; Giordano, G.; Nagy, J.B. Effect of support surface on methane dry-reforming catalyst preparation. Catal. Today 2013, 218–219, 18–29. [Google Scholar] [CrossRef]
- Candamano, S.; Frontera, P.; Macario, A.; Crea, F.; Nagy, J.B. Preparation and characterization of active Ni-supported catalyst for syngas production. Chem. Eng. Res. Des. 2015, 96, 78–86. [Google Scholar] [CrossRef]
- Breen, J.P.; Burch, R.; Coleman, H.M. Metal-catalyzed steam reforming of ethanol in the production of hydrogen for fuel cell application. Appl. Catal. B Environ. 2002, 39, 65–74. [Google Scholar] [CrossRef]
- Pompeo, F.; Nichio, N.N.; Feretti, O.A.; Resasco, D. Study of Ni catalysts on different supports to obtain synthesis gas. Int. J. Hydrogen Energy 2005, 30, 1399–1405. [Google Scholar] [CrossRef]
- Lo Faro, M.; Frontera, P.; Antonucci, P.L.; Aricò, A.S. Ni-Cu based catalysts prepared by two different methods and their catalytic activity toward the ATR of methane. Chem. Eng. Res. Des. 2015, 93, 269–277. [Google Scholar] [CrossRef]
- Jacquemin, M.; Beuls, A.; Ruiz, P. Catalytic production of methane from CO2 and H2 at low temperature: Insight on the reaction mechanism. Catal. Today 2010, 157, 462–466. [Google Scholar] [CrossRef]
- Beuls, A.; Swalus, C.; Jacquemin, M.; Heyen, G.; Karelovic, A.; Ruiz, P. Methanation of CO2: Further insight into the mechanism over Rh/γ-Al2O3 catalyst. Appl. Catal. B Environ. 2012, 113–114, 2–10. [Google Scholar] [CrossRef]
- Karelovic, A.; Ruiz, P. CO2 hydrogenation at low temperature over Rh/γ-Al2O3 catalysts: Effect of the metal particle size on catalytic performances and reaction mechanism. Appl. Catal. B Environ. 2012, 113–114, 237–249. [Google Scholar] [CrossRef]
- Wijayapalaa, R.; Yu, F.; Pittman, C.U., Jr.; Mlsna, T.T. K-promoted Mo/Co- and Mo/Ni-catalyzed Fischer–Tropsch synthesis of aromatic hydrocarbons with and without a Cu water gas shift catalyst. Appl. Catal. A Gen. 2014, 480, 93–99. [Google Scholar] [CrossRef]
- Swalus, C.; Jacquemin, M.; Poleunis, C.; Bertrand, P.; Ruiz, P. CO2 methanation on Rh/γ-Al2O3 catalyst at low temperature: “In situ” supply of hydrogen by Ni/activated carbon catalyst. Appl. Catal. B Environ. 2012, 125, 41–50. [Google Scholar] [CrossRef]
- Karelovic, A.; Ruiz, P. Mechanistic study of low temperature CO2 methanation over Rh/TiO2 catalysts. J. Catal. 2013, 301, 141–153. [Google Scholar] [CrossRef]
- Solymosi, F.; Erdöhelyi, A.; Bánsági, T. Methanation of CO2 on supported rhodium catalyst. J. Catal. 1981, 68, 371–382. [Google Scholar] [CrossRef]
- Zhang, Z.L.; Kladi, A.; Verykios, X.E. Effects of Carrier Doping on Kinetic Parameters of CO2 Hydrogenation on Supported Rhodium Catalysts. J. Catal. 1994, 148, 737–747. [Google Scholar] [CrossRef]
- Solymosi, F.; Tombácz, I.; Koszta, J. Effects of variation of electric properties of TiO2 support on hydrogenation of CO and CO2 over Rh catalysts. J. Catal. 1985, 95, 578–586. [Google Scholar] [CrossRef]
- Bell, A.T. The influence of metal oxides on the activity and selectivity of transition metal catalysts. J. Mol. Catal. A 1995, 100, 1–11. [Google Scholar]
- Williams, K.J.; Boffa, A.B.; Salmeron, M.; Bell, A.T.; Somorjai, G.A. The kinetics of CO2 hydrogenation on a Rh foil promoted by Titania overlayers. Catal. Lett. 1991, 9, 415426. [Google Scholar] [CrossRef]
- Trovarelli, A.; Deleitenburg, C.; Dolcetti, G.; Lorca, J.L. CO2 Methanation under Transient and Steady-State Conditions over Rh/CeO2 and CeO2-Promoted Rh/SiO2: The Role of Surface and Bulk Ceria. J. Catal. 1995, 151, 111–124. [Google Scholar] [CrossRef]
- Deleitenburg, C.; Trovarelli, A. Metal-Support Interactions in Rh/CeO2, Rh/TiO2, and Rh/Nb2O5 Catalysts as Inferred from CO2 Methanation Activity. J. Catal. 1995, 156, 171–174. [Google Scholar] [CrossRef]
- Ma, S.; Song, W.; Liu, B.; Zheng, H.; Deng, J.; Zhong, W.; Liu, J.; Gong, X.Q.; Zhao, Z. Elucidation of the high CO2 reduction selectivity of isolated Rh supported on TiO2: A DFT study. Catal. Sci. Technol. 2016, 6, 6128–6136. [Google Scholar] [CrossRef]
- Fukui, K.; Yonezawa, T.; Shingu, H. A molecular Orbital Theory of Reactivity in Aromatic Hydrocarbons. J. Chem. Phys. 1952, 20, 722–725. [Google Scholar] [CrossRef]
- Kusmierz, M. Kinetic study on carbon dioxide hydrogenation over Ru/γ-Al2O3 catalysts. Catal. Today 2008, 137, 429–432. [Google Scholar] [CrossRef]
- Mills, G.A.; Steffgen, F.W. Catalytic Methanation. Catal. Rev. 1974, 8, 159–210. [Google Scholar] [CrossRef]
- Weatherbee, G.D.; Bartholomew, C.H. Hydrogenation of CO2 on group VIII metals: IV. Specific activities and selectivities of silica-supported Co, Fe, and Ru. J. Catal. 1984, 87, 352–362. [Google Scholar] [CrossRef]
- Sugawa, S.; Sayama, K.; Okabe, K.; Arakawa, H. Methanol synthesis from CO2 and H2 over silver catalyst. Energy Convers. Manag. 1995, 36, 665–668. [Google Scholar] [CrossRef]
- Scirè, S.; Crisafulli, C.; Maggiore, R.; Minicò, S.; Galvagno, S. Influence of the support on CO2 methanation over Ru catalysts: An FT-IR study. Catal. Lett. 1998, 51, 41–45. [Google Scholar] [CrossRef]
- Li, D.; Ichikuni, N.; Shimazu, S.; Uematsu, T. Catalytic properties of sprayed Ru/Al2O3 and promoter effects of alkali metals in CO2 hydrogenation. Appl. Catal. A Gen. 1998, 172, 351–358. [Google Scholar] [CrossRef]
- Li, D.; Ichikuni, N.; Shimazu, S.; Uematsu, T. Hydrogenation of CO2 over sprayed Ru/TiO2 fine particles and strong metal-support interaction. Appl. Catal. A Gen. 1999, 180, 227–235. [Google Scholar] [CrossRef]
- Toemen, S.; Bakar, W.A.W.A.; Ali, R. Effect of ceria and strontian over Ru/Mn/Al2O3 catalyst: Catalytic methanation, physicochemical and mechanistic studies. J. CO2 Util. 2016, 13, 38–49. [Google Scholar] [CrossRef]
- Garbarino, G.; Bellotti, D.; Riani, P.; Magistri, L.; Busca, G. Methanation of carbon dioxide on Ru/Al2O3 and Ni/Al2O3 catalysts at atmospheric pressure: Catalysts activation, behaviour and stability. Int. J. Hydrogen Energy 2015, 40, 9171–9182. [Google Scholar] [CrossRef]
- Garbarino, G.; Bellotti, D.; Finocchio, E.; Magistri, L.; Busca, G. Methanation of carbon dioxide on Ru/Al2O3: Catalytic activity and infrared study. Catal. Today 2016, 277, 21–28. [Google Scholar] [CrossRef]
- Kowalczyk, Z.; Stołecki, K.; Raròg-Pilecka, W.; Miskiewicz, E.; Wilczkowska, E.; Karpinski, Z. Supported ruthenium catalysts for selective methanation of carbon oxides at very low COx/H2 ratios. Appl. Catal. A Gen. 2008, 342, 35–39. [Google Scholar] [CrossRef]
- Li, T.; Wang, S.; Gao, D.; Wang, S. Effect of support calcination temperature on the catalytic properties of Ru/Ce0.8Zr0.2O2 for methanation of carbon dioxide. J. Fuel Chem. Technol. 2014, 42, 1440–1446. [Google Scholar] [CrossRef]
