
Effect of Copper Particle Size on the Surface Structure and Catalytic Activity of Cu–CeO2 Nanocomposites Prepared by Mechanochemical Synthesis in the Preferential CO Oxidation in a H2-Rich Stream (CO-PROX)
 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Results and Discussion
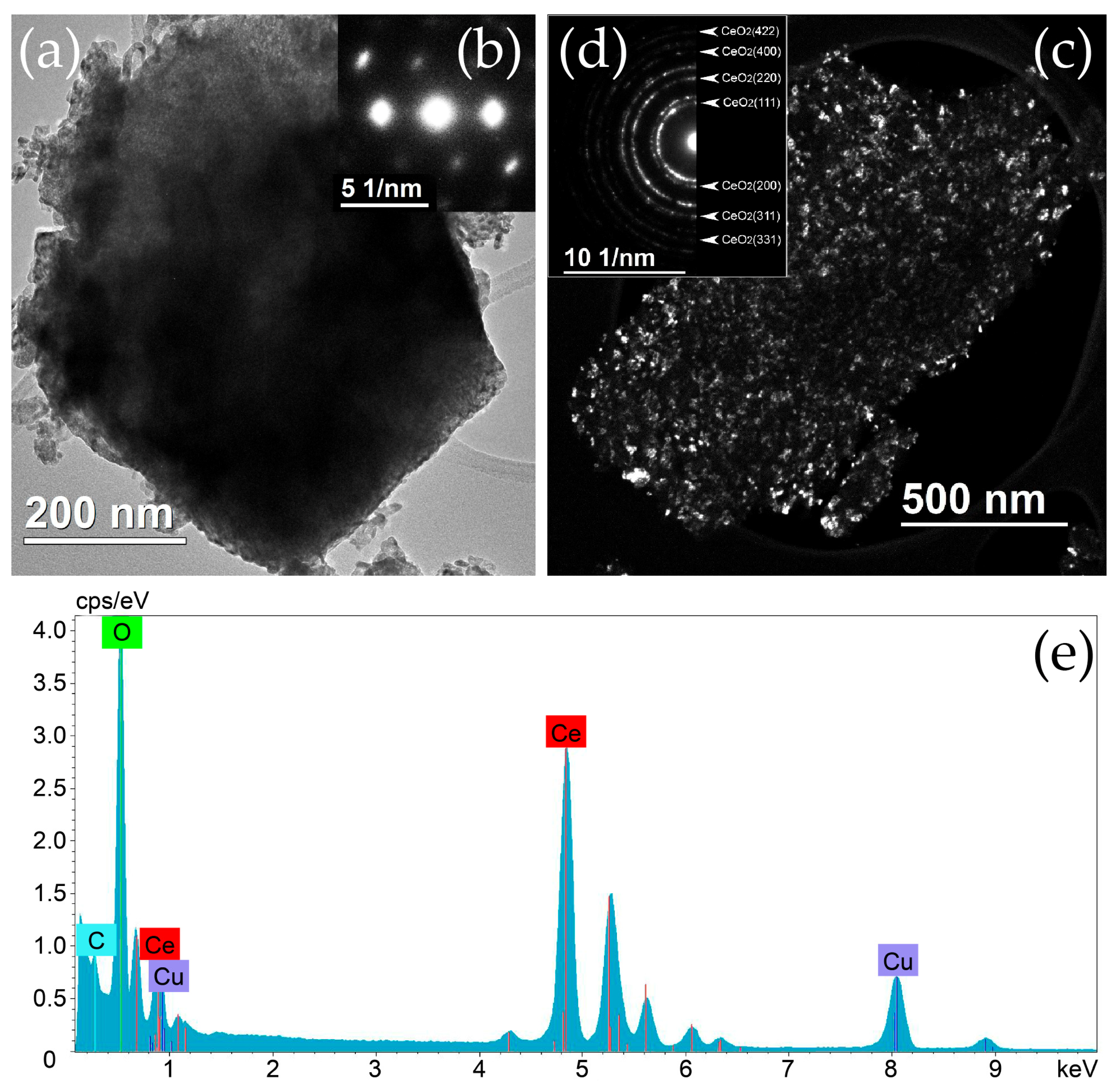
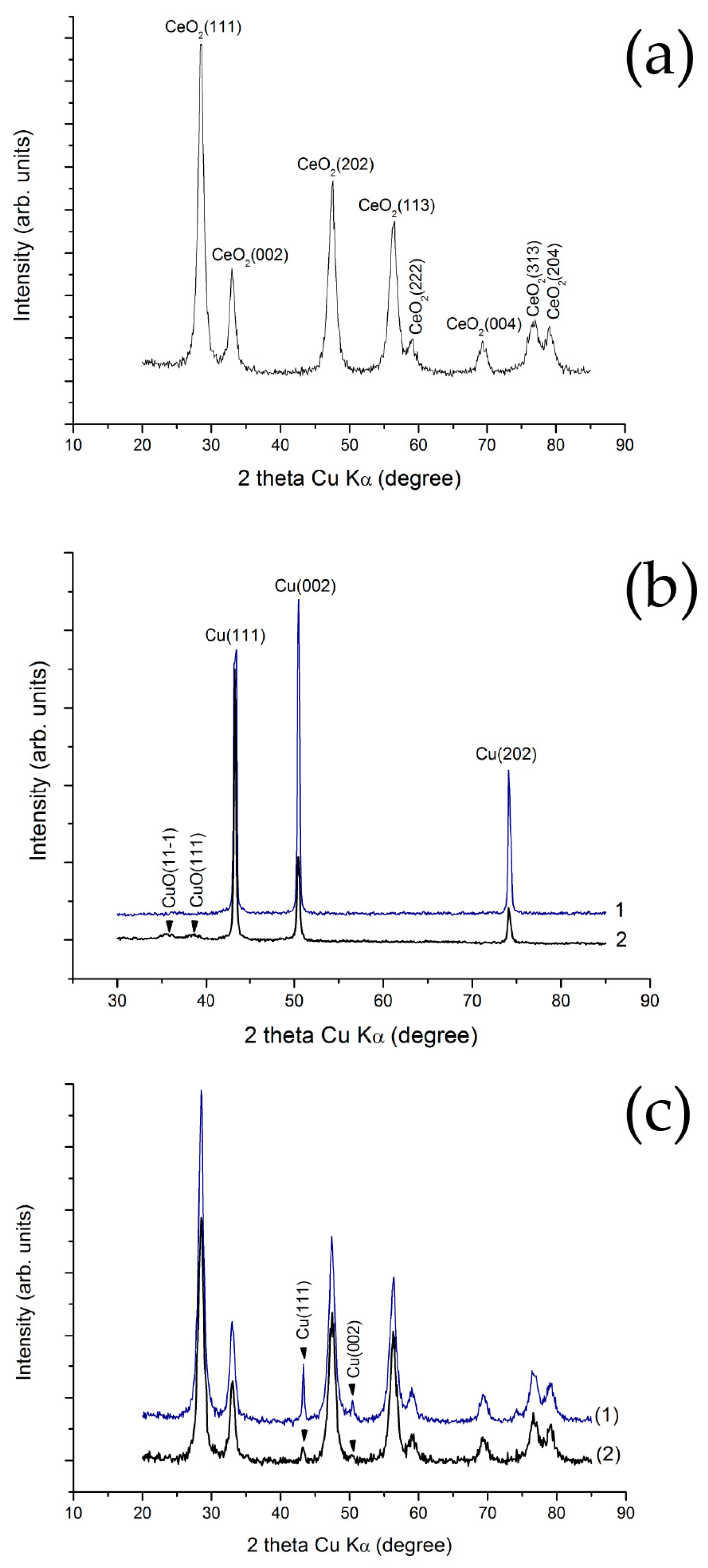