- Akamarua, S.; Shimazaki, T.; Kuboc, M.; Abe, T. Density functional theory analysis of methanation reaction of CO2 on Ru nanoparticle supported on TiO2 (101). Appl. Catal. A Gen. 2014, 470, 405–411. [Google Scholar] [CrossRef]
- Ravindranathan Thampi, K.; Kiwi, J.; Grätzel, M. Methanation and photo-methanation of carbon dioxide at room temperature and atmospheric pressure. Nature 1987, 327, 506–508. [Google Scholar] [CrossRef]
- Gupta, N.M.; Kamble, V.S.; Kartha, V.B.; Iyer, R.M.; Ravindranathan Thampi, K.; Gratzel, M. FTIR spectroscopic study of the interaction of CO2 and CO2 + H2 over partially oxidized RuTiO2 catalyst. J. Catal. 1994, 146, 173–184. [Google Scholar] [CrossRef]
- Abe, T.; Tanizawa, M.; Watanabe, K.; Taguchi, A. CO2 methanation property of Ru nanoparticle-loaded TiO2 prepared by a polygonal barrel-sputtering method. Energy Environ. Sci. 2009, 2, 315–321. [Google Scholar] [CrossRef]
- Xu, J.; Su, X.; Duan, H.; Hou, B.; Lin, Q.; Liu, X.; Pan, X.; Pei, G.; Geng, H.; Huang, Y.; et al. Influence of pretreatment temperature on catalytic performance of rutile TiO2-supported ruthenium catalysts in CO2 methanation. J. Catal. 2016, 333, 227–237. [Google Scholar] [CrossRef]
- Tada, S.; Ochienga, O.J.; Kikuchi, R.; Haneda, T.; Kameyama, H. Promotion of CO2 methanation activity and CH4 selectivity at low temperatures over Ru/CeO2/Al2O3 catalysts. Int. J. Hydrogen Energy 2014, 39, 10090–10100. [Google Scholar] [CrossRef]
- Sharma, S.; Hu, Z.; Zhang, P.; McFarland, E.W.; Metiu, H. CO2 methanation on Ru-doped ceria. J. Catal. 2011, 278, 297–309. [Google Scholar] [CrossRef]
- Xu, J.; Lin, Q.; Su, X.; Duan, H.; Geng, H.; Huang, Y. CO2 Methanation over TiO2-Al2O3 Binary Oxides Supported Ru Catalysts. Chin. J. Chem. Eng. 2016, 24, 140–145. [Google Scholar] [CrossRef]
- Zamani, A.H.; Ali, R.; Bakar, W.A.W.A. Optimization of CO2 methanation reaction over M*/Mn/Cu–Al2O3 (M*: Pd, Rh and Ru) catalysts. J. Ind. Eng. Chem. 2015, 29, 238–248. [Google Scholar] [CrossRef]
- Zamani, A.H.; Ali, R.; Bakar, W.A.W.A. The investigation of Ru/Mn/Cu–Al2O3 oxide catalysts for CO2/H2 methanation in natural gas. J. Tawain Inst. Chem. Eng. 2014, 45, 143–152. [Google Scholar] [CrossRef]
- Toemen, S.; Bakar, W.A.W.A.; Ali, R. Investigation of Ru/Mn/Ce/Al2O3 catalyst for carbon dioxide methanation: Catalytic optimization, physicochemical studies and RSM. J. Tawain Inst. Chem. Eng. 2014, 45, 2370–2378. [Google Scholar] [CrossRef]
- Borodziński, A.; Bond, G.C. Selective Hydrogenation of Ethyne in Ethene-Rich Streams on Palladium Catalysts. Part 1. Effect of Changes to the Catalyst During Reaction. Catal. Rev. Sci. Eng. 2006, 48, 91–144. [Google Scholar] [CrossRef]
- Albers, P.; Pietsch, J.; Parker, S.F. Poisoning and deactivation of palladium catalysts. J. Mol. Catal. A Chem. 2001, 173, 275–286. [Google Scholar] [CrossRef]
- Schuurman, Y.; Mirodatos, C.; Ferreira-Aparicio, P.; Rodriguez-Ramos, I.; Guerrero-Ruiz, A. Bifunctional pathways in the carbon dioxide reforming of methane over MgO-promoted Ru/C catalysts. Catal. Lett. 2000, 66, 33–37. [Google Scholar] [CrossRef]
- Galuszka, J. Carbon dioxide chemistry during oxidative coupling of methane on a Li/MgO catalyst. Catal. Today 1994, 21, 321–331. [Google Scholar] [CrossRef]
- Chen, Y.; Tomishige, K.; Yokoyama, K.; Fujimoto, K. Promoting effect of Pt, Pd and Rh noble metals to the Ni0.03Mg0.97O solid solution catalysts for the reforming of CH4 with CO2. Appl. Catal. A Gen. 1997, 165, 335–347. [Google Scholar] [CrossRef]
- Veith, G.M.; Lupini, A.R.; Rashkeev, S.; Pennycook, S.J.; Mullins, D.R.; Schwartz, V.; Bridges, C.A.; Dudney, N.J. Thermal stability and catalytic activity of gold nanoparticles supported on silica. J. Catal. 2009, 262, 92–101. [Google Scholar] [CrossRef]
- Martins, J.; Batail, N.; Silva, S.; Rafik-Clement, S.; Karelovic, A.; Debecker, D.P.; Chaumonnot, A.; Uzio, D. CO2 hydrogenation with shape-controlled Pd nanoparticles embedded in mesoporous silica: Elucidating stability and selectivity issues. Catal. Commun. 2015, 58, 11–15. [Google Scholar] [CrossRef]
- Delmelle, R.; Duarte, R.B.; Franken, T.; Burnat, D.; Holzer, L.; Borgschulte, A. Development of improved nickel catalysts for sorption enhanced CO2 methanation. Int. J. Hydrogen Energy 2015, 41, 20185–20191. [Google Scholar] [CrossRef]
- Vaudagna, S.R.; Comelli, R.A.; Figoli, N.S. Influence of the tungsten oxide precursor on WOx-ZrO2 and Pt/WOx-ZrO2 properties. Appl. Catal. A Gen. 1997, 164, 265–280. [Google Scholar] [CrossRef]
- Chang, F.W.; Kuo, M.S.; Tsay, M.T.; Hsieh, M.C. Hydrogenation of CO2 over nickel catalysts on rice husk ash-alumina prepared by incipient wetness impregnation. Appl. Catal. A Gen. 2003, 247, 309–320. [Google Scholar] [CrossRef]
- Gao, J.; Jia, C.; Li, J.; Zhang, M.; Gua, F.; Xua, G.; Zhong, Z.; Sua, F. Ni/Al2O3 catalysts for CO methanation: Effect of Al2O3 supports calcined at different temperatures. J. Energy Chem. 2013, 22, 919–927. [Google Scholar] [CrossRef]
- Takenaka, S.; Shimizu, T.; Otsuka, K. Complete removal of carbon monoxide in hydrogen-rich gas stream through methanation over supported metal catalysts. Int. J. Hydrogen Energy 2004, 29, 1065–1073. [Google Scholar] [CrossRef]
- Danaci, S.; Protasova, L.; Lefevere, J.; Bedel, L.; Guilet, R.; Marty, P. Efficient CO2 methanation over Ni/Al2O3 coated structured catalysys. Catal. Today 2016, 273, 234–243. [Google Scholar] [CrossRef]
- Tada, S.; Shimizu, T.; Kameyama, H.; Haneda, T.; Kikuchi, R. Ni/CeO2 catalysts with high CO2 methanation activity and high CH4 selectivity at low temperatures. Int. J. Hydrogen Energy 2012, 37, 5527–5531. [Google Scholar] [CrossRef]
- Song, H.; Yang, J.; Zhao, J.; Chou, L. Methanation of carbon dioxide over a highly dispersed Ni/La2O3 catalyst. Chin. J. Catal. 2010, 31, 21–23. [Google Scholar] [CrossRef]
- Zhang, L.; Li, L.; Zhang, Y.; Zhao, Y.; Li, J. Nickel catalysts supported on MgO with different specific surface area for carbon dioxide reforming of methane. J. Energy Chem. 2014, 23, 66–72. [Google Scholar] [CrossRef]
- Fukuhara, C.; Hayakawa, K.; Suzuki, Y.; Kawasaki, W.; Watanabe, R. A novel nickel-based structured catalyst for CO2 methanation: A honeycomb-type Ni/CeO2 catalyst to transform greenhouse gas into useful resource. Appl. Catal. A Gen. 2017, 532, 12–18. [Google Scholar] [CrossRef]
- Aksoylu, A.E.; Önsan, Z.I. Hydrogenation of carbon oxides using coprecipitated and impregnated Ni/Al2O3 catalysts. Appl. Catal. A Gen. 1997, 164, 1–11. [Google Scholar] [CrossRef]
- Zhou, R.; Rui, N.; Fan, Z.; Liu, C. Effect of the structure of Ni/TiO2 catalyst on CO2 methanation. Int. J. Hydrogen Energy. 2016, 41, 22017–22025. [Google Scholar] [CrossRef]
- Da Silva, D.C.D.; Letichevsky, S.; Borges, L.E.P.; Appel, L.G. The Ni/ZrO2 catalyst and the methanation of CO and CO2. Int. J. Hydrogen Energy 2012, 37, 8923–8928. [Google Scholar] [CrossRef]
- Muroyama, H.; Tsuda, Y.; Asakoshi, T.; Masitah, H.; Okanishi, T.; Matsui, T.; Eguchi, K. Carbon dioxide methanation over Ni catalysts supported on various metal oxides. J. Catal. 2016, 343, 178–184. [Google Scholar] [CrossRef]
- Takano, H.; Kirihata, Y.; Izumiya, K.; Kumagai, N.; Habazaki, H.; Hashimoto, K. Higly active Ni/Y doped Zr2O catalyst for CO2 methanation. App. Surf. Sci. 2016, 358, 653–663. [Google Scholar] [CrossRef]
- Wang, S.; Lu, G.Q.M. Role of CeO2 in Ni/CeO2-Al2O3 catalysts for carbon dioxide reforming of methane. Appl. Catal. B Environ. 1998, 19, 267–277. [Google Scholar] [CrossRef]
- Aksoylu, A.E.; Akin, A.N.; Önsan, Z.I.; Trimm, D.L. Structure/activity relationships in coprecipitated nickel-alumina catalysts using CO2 adsorption and methanation. Appl. Catal. A Gen. 1996, 145, 185–193. [Google Scholar] [CrossRef]
- Rahmani, S.; Rezaei, M.; Meshkania, F. Preparation of highly active nickel catalysts supported on mesoporous nanocrystalline γ-Al2O3 for CO2 methanation. J. Ind. Eng. Chem. 2014, 20, 1346–1352. [Google Scholar] [CrossRef]
- Garbarino, G.; Riani, P.; Magistri, L.; Busca, G. A study of the methanation of carbon dioxide on Ni/Al2O3 catalysts at atmospheric pressure. Int. J. Hydrogen Energy 2014, 39, 11557–11565. [Google Scholar] [CrossRef]
- Mutz, B.; Carvalho, H.W.P.; Mangold, S.; Kleist, W.; Grunwaldt, J.D. Methanation of CO2: Structural response of a Ni-based catalyst under fluctuating reaction conditions unraveled by operando spectroscopy. J. Catal. 2015, 327, 48–53. [Google Scholar] [CrossRef]
- Weckhuysen, B.M. Determining the active site in a catalytic process: Operando spectroscopy is more than a buzzword. Phys. Chem. Chem. Phys. 2003, 5, 4351–4360. [Google Scholar] [CrossRef]
- Topsøe, H. Developments in operando studies and in situ characterization of heterogeneous catalysts. J. Catal. 2003, 216, 155–164. [Google Scholar] [CrossRef]
- Grunwaldt, J.D.; Clausen, B.S. Combining XRD and EXAFS with on-line catalytic studies for in situ characterization of catalysts. Top. Catal. 2002, 18, 37–43. [Google Scholar] [CrossRef]
- Banares, M.A. Operando methodology: Combination of in situ spectroscopy and simultaneous activity measurements under catalytic reaction conditions. Catal. Today 2005, 100, 71–77. [Google Scholar] [CrossRef]
- Prakash, A.S.; Shivakumara, C.; Hegde, M.S. Single step preparation of CeO2/CeAlO3/-Al2O3 by solution combustion method: Phase evolution, thermal stability and surface modification. Mater. Sci. Eng. B 2007, 139, 55–61. [Google Scholar] [CrossRef]
- Liang, H.; Zhang, Y.; Liu, Y. Ceria modified three-dimensionally ordered macro-porous Pt/TiO2 catalysts for water-gas shift reaction. J. Rare Earth 2009, 27, 425–430. [Google Scholar] [CrossRef]
- Laosiripojana, N.; Assabumrungrat, S. Catalytic steam reforming of ethanol over high surface area CeO2: The role of CeO2 as an internal pre-reforming catalyst. Appl. Catal. B Environ. 2006, 66, 29–39. [Google Scholar] [CrossRef]
- Yao, H.C.; Yu Yao, Y.F. Ceria in automotive exhaust catalysts: I. Oxygen storage. J. Catal. 1984, 86, 254–265. [Google Scholar] [CrossRef]
- Liu, H.; Zou, X.; Wang, X.; Lu, X.; Ding, W. Effect of CeO2 addition on Ni/Al2O3 catalysts for methanation of carbon dioxide with hydrogen. J. Nat. Gas Chem. 2012, 21, 703–707. [Google Scholar] [CrossRef]
- Rahmani, S.; Rezaei, M.; Meshkani, F. Preparation of promoted nickel catalysts supported on mesoporous nanocrystalline gamma alumina for carbon dioxide methanation reaction. J. Ind. Eng. Chem. 2014, 20, 4176–4182. [Google Scholar] [CrossRef]
- Saha, N.C.; Wolf, E.E. Co methanation activity and XPS studies of Pd supported on ZSM5 and Y-zeolites. Appl. Catal. 1984, 13, 101–112. [Google Scholar] [CrossRef]
- Eckle, S.; Anfang, H.G.; Behm, R.J. Reaction Intermediates and side products in the methanation of CO and CO2 over supported Ru catalysts in H2-rich reformate gases. J. Phys. Chem. C 2010, 115, 1361–1367. [Google Scholar] [CrossRef]
- Patzelová, V.; Zukal, A.; Tvarůžková, Z.; Malíček, O. Hydrogenation of CO and CO2 Over Stabilized NiY Catalysts. Stud. Surf. Sci. Catal. 1984, 18, 367–374. [Google Scholar]
- Graca, I.; González, L.V.; Bacariza, M.C.; Fernandes, A.; Henriques, C.; Lopes, J.M.; Ribeiro, M.F. CO2 hydrogenation into CH4 on NiHNaUSY zeolites. Appl. Catal. B Environ. 2014, 147, 101–110. [Google Scholar] [CrossRef]
- Xavier, K.O.; Sreekala, R.; Rashid, K.K.A.; Yusuff, K.K.M.; Sen, B. Doping effects of cerium oxide on Ni/Al2O3 catalysts for methanation. Catal. Today 1999, 49, 17–21. [Google Scholar] [CrossRef]
- Rynkowski, J.M.; Paryjczak, T. Characterization of Ru/CeO2-Al2O3 catalysts and their Performance in CO2 Methanation. React. Kinet. Catal. Lett. 2000, 71, 55–64. [Google Scholar] [CrossRef]
- Westermann, A.; Azambre, B.; Bacariza, M.C.; Graca, I.; Ribeiroa, M.F.; Lopes, J.M.; Henriquesapp, C. Insight into CO2 methanation mechanism over NiUSY zeolites: An operando IR study. Appl. Catal. B Environ. 2015, 174–175, 120–125. [Google Scholar] [CrossRef]
- Rivero-Mendoza, D.E.; Stanley, J.N.G.; Scott, J.; Aguey-Zinsou, K.F. An alumina-supported Ni-La-based catalyst for producing synthetic natural gas. Catalysts 2016, 6, 170. [Google Scholar] [CrossRef]
- Sokolov, S.; Kondratenko, E.V.; Pohl, M.M.; Barkschat, A.; Rodemerck, U. Stable low-temperature dry reforming of methane over mesoporous La2O3-ZrO2 supported Ni catalyst. Appl. Catal. B Environ. 2012, 113–114, 19–30. [Google Scholar] [CrossRef]
- Rossetti, I.; Biffi, C.; Bianchi, C.L.; Nichele, V.; Signoretto, M.; Menegazzo, F.; Finocchio, E.; Ramis, G.; Di Michele, A. Ni/SiO2 and Ni/ZrO2 catalysts for the steam reforming of ethanol. Appl. Catal. B Environ. 2012, 117–118, 384–396. [Google Scholar] [CrossRef]
- Nichele, V.; Signoretto, M.; Menegazzo, F.; Gallo, A.; Dal Santo, V.; Cruciani, G.; Cerrato, G. Glycerol steam reforming for hydrogen production: Design of Ni supported catalysts. Appl. Catal. B Environ. 2012, 111–112, 225–232. [Google Scholar] [CrossRef]
- Zheng, H.; Gao, C.; Peng, B.; Shu, M.; Che, S. pH-Responsive Drug Delivery System Based on Coordination Bonding in a Mesostructured Surfactant/Silica Hybrid. J. Phys. Chem. C 2011, 115, 7230–7237. [Google Scholar] [CrossRef]