2.1. Structure and Morphology of Initial Materials
2.2. Structure and Morphology of Composite Materials
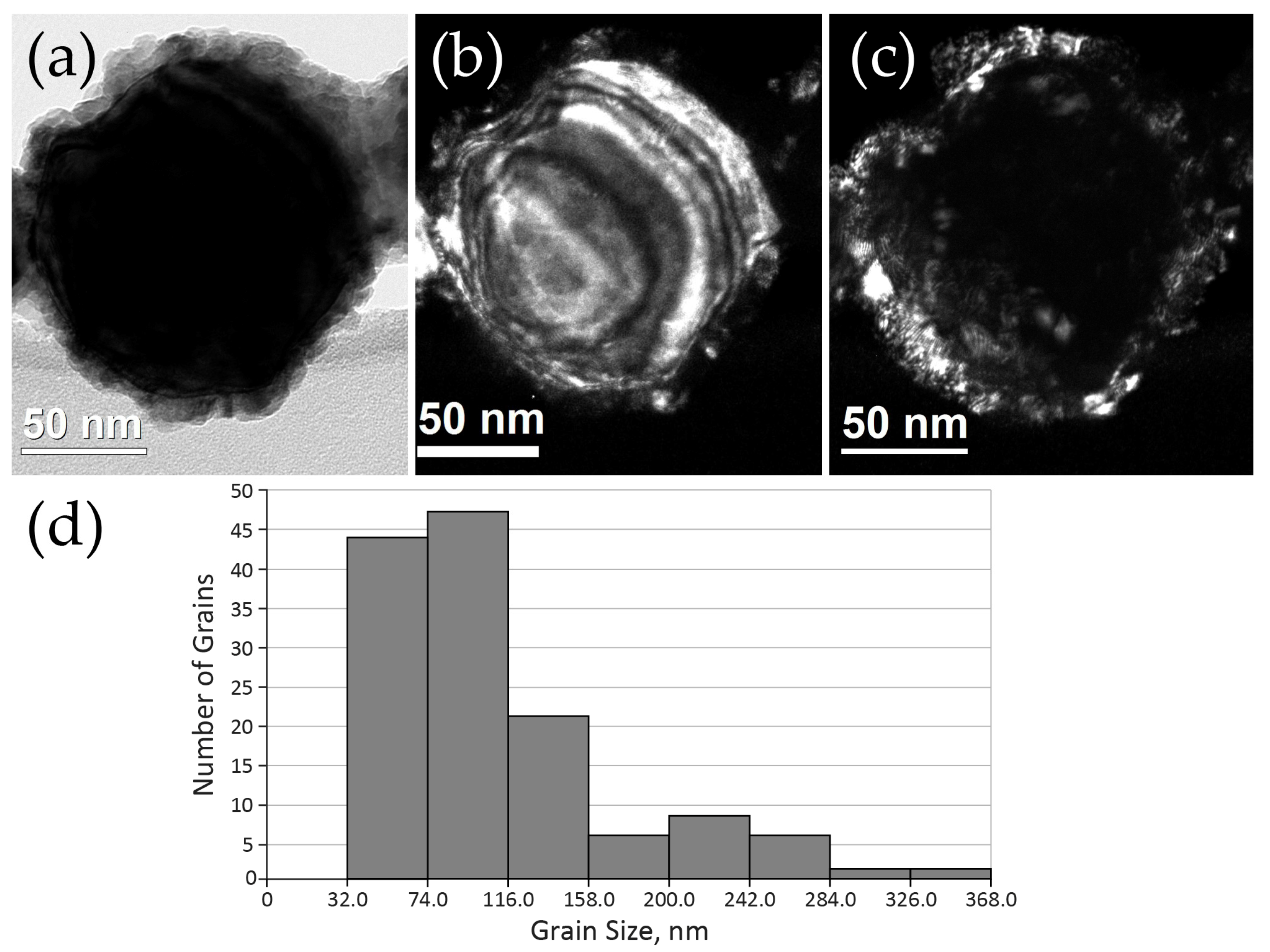