- Lin, Y.S.; Haynes, C.L. Impacts of Mesoporous Silica Nanoparticle Size, Pore Ordering, and Pore Integrity on Hemolytic Activity. J. Am. Chem. Soc. 2010, 132, 4834–4842. [Google Scholar] [CrossRef] [PubMed]
- Liong, M.; Angelos, S.; Choi, E.; Patel, K.; Fraser Stoddart, J.; Zink, J.I. Mesostructured multifunctional nanoparticles for imaging and drug delivery. J. Mater. Chem. 2009, 19, 6251–6257. [Google Scholar] [CrossRef]
- Sakamoto, Y.; Kaneda, M.; Terasaki, O.; Zhao, D.Y.; Kim, J.M.; Stucky, G.; Shin, H.J.; Ryoo, R. Direct imaging of the pores and cages of three-dimensional mesoporous materials. Nature 2000, 408, 449–453. [Google Scholar] [PubMed]
- Dou, J.; Zeng, H.C. Targeted Synthesis of Silicomolybdic Acid (Keggin Acid) inside Mesoporous Silica Hollow Spheres for Friedel–Crafts Alkylation. J. Am. Chem. Soc. 2012, 134, 16235–16246. [Google Scholar] [CrossRef] [PubMed]
- Jiao, F.; Frei, H. Nanostructured Cobalt Oxide Clusters in Mesoporous Silica as Efficient Oxygen-Evolving Catalysts. Angew. Chem. Int. Ed. 2009, 48, 1841–1844. [Google Scholar] [CrossRef] [PubMed]
- Hulea, V.; Brunel, D.; Galarneau, A.; Philippot, K.; Chaudret, B.; Kooyman, P.J.; Fajula, F. Synthesis of well-dispersed ruthenium nanoparticles inside mesostructured porous silica under mild conditions. Microporous Mesoporous Mater. 2005, 79, 185–194. [Google Scholar] [CrossRef]
- Aziz, M.A.A.; Jalil, A.A.; Triwahyono, S.; Mukti, R.R.; Taufiq-Yap, Y.H.; Sazegar, M.R. Highly active Ni-promoted mesostructured silica nanoparticles for CO2 methanation. Appl. Catal. B Environ. 2014, 147, 359–368. [Google Scholar] [CrossRef]
- Aziz, M.A.A.; Jalil, A.A.; Triwahyono, S.; Sidik, S.M. Methanation of carbon dioxide on metal-promoted mesostructured silica nanoparticles. Appl. Catal. A Gen. 2014, 486, 115–122. [Google Scholar] [CrossRef]
- Aziz, M.A.A.; Jalil, A.A.; Triwahyono, S.; Saad, M.W.A. CO2 methanation over Ni-promoted mesostructured silica nanoparticles: Influence of Ni loading and water vapor on activity and response surface methodology studies. Chem. Eng. J. 2015, 260, 757–764. [Google Scholar] [CrossRef]
- Du, G.; Lim, S.; Yang, Y.; Wang, C.; Pfefferle, L.; Haller, G.L. Methanation of carbon dioxide on Ni-incorporated MCM-41 catalysts: The influence of catalyst pretreatment and study of steady-state reaction. J. Catal. 2007, 249, 370–379. [Google Scholar] [CrossRef]
- Pan, Q.; Peng, J.; Wang, S.; Wang, S. In situ FTIR spectroscopic study of the CO2 methanation mechanism on Ni/Ce0.5Zr0.5O2. Catal. Sci. Technol. 2014, 4, 502–509. [Google Scholar] [CrossRef]
- Fujita, S.I.; Nakamura, M.; Doi, T.; Takezawa, N. Mechanisms of methanation of carbon dioxide and carbon monoxide over nickel/alumina catalysts. Appl. Catal. A Gen. 1993, 104, 87–100. [Google Scholar] [CrossRef]
- Kwak, J.H.; Kovarik, L.; Szanyi, J. CO2 Reduction on Supported Ru/Al2O3 Catalysts: Cluster Size Dependence of Product Selectivity. ACS Catal. 2013, 3, 2449–2455. [Google Scholar] [CrossRef]
- Benítez, J.J.; Alvero, R.; Capitán, M.J.; Carrizosa, I.; Odriozola, J.A. DRIFTS study of adsorbed formate species in the carbon dioxide and hydrogen reaction over rhodium catalysts. Appl. Catal. 1991, 71, 219–231. [Google Scholar] [CrossRef]
- Dalla Betta, R.A.; Shelef, M. Heterogeneous methanation: In situ infrared spectroscopic study of RuAl2O3 during the hydrogenation of CO. J. Catal. 1977, 48, 111–119. [Google Scholar] [CrossRef]
- Bartholomew, C.H.; Pannell, R.B.; Fowler, R.W. Sintering of alumina-supported nickel and nickel bimetallic methanation catalysts in H2H2O atmospheres. J. Catal. 1983, 79, 34–46. [Google Scholar] [CrossRef]
- Zhang, J.; Xin, Z.; Meng, X.; Tao, M. Synthesis, characterization and properties of anti-sintering nickel incorporated MCM-41 methanation catalysts. Fuel 2013, 109, 693–701. [Google Scholar] [CrossRef]
- Huber, G.W.; Iborra, S.; Corma, A. Synthesis of Transportation Fuels from Biomass: Chemistry, Catalysts and Engineering. Chem. Rev. 2006, 106, 4044–4098. [Google Scholar] [CrossRef] [PubMed]
- Yung, M.M.; Jablonski, W.S.; Magrini-Bair, K.A. Review of Catalytic Conditioning of Biomass-Derived Syngas. Energy Fuels 2009, 23, 1874–1887. [Google Scholar] [CrossRef]
- Zhao, D.; Feng, J.; Huo, Q.; Melosh, N.; Fredrickson, G.H.; Chmelka, B.F.; Stucky, G.D. Triblock Copolymer Syntheses of Mesoporous Silica with Periodic 50 to 300 Angstrom Pores. Science 1998, 279, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Huo, Q.; Feng, J.; Chmelka, B.F.; Stucky, G.D. Nonionic Triblock and Star Diblock Copolymer and Oligomeric Surfactant Syntheses of Highly Ordered, Hydrothermally Stable, Mesoporous Silica Structures. J. Am. Chem. Soc. 1998, 120, 6024–6036. [Google Scholar] [CrossRef]
- Cheng, M.Y.; Pan, C.J.; Hwang, B.J. Highly-dispersed and thermally-stable NiO nanoparticles exclusively confined in SBA-15: Blockage-free nanochannels. J. Mater. Chem. 2009, 19, 5193–5200. [Google Scholar] [CrossRef]
- Vradman, L.; Landau, M.V.; Kantorovich, D.; Koltypin, Y.; Gedanken, A. Evaluation of metal oxide phase assembling mode inside the nanotubular pores of mesostructured silica. Microporous Mesoporous Mater. 2005, 79, 307–318. [Google Scholar] [CrossRef]
- Lu, B.; Kawamoto, K. Preparation of the highly loaded and well-dispersed NiO/SBA-15 for methanation of producer gas. Fuel 2013, 103, 699–704. [Google Scholar] [CrossRef]
- Köck, E.M.; Kogler, M.; Bielz, T.; Klötzer, B.; Penner, S. In Situ FT-IR Spectroscopic Study of CO2 and CO Adsorption on Y2O3, ZrO2, and Yttria-Stabilized ZrO2. J. Phys. Chem. C 2013, 117, 17666–17673. [Google Scholar] [CrossRef] [PubMed]
- Pokrovski, K.; Jung, K.T.; Bell, A.T. Investigation of CO and CO2 adsorption on tetragonal and monoclinic zirconia. Langmuir 2001, 17, 4297–4303. [Google Scholar] [CrossRef]
- Baeza, B.B.; Ramos, I.R.; Ruiz, A.G. Interaction of Carbon Dioxide with the Surface of Zirconia Polymorphs. Langmuir 1998, 14, 3556–3564. [Google Scholar] [CrossRef]
- Yamasaki, M.; Habazaki, H.; Yoshida, T.; Akiyama, E.; Kawashima, A.; Asami, K.; Hashimoto, K.; Komori, M.; Shimamura, K. Compositional dependence of the CO2 methanation activity of Ni/ZrO2 catalysts prepared from amorphous Ni-Zr alloy precursors. Appl. Catal. A Gen. 1997, 163, 187–197. [Google Scholar] [CrossRef]
- Habazaki, H.; Tada, T.; Wakuda, K.; Kawashima, A.; Asami, K.; Hashimoto, K. Corrosion, Electrochemistry and Catalysis of Metastable Metals and Intermetallics; Clayton, C.R., Hashimoto, K., Eds.; The Electrochemical Society: Washington, DC, USA, 1993; p. 393. [Google Scholar]
- Yamasaki, M.; Habazaki, H.; Asami, K.; Izumiya, K.; Hashimoto, K. Effect of tetragonal ZrO2 on the catalytic activity of Ni/ZrO2 catalyst prepared from amorphous Ni-Zr alloys. Catal. Commun. 2006, 7, 24–28. [Google Scholar] [CrossRef]
- Yamasaki, M.; Komori, M.; Akiyama, E.; Habazaki, H.; Kawashima, A.; Asami, K.; Hashimoto, K. CO2 methanation catalysts prepared from amorphous Ni-Zr-Sm and Ni-Zr-misch metal alloy precursors. Mater. Sci. Eng. A 1999, 267, 220–226. [Google Scholar] [CrossRef]
- Habazaki, H.; Yamasaki, M.; Kawashima, A.; Hashimoto, K. Methanation of carbon dioxide on Ni/(Zr-Sm)Ox catalysts. Appl. Organomet. Chem. 2000, 14, 803–808. [Google Scholar] [CrossRef]
- Huang, Y.H.; Wang, J.J.; Liu, Z.M.; Lin, G.D.; Zhang, H.B. Highly efficient Ni-ZrO2 catalyst doped with Yb2O3 for co-methanation of CO and CO2. Appl. Catal. A Gen. 2013, 466, 300–306. [Google Scholar] [CrossRef]
- Takano, H.; Izumiyaa, K.; Kumagai, N.; Hashimoto, K. The effect of heat treatment on the performance of the Ni/(Zr-Sm oxide) catalysts for carbon dioxide methanation. Appl. Surf. Sci. 2011, 257, 8171–8176. [Google Scholar] [CrossRef]
- Qin, Z.; Ren, J.; Miao, M.; Li, Z.; Lin, J.; Xie, K. The catalytic methanation of coke oven gas over Ni-Ce/Al2O3 catalyst prepared by microwave heating: Effect of amorphous NiO formation. Appl. Catal. B 2015, 164, 18–30. [Google Scholar] [CrossRef]
- Zhou, G.; Liu, H.; Cui, K.; Jia, A.; Hu, G.; Jiao, Z.; Liu, Y.; Zhang, X. Role of surface Ni and Ce species of Ni/CeO2 catalyst in CO2 methanation. Appl. Surf. Sci. 2016, 383, 248–252. [Google Scholar] [CrossRef]
- Konishcheva, M.V.; Potemkin, D.I.; Badmaev, S.D.; Snytnikov, P.V.; Paukshtis, E.A.; Sobyanin, V.A.; Parmon, V.N. On the mechanism of CO and CO2 methanation over Ni/CeO2 catalysts. Top. Catal. 2016, 59, 1424–1430. [Google Scholar] [CrossRef]
- Nematollahi, B.; Rezaei, M.; Nemati Lay, E. Selective methanation of carbon monoxide in hydrogen rich stream over Ni/CeO2 nanocatalysts. J. Rare Earth 2015, 33, 619–628. [Google Scholar] [CrossRef]
- Ocampo, F.; Louis, B.; Roger, A.C. Methanation of carbon dioxide over nickel-based Ce0.72Zr0.28O2 mixed oxide catalysts prepared by sol–gel method. Appl. Catal. A Gen. 2009, 369, 90–96. [Google Scholar] [CrossRef]
- Laosiripojana, N.; Assabumrungrat, S. Methane steam reforming over Ni/Ce–ZrO2 catalyst: Influences of Ce-ZrO2 support on reactivity, resistance toward carbon formation and intrinsic reaction kinetics. Appl. Catal. A Gen. 2005, 290, 200–211. [Google Scholar] [CrossRef]
- Di Monte, R.; Fornasiero, P.; Kašpar, J.; Rumori, P.; Gubitosa, G.; Graziani, M. Pd/Ce0.6Zr0.4O2/Al2O3 as advanced materials for three-way catalysts Part 1. Catalyst characterisation, thermal stability and catalytic activity in the reduction of NO by CO. Appl. Catal. B Environ. 2000, 24, 157–167. [Google Scholar] [CrossRef]
- Strobel, R.; Krumeich, F.; Pratsinis, S.E.; Baiker, A. Flame-derived Pt/Ba/CexZr1−xO2: Influence of support on thermal deterioration and behavior as NOx storage-reduction catalysts. J. Catal. 2006, 243, 229–238. [Google Scholar] [CrossRef]
- Ocampo, F.; Louis, B.; Kiwi-Minsker, L.; Roger, A.C. Effect of Ce/Zr composition and noble metal promotion on nickel based CexZr1−xO2 catalysts for carbon dioxide methanation. Appl. Catal. A Gen. 2011, 392, 36–44. [Google Scholar] [CrossRef]
- Haneda, M.; Shinoda, K.; Nagane, A.; Houshito, O.; Takagi, H.; Nakahara, Y.; Hiroe, K.; Fujitani, T.; Hamada, H. Catalytic performance of rhodium supported on ceria–zirconia mixed oxides for reduction of NO by propene. J. Catal. 2008, 259, 223–231. [Google Scholar] [CrossRef]
- Silva, P.P.; Silva, F.A.; Portela, L.S.; Mattos, L.V.; Noronha, F.B.; Hori, C.E. Effect of Ce/Zr ratio on the performance of Pt/CeZrO2/Al2O3 catalysts for methane partial oxidation. Catal. Today 2005, 107–108, 734–740. [Google Scholar] [CrossRef]
- Koubaissy, B.; Pietraszek, A.; Roger, A.C.; Kiennemann, A. CO2 reforming of methane over Ce-Zr-Ni-Me mixed catalysts. Catal. Today 2010, 157, 436–439. [Google Scholar] [CrossRef]
- Youn, M.H.; Gil Seo, J.; Min Cho, K.; Park, S.; Park, D.R.; Jung, J.C.; Song, I.K. Hydrogen production by auto-thermal reforming of ethanol over nickel catalysts supported on Ce-modified mesoporous zirconia: Effect of Ce/Zr molar ratio. Int. J. Hydrogen Energy 2008, 33, 5052–5059. [Google Scholar] [CrossRef]
- Ussa Aldana, P.A.; Ocampo, F.; Kobl, K.; Louis, B.; Thibault-Starzyka, F.; Daturi, M.; Bazina, P.; Thomas, S.; Roger, A.C. Catalytic CO2 valorization into CH4 on Ni-based ceria-zirconia. Reaction mechanism by operando IR spectroscopy. Catal. Today 2013, 215, 201–207. [Google Scholar] [CrossRef]
- Pan, Q.; Peng, J.; Sun, T.; Wang, S.; Wang, S. Insight into the reaction route of CO2 methanation: Promotion effect of medium basic sites. Catal. Commun. 2014, 45, 74–78. [Google Scholar] [CrossRef]
- Korhonen, S.T.; Calatayud, M.; Krause, A.O.I. Structure and Stability of Formates and Carbonates on Monoclinic Zirconia: A Combined Study by Density Functional Theory and Infrared Spectroscopy. J. Phys. Chem. C 2008, 112, 16096–16102. [Google Scholar] [CrossRef]
- Nizio, M.; Albarazi, A.; Cavadias, S.; Amouroux, J.; Galvez, M.E.; Da Costa, P. Hybrid plasma-catalytic methanation of CO2 at low temperature over ceria zirconia supported Ni catalysts. Int. J. Hydrogen Energy 2016, 41, 11584–11592. [Google Scholar] [CrossRef]
- Li, C.; Sakata, Y.; Arai, T.; Domen, K.; Maruya, K.; Onishi, T. Carbon monoxide and carbon dioxide adsorption on cerium oxide studied by Fourier-transform infrared spectroscopy. Part 1.Formation of carbonate species on dehydroxylated CeO2, at room temperature. J. Chem. Soc. Faraday Trans. 1989, 85, 929–943. [Google Scholar] [CrossRef]
- Jin, T.; Zhou, Y.; Mains, G.J.; White, J.M. Infrared and X-ray Photoelectron Spectroscopy Study of CO and CO2 on Pt/CeO2. J. Phys. Chem. 1987, 91, 5931–5937. [Google Scholar] [CrossRef]
- Marwood, M.; Doepper, R.; Renken, A. In-situ surface and gas phase analysis for kinetic studies under transient conditions the catalytic hydrogenation of CO2. Appl. Catal. A Gen. 1997, 151, 223–246. [Google Scholar] [CrossRef]
- Prairie, M.R.; Highfield, J.G.; Renken, A. Diffuse-reflectance FTIR spectroscopy for kinetic and mechanistic studies of CO2 hydrogenation in a continuous recycle reactor. Chem. Eng. Sci. 1991, 46, 113–121. [Google Scholar] [CrossRef]
- Prairie, M.R.; Renken, A.; Highfield, J.G.; Ravindranathan Thampi, K.; Grätzel, M. A Fourier transform infrared spectroscopic study of CO2 methanation on supported ruthenium. J. Catal. 1991, 129, 130–144. [Google Scholar] [CrossRef]