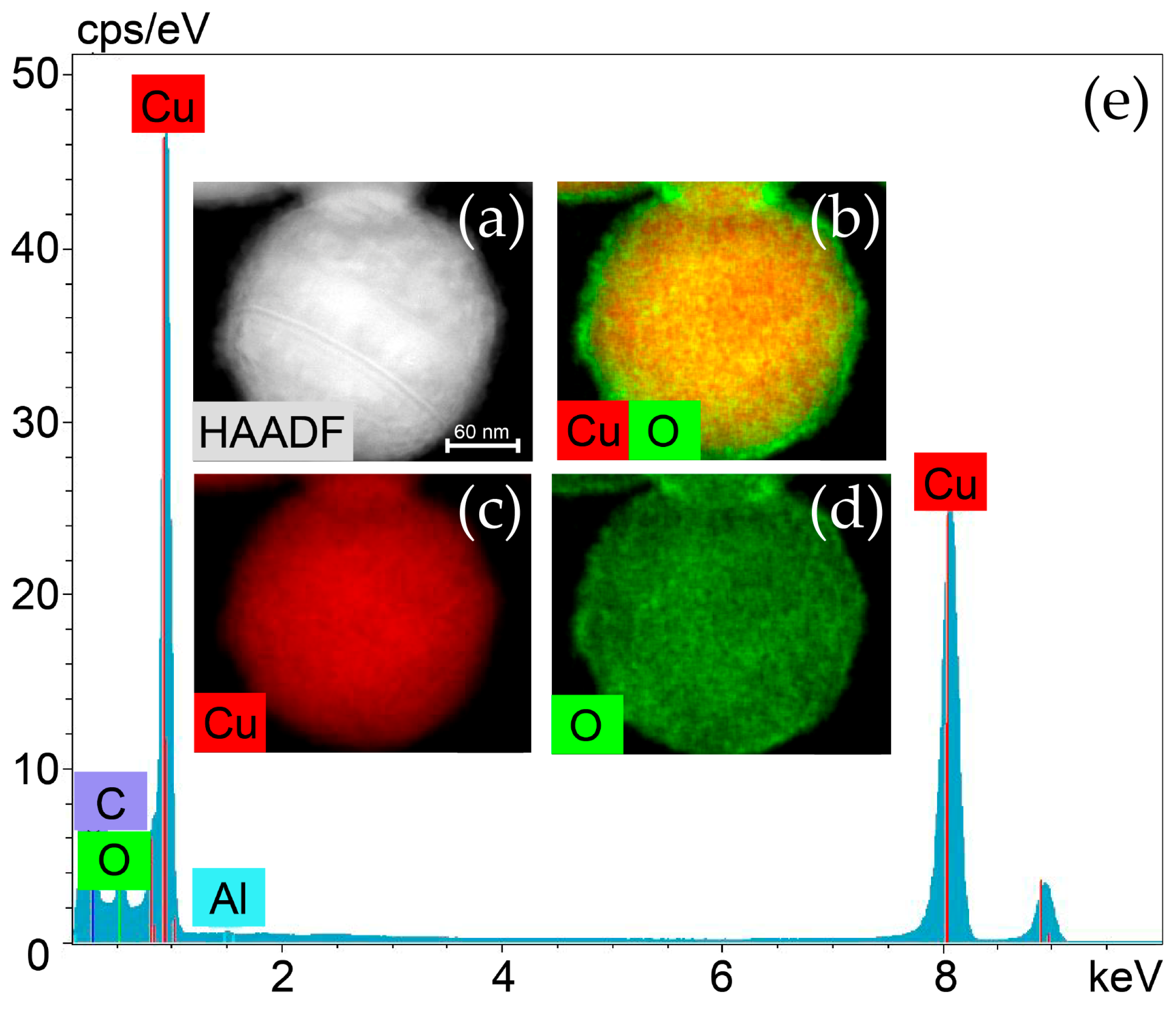
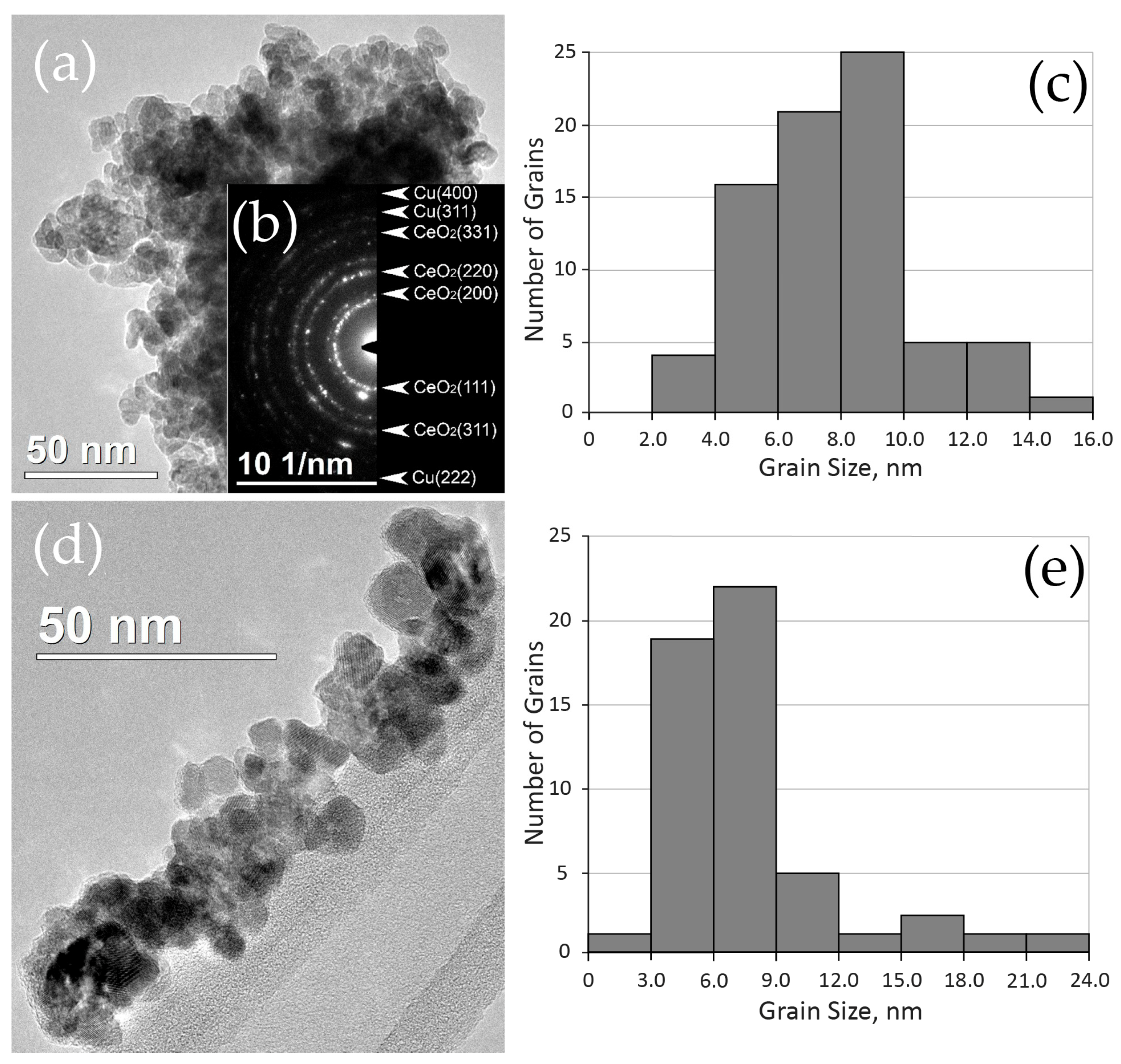
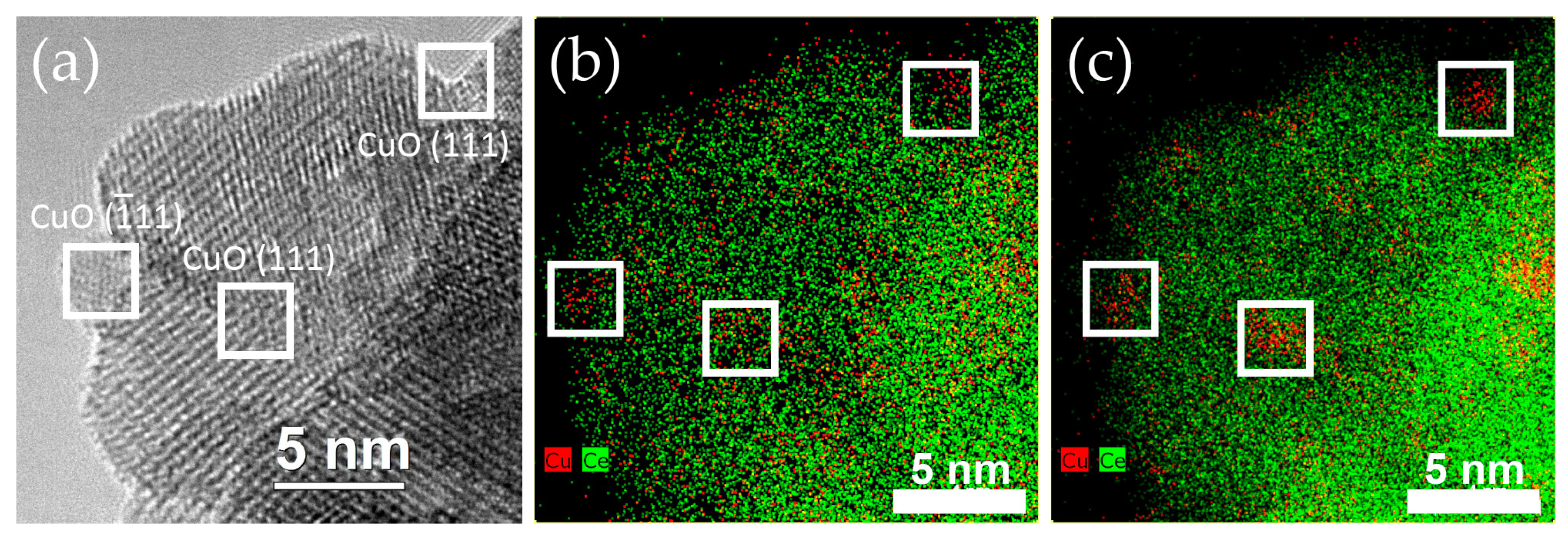