- Schild, C.; Wokaun, A. CO2 hydrogenation over Nickel/Zirconia catalysts from amorphous precursors: On the mechanism of methane formation. J. Phys. Chem. 1991, 95, 6341–6346. [Google Scholar] [CrossRef]
- Schild, C.; Wokaun, A.; Baiker, A. Surface species in CO2 methanation over amorphous palladium/zirconia catalysts. J. Mol. Catal. 1991, 69, 347–357. [Google Scholar] [CrossRef]
- Benitez, J.J.; Carrizosa, I.; Odriozola, J.A. HCOOH hydrogenation over lanthanide-oxide-promoted Rh/Al2O3 catalysts. Appl. Surf. 1993, 68, 565–573. [Google Scholar] [CrossRef]
- Yashima, M.; Morimoto, K.; Ishizawa, N.; Yoshimura, M. Diffusionless Tetragonal–Cubic Transformation Temperature in Zirconia–Ceria Solid Solutions. J. Am. Ceram. Soc. 1993, 76, 2865–2868. [Google Scholar] [CrossRef]
- Yashima, M.; Morimoto, K.; Ishizawa, N.; Yoshimura, M. Zirconia–Ceria Solid Solution Synthesis and the Temperature–Time–Transformation Diagram for the 1:1 Composition. J. Am. Chem. Soc. 1993, 76, 1745–1750. [Google Scholar] [CrossRef]
- Yashima, M.; Ohtake, K.; Arashi, H.; Kakihana, M.; Yoshimura, M. Determination of cubic-tetragonal phase boundary in Zr1−xYxO2−x/2 solid solutions by Raman spectroscopy. J. Appl. Phys. 1993, 74, 7603–7605. [Google Scholar] [CrossRef]
- Yashima, M.; Arashi, H.; Kakihana, M.; Yoshimura, M. Raman Scattering Study of Cubic–Tetragonal Phase Transition in Zr1−xCexO2 Solid Solution. J. Am. Ceram. Soc. 1994, 77, 1067–1071. [Google Scholar] [CrossRef]
- Pan, Q.; Peng, J.; Sun, T.; Gao, D.; Wang, S.; Wang, S. CO2 methanation on Ni/Ce0.5Zr0.5O2 catalysts for the production of synthetic natural gas. Fuel Proc. Technol. 2014, 123, 166–171. [Google Scholar] [CrossRef]
- Kozlov, A.I.; Kim, D.H.; Yezerets, A.; Andersen, P.; Kung, H.H.; Kung, M.C. Effect of Preparation Method and Redox Treatment on the Reducibility, and Structure of Supported Ceria-Zirconia Mixed Oxide. J. Catal. 2002, 209, 417–426. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, S.; Liu, Y. Reverse water gas shift reaction over Co-precipitated Ni-CeO2 catalysts. J. Rare Earth 2008, 26, 66–70. [Google Scholar] [CrossRef]
- Roh, H.S.; Potdar, H.S.; Jun, K.W.; Kim, J.W.; Oh, Y.S. Carbon dioxide reforming of methane over Ni incorporated into Ce-ZrO2 catalysts. Appl. Catal. A 2004, 276, 231–239. [Google Scholar] [CrossRef]
- Takano, H.; Shinomiya, H.; Izumiya, K.; Kumagai, N.; Habazaki, H.; Hashimoto, K. CO2 methanation of Ni catalysts supported on tetragonal ZrO2 doped with Ca2+ and Ni2+ ions. Int. J. Hydrogen Energy 2015, 40, 8347–8355. [Google Scholar] [CrossRef]
- Perkas, N.; Amirian, G.; Zhong, Z.; Teo, J.; Gofer, Y.; Gedanken, A. Methanation of Carbon Dioxide on Ni Catalysts on Mesoporous ZrO2 Doped with Rare Earth Oxides. Catal. Lett. 2009, 130, 455–462. [Google Scholar] [CrossRef]
- Cai, M.; Wen, J.; Chu, W.; Cheng, X.; Li, Z. Methanation of carbon dioxide on Ni/ZrO2-Al2O3 catalysts: Effects of ZrO2 promoter and preparation method of novel ZrO2-Al2O3 carrier. J. Nat. Gas Chem. 2011, 20, 318–324. [Google Scholar] [CrossRef]
- Lu, H.; Yang, X.; Gao, G.; Wang, K.; Shi, Q.; Wang, J.; Han, C.; Liu, J.; Tong, M.; Liang, X.; et al. Mesoporous zirconia-modified clays supported nickel catalysts for CO and CO2 methanation. Int. J. Hydrogen Energy 2014, 39, 18894–18907. [Google Scholar] [CrossRef]
- Abate, S.; Mebrahtu, C.; Giglio, E.; Deorsola, F.; Bensaid, S.; Perathoner, S.; Pirone, R.; Centi, G. Catalytic performance of γ-Al2O3-ZrO2-TiO2-CeO2 composite oxide supported Ni-based catalysts for CO2 methanation. Int. Eng. Chem. Res. 2016, 55, 4451–4460. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, Y.F.; Qian, G.; Xu, Z.P.; Chen, C.; Liu, Q. Reinvestigation of dehydration and dehydroxylation of hydrotalcite-like compounds through combined TG-DTA-MS analyses. J. Phys. Chem. 2010, 114, 10768–10774. [Google Scholar] [CrossRef]
- Abate, S.; Barbera, K.; Giglio, E.; Deorsola, F.; Bensaid, S.; Perathoner, S.; Pirone, R.; Centi, G. Synthesis, characterization and activity pattern of Ni-Al Hydrotalcite catalysts in CO2 methanation. Ind. Eng. Chem. Res. 2016, 55, 8299–8308. [Google Scholar] [CrossRef]
- He, L.; Lin, Q.; Liu, Y.; Huang, Y. Unique catalysis of Ni-Al hydrotalcite derived catalyst in CO2 methanation: Cooperative effect between Ni nanoparticle and a basic support. J. Energy Chem. 2014, 23, 587–592. [Google Scholar] [CrossRef]
- Gabrovska, M.; Edreva-Kardjieva, R.; Crisan, D.; Tzvetkov, P.; Shopska, M.; Shtereva, I. Ni-Al layered double hydroxides as catalyst precursors for CO2 removal by methanation. React. Kinet. Mech. Catal. 2012, 105, 79–99. [Google Scholar] [CrossRef]
- Wierzbicki, D.; Debek, R.; Motak, M.; Grzybek, T.; Galvez, M.E.; Da Costa, P. Novel Ni-La-hydrotalcite derived catalysts for CO2 methanation. Catal. Commun. 2016, 83, 5–8. [Google Scholar] [CrossRef]
- Feng, Y.; Yang, W.; Chen, S.; Chu, W. Cerium promoted nano nickel catalysts Ni-Ce/CNTs and Ni-Ce/Al2O3 for CO2 methanation. Integr. Ferroelectr. 2014, 151, 116–125. [Google Scholar] [CrossRef]
- Yan, Y.; Dai, Y.; He, H.; Yu, Y.; Yang, Y. A novel W-doped Ni-Mg mixed oxide catalysts for CO2 methanation. Appl. Catal. B Environ. 2016, 196, 108–116. [Google Scholar] [CrossRef]
- Merkache, R.; Fechete, I.; Maamache, M.; Bernard, M.; Turek, P.; Al-Dalama, K.; Garin, F. 3D ordered mesoporous Fe-KIT-6 catalysts for methylcyclopentane (MCP) conversion and carbon dioxide (CO2) hydrogeneation for energy and environmental applications. Appl. Catal. A Gen. 2015, 504, 672–681. [Google Scholar] [CrossRef]
- Tsuji, M.; Kodama, T.; Yoshida, T.; Kitayama, Y.; Tamaura, Y. Preparation and CO2 Methanation Activity of an ultrafine Ni(II) Ferrite Catalyst. J. Catal. 1996, 164, 315–321. [Google Scholar] [CrossRef]
- Nishizawa, K.; Kodama, T.; Tabata, M.; Yoshida, T.; Tsuji, M.; Tamaura, Y. Adsorption of CO2 on oxygen-deficient magnetite: Adsorption enthalpy and adsorption isotherm. J. Chem. Soc. Faraday Trans. 1992, 88, 2771–2773. [Google Scholar] [CrossRef]
- Yoshida, T.; Nishizawa, K.; Tabata, M.; Abe, H.; Kodama, T.; Tsuji, M.; Tamaura, Y. Methanation of CO2 with H2-reduced magnetite. J. Mater. Sci. 1993, 28, 1220–1226. [Google Scholar] [CrossRef]
- Nishizawa, K.; Kato, H.; Mimori, K.; Yoshida, T.; Hasegawa, N.; Tsuji, M.; Tamaura, Y. Methanation of carbon deposited directly from CO2 on rhodium-bearing activated magnetite. J. Mater. Sci. 1994, 29, 768–772. [Google Scholar] [CrossRef]
- Tsuji, M.; Nishizawa, K.; Yoshida, T.; Tamaura, Y. Methanation reactivity of carbon deposited directly from CO2 on to the oxygen-deficient magnetite. J. Mater. Sci. 1994, 29, 5481–5484. [Google Scholar] [CrossRef]
- Tsuji, M.; Kato, H.; Kodama, T.; Chang, S.G.; Hesegawa, N.; Tamaura, Y. Methanation of CO2 on H2-reduced Ni(II)-or Co(II)-bearing ferrites at 200 °C. J. Mater. Sci. 1994, 29, 6227–6230. [Google Scholar] [CrossRef]
- Kato, H.; Sano, T.; Wada, Y.; Tamaura, Y.; Tsuji, M.; Tsuji, T.; Miyazaki, S. Methanation of CO2 with the oxygen-deficient Ni(II)-ferrite under dynamic conditions. J. Mater. Sci. 1995, 30, 6350–6354. [Google Scholar] [CrossRef]
- Kodama, T.; Kitayama, Y.; Tsuji, M.; Tamaura, Y. Methanation of CO2 using ultrafine NixFe3−xO4. Energy 1997, 22, 183–187. [Google Scholar] [CrossRef]
- Yoshida, T.; Tsuji, M.; Tamaura, Y.; Hurue, T.; Hayashida, T.; Ogawa, K. Carbon recycling system through methanation of CO2 in flue gas in LNG power plant. Energy Convers. Manag. 1997, 38, 443–448. [Google Scholar] [CrossRef]
- Bligaard, T.; Nørskov, J.K.; Dahl, S.; Matthiesen, J.; Christensen, C.H.; Sehested, J. The Brønsted–Evans–Polanyi relation and the volcano curve in heterogeneous catalysis. J. Catal. 2004, 224, 206–217. [Google Scholar] [CrossRef]
- Andersson, M.P.; Bligaard, T.; Kustov, A.; Larsen, K.E.; Greeley, J.; Johannessen, T.; Christensen, C.H.; Nørskov, J.K. Toward computational screening in heterogeneous catalysis: Pareto-optimal methanation catalysts. J. Catal. 2006, 239, 501–506. [Google Scholar] [CrossRef]
- Sehesteda, J.; Larsen, K.E.; Kustov, A.L.; Frey, A.M.; Johannessen, T.; Bligaard, T.; Andersson, M.P.; Nørskov, J.K.; Christensen, C.H. Discovery of technical methanation catalysts based on computational Screening. Top. Catal. 2007, 45, 9–13. [Google Scholar] [CrossRef]
- Kustov, A.L.; Frey, A.M.; Larsen, K.E.; Johannessen, T.; Nørskov, J.K.; Christensen, C.H. CO methanation over supported bimetallic Ni-Fe catalysts: From computational studies towards catalyst optimization. Appl. Catal. A Gen. 2007, 320, 98–104. [Google Scholar] [CrossRef]
- Ertl, G.; Knozinger, H.; Schüth, F.; Weitkamp, J. Handbook of Heterogenous Catalysis; Wiley-VCH: Weinheim, Germany, 1997. [Google Scholar]
- Dumesic, J.A.; Rudd, D.F.; Aparicio, L.M.; Rekoske, J.E.; Treviño, A.A. The Microkinetics of Heterogeneous Catalysis; ACS Professional Reference Book; American Chemical Society: Washington, DC, USA, 1993; p. 315. [Google Scholar]
- Boudart, M.; Djega-Mariadassou, G. Kinetics of Heterogeneous Catalytic Reactions; Pricenton University: Princeton, NJ, USA, 1984. [Google Scholar]
- Nørskov, J.K.; Bligaard, T.; Logadottir, A.; Bahn, S.; Hansen, L.B.; Bollinger, M.; Bengaard, H.; Hammer, B.; Sljivancanin, Z.; Mavrikakis, M.; et al. Universality in Heterogeneous Catalysis. J. Catal. 2002, 209, 275–278. [Google Scholar] [CrossRef]
- Linic, S.; Barteau, M.A. Control of Ethylene Epoxidation Selectivity by Surface Oxametallacycles. J. Am. Chem. Soc. 2003, 125, 4034–4035. [Google Scholar] [CrossRef] [PubMed]
- Reuter, K.; Frenkel, D.; Scheffler, M. The Steady State of Heterogeneous Catalysis, Studied by First-Principles Statistical Mechanics. Phys. Rev. Lett. 2004, 93, 116105. [Google Scholar] [CrossRef] [PubMed]
- Honkala, K.; Hellman, A.; Remediakis, I.N.; Logadottir, A.; Carlsson, A.; Dahl, S.; Christensen, C.H.; Nørskov, J.K. Ammonia Synthesis from First-Principles Calculations. Science 2005, 307, 555–558. [Google Scholar] [CrossRef] [PubMed]
- Pareto, V.; Montesano, A.; Zanni, A.; Bruni, L.; Chipman, J.S.; McLure, M. Manual of Political Economy: A Critical and Variorum Edition, 1st ed.; Oxford University Press: Oxford, UK, 2014. [Google Scholar]
- Bligaard, T.; Jóhannesson, G.H.; Ruban, A.V.; Skriver, H.L.; Jacobsen, K.W.; Nørskov, J.K. Pareto-optimal alloys. Appl. Phys. Lett. 2003, 83, 4527–4529. [Google Scholar] [CrossRef] [Green Version]
- Mori, S.; Xu, W.C.; Ishidzuki, T.; Ogasawara, N.; Imai, J.; Kobayashi, K. Mechanochemical activation of catalysts for CO2 methanation. Appl. Catal. A Gen. 1996, 137, 255–268. [Google Scholar] [CrossRef]
- Yaccato, K.; Carhart, R.; Hagemeyer, A.; Lesik, A.; Strasser, P.; Volpe, A.F., Jr.; Turner, H.; Weinberg, H.; Grasselli, R.K.; Brooks, C. Competitive CO and CO2 methanation over supported noble metal catalysts in high throughput scanning mass spectrometer. Appl. Catal. A Gen. 2005, 296, 30–48. [Google Scholar] [CrossRef]
- Ren, J.; Guo, H.; Yang, J.; Qin, Z.; Lin, J.; Li, Z. Insights into the mechanisms of CO2 methanation on Ni(111) surfaces by density functional theory. Appl. Surf. Sci. 2015, 351, 504–516. [Google Scholar] [CrossRef]
- Goodman, D.J. Methanation of Carbon Dioxide. Mc.S. Thesis, University of California, Los Angeles, CA, USA, 2013; pp. 1–38. [Google Scholar]
- Lapidus, A.L.; Gaidai, N.A.; Nekrasov, N.V.; Tishkova, L.A.; Agafonov, Y.A.; Myshenkova, T.N. The mechanism of carbon dioxide hydrogenation on copper and nickel catalysts. Pet. Chem. 2007, 47, 75–82. [Google Scholar] [CrossRef]
- Miao, B.; Ma, S.S.K.; Wang, X.; Su, H.; Chan, S.H. Catalysis mechanism of CO2 and CO methanation. Catal. Sci. Technol. 2016, 6, 4048–4058. [Google Scholar] [CrossRef]
- Choe, S.J.; Kang, H.J.; Kim, S.J.; Park, S.B.; Park, D.H.; Huh, D.S. Adsorbed carbon formation and carbon hydrogenation for CO2 methanation on the Ni(111) surface: ASED-MO study. Bull. Korean Chem. Soc. 2005, 26, 1682–1688. [Google Scholar]
- Panagiotopoulou, P.; Kondarides, D.I.; Verykios, X.E. Mechanistic aspects of the selective methanation of CO over Ru/TiO2 catalyst. Catal. Today 2012, 181, 138–147. [Google Scholar] [CrossRef]
- Medsforth, S. CLXIX—Promotion of catalytic reactions: Part I. J. Chem. Soc. Trans. 1923, 123, 1452–1469. [Google Scholar] [CrossRef]
- Schild, C.; Wokaun, A.; Baiker, A. On the mechanism of CO and CO2 hydrogenation reactions on zirconia-supported catalysts: A diffuse reflectance FTIR study: Part II. Surface species on copper/zirconia catalysts: Implications for methanoi synthesis selectivity. J. Mol. Catal. 1990, 63, 243–255. [Google Scholar] [CrossRef]
- Kang, S.H.; Ryu, J.H.; Kim, J.H.; Seo, S.J.; Yoo, Y.D.; Prasad, P.S.S.; Lim, H.J.; Byun, C.D. Co-methanation of CO and CO2 on the Nix-Fe1−x/Al2O3 catalysts: Effect of Fe contents. Korean J. Chem. Eng. 2011, 28, 2282–2286. [Google Scholar] [CrossRef]
- Hwang, S.; Lee, J.; Hong, U.G.; Jung, J.C.; Koh, D.J.; Lim, H.; Byun, C.; Song, I.K. Hydrogenation of carbon monoxide to methane over mesoporous nickel-M-alumina (M = Fe, Ni, Co, Ce, and La) xerogel catalysts. Ind. Eng. Chem. 2012, 18, 243–248. [Google Scholar] [CrossRef]
- Hwang, S.; Hong, U.G.; Lee, J.; Seo, J.G.; Baik, J.H.; Koh, D.J.; Lim, H.; Song, I.K. Methanation of carbon dioxide over mesoporous Ni-Fe-Al2O3 catalysts prepared by a coprecipitation method: Effect of precipitation agent. Ind. Eng. Chem. 2013, 19, 2016–2021. [Google Scholar] [CrossRef]
- Hwang, S.; Lee, J.; Hong, U.G.; Baik, J.H.; Koh, D.J.; Lim, H.; Song, I.K.S. Methanation of carbon dioxide over mesoporous Ni-Fe-Ru-Al2O3 xerogel catalysts: Effect of ruthenium content. Ind. Eng. Chem. 2013, 19, 698–703. [Google Scholar] [CrossRef]
- Kopyscinski, J.; Schildhauer, T.J.; Biollaz, S.M.A. Employing Catalyst Fluidization to Enable Carbon Management in the Synthetic Natural Gas Production from Biomass. Chem. Eng. Technol. 2009, 32, 343–347. [Google Scholar] [CrossRef]
- Panagiotopoulou, P.; Kondarides, D.I.; Verykios, X.E. Selective methanation of CO over supported Ru catalysts. Appl. Catal. B Environ. 2009, 88, 470–478. [Google Scholar] [CrossRef]
- Park, E.D.; Lee, D.; Lee, H.C. Recent progress in selective CO removal in a H2-rich stream. Catal. Today 2009, 139, 280–290. [Google Scholar] [CrossRef]
- Snel, R. Supported iron catalysts in Fischer-Tropsch synthesis: Influence of the preparation method. Ind. Eng. Chem. Res. 1989, 28, 654–659. [Google Scholar] [CrossRef]
- Tsuji, M.; Wada, Y.; Yamamoto, T.; Sano, T.; Tamaura, Y. CO2 decomposition by metallic phase on oxygen-deficient Ni(II)-bearing ferrite. J. Mater. Sci. Lett. 1996, 15, 156–158. [Google Scholar] [CrossRef]
- Choung, J.W.; Xu, Z. Role of complexing agents in ferrite formation under ambient conditions. Ind. Eng. Chem. Res. 1999, 38, 4689–4693. [Google Scholar] [CrossRef]
- Rondinone, A.J.; Samia, A.C.S.; Zhang, Z.J. A chemometric approach for predicting the size of magnetic spinel ferrite nanoparticles from the synthesis conditions. J. Phys. Chem. B 2000, 104, 7919–7922. [Google Scholar] [CrossRef]
- Luo, H.Y.; Yue, Z.X.; Zhou, J. Synthesis and high-frequency magnetic properties of sol-gel derived Ni-Zn ferrite forsterite composites. J. Magn. Magn. Mater. 2000, 210, 104–108. [Google Scholar] [CrossRef]
- Tamaura, Y.; Tahata, M. Complete reduction of carbon dioxide to carbon using cation-excess magnetite. Nature 1990, 346, 255–256. [Google Scholar] [CrossRef]
- Kato, H.; Kodama, T.; Tsuji, M.; Tamaura, Y. Decomposition of carbon dioxide to carbon by hydrogen-reduced Ni(II)-bearing ferrite. J. Mater. Sci. 1994, 29, 5689–5692. [Google Scholar] [CrossRef]
- Tabata, M.; Nishida, Y.; Kodama, T.; Mimori, K.; Yoshida, T.; Tamaura, Y. CO2 decomposition with oxygen-deficient Mn(II) ferrite. J. Mater. Sci. 1993, 28, 971–974. [Google Scholar] [CrossRef]
- Tabata, M.; Akanuma, K.; Nishizawa, K.; Mimori, K.; Yoshida, T.; Tsuji, M.; Tamaura, Y. Reactivity of oxygen-deficient Mn(II)-bearing ferrites (MnxFe3−xO4−δ, 0 ≤ x ≥ δ > 0) toward CO2 decomposition to carbon. J. Mater. Sci. 1993, 28, 6753–6760. [Google Scholar] [CrossRef]
- Kodama, T.; Tabata, M.; Sano, T.; Tsuji, M.; Tamaura, Y. XRD and Mössbauer Studies on Oxygen-Deficient Ni(II)-Bearing Ferrite with a High Reactivity for CO2 Decomposition to Carbon. J. Solid State Chem. 1995, 120, 64–69. [Google Scholar] [CrossRef]
- Lin, K.S.; Adhikari, A.K.; Tsai, Z.Y.; Chen, Y.P.; Chien, T.T.; Tsai, H.B. Synthesis and characterization of nickel ferrite nanocatalysts for CO2 decomposition. Catal. Today 2011, 174, 88–96. [Google Scholar] [CrossRef]
- Tabata, M.; Akanuma, K.; Togawa, T.; Tsuji, M.; Tamaura, Y. Mössbauer study of oxygen-deficient ZnII-bearing ferrites (ZnxFe3−xO4−δ, 0 ≤ x ≤ 1) and their reactivity toward CO2 decomposition to carbon. J. Chem. Soc. Faraday Trans. 1994, 90, 1171–1175. [Google Scholar] [CrossRef]
- Sarangi, P.P.; Vadera, S.R.; Patra, M.K.; Ghosh, N.N. Synthesis and characterization of pure single phase Ni-Zn ferrite nanopowders by oxalate based precursor method. Powder Technol. 2010, 203, 348–353. [Google Scholar] [CrossRef]
- Shin, H.C.; Choi, S.C.; Jung, K.D.; Han, S.H. Mechanism of M Ferrites (M = Cu and Ni) in the CO2 decomposition reaction. Chem. Mater. 2001, 13, 1238–1242. [Google Scholar] [CrossRef]
- Nordhei, C.; Mathisen, K.; Safonova, O.; van Beek, W.; Nicholson, D.G. Decomposition of Carbon Dioxide at 500 °C over Reduced Iron, Cobalt, Nickel, and Zinc Ferrites: A Combined XANES−XRD Study. J. Phys. Chem. C 2009, 113, 19568–19577. [Google Scholar] [CrossRef]
- Nordhei, C.; Mathisen, K.; Bezverkhyy, I.; Nicholson, D. Decomposition of Carbon Dioxide over the Putative Cubic Spinel Nanophase Cobalt, Nickel, and Zinc Ferrites. J. Phys. Chem. C 2008, 112, 6531–6537. [Google Scholar] [CrossRef]
- Wang, G.; Xu, S.; Jiang, L.; Wang, C. Nickel supported on iron-bearing olivine for CO2 methanation. Int. J. Hydrogen Energy 2016, 41, 12910–12919. [Google Scholar] [CrossRef]
- Swierczynski, D.; Courson, C.; Bedel, L.; Kiennemann, A.; Guille, J. Characterization of Ni-Fe/MgO/Olivine catalyst for fluidized bed steam gasification of biomass. Chem. Mater. 2006, 18, 4025–4032. [Google Scholar] [CrossRef]
- Yang, X.; Xu, S.; Xu, H.; Liu, X.; Liu, C. Nickel supported on modified olivine catalyst for steam reforming of biomass gasification tar. Catal. Commun. 2010, 11, 383–386. [Google Scholar] [CrossRef]
- Lu, H.; Yang, X.; Gao, G.; Wang, J.; Han, C.; Liang, X.; Li, C.; Li, Y.; Zhang, W.; Chen, X. Metal (Fe, Co, Ce or La) doped nickle catalyst supported on ZrO2 modified mesoporous clays for CO and CO2 methanation. Fuel 2016, 183, 335–344. [Google Scholar] [CrossRef]
- Pandey, D.; Deo, G. Effect of support on the catalytic activity of Ni-Fe catalysts for CO2 methanation reaction. J. Ind. Eng. Chem. 2016, 33, 99–107. [Google Scholar] [CrossRef]
- Project Homepage—HELMETH. Available online: http://www.helmeth.eu/index.php/project (accessed on 16 December 2016).
- Project Homepage—BioCat Project, Electrochaea. Available online: http://www.electrochaea.com/technology/ (accessed on 16 December 2016).
- Kristjanpoller, F.; Crespo, A.; Barbera, L.; Viveros, P. Biomethanation plant assessment based on reliability impact on operational effectiveness. Renew. Energy 2017, 101, 301–310. [Google Scholar] [CrossRef]






© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frontera, P.; Macario, A.; Ferraro, M.; Antonucci, P. Supported Catalysts for CO2 Methanation: A Review. Catalysts 2017, 7, 59. https://doi.org/10.3390/catal7020059
Frontera P, Macario A, Ferraro M, Antonucci P. Supported Catalysts for CO2 Methanation: A Review. Catalysts. 2017; 7(2):59. https://doi.org/10.3390/catal7020059
Chicago/Turabian StyleFrontera, Patrizia, Anastasia Macario, Marco Ferraro, and PierLuigi Antonucci. 2017. "Supported Catalysts for CO2 Methanation: A Review" Catalysts 7, no. 2: 59. https://doi.org/10.3390/catal7020059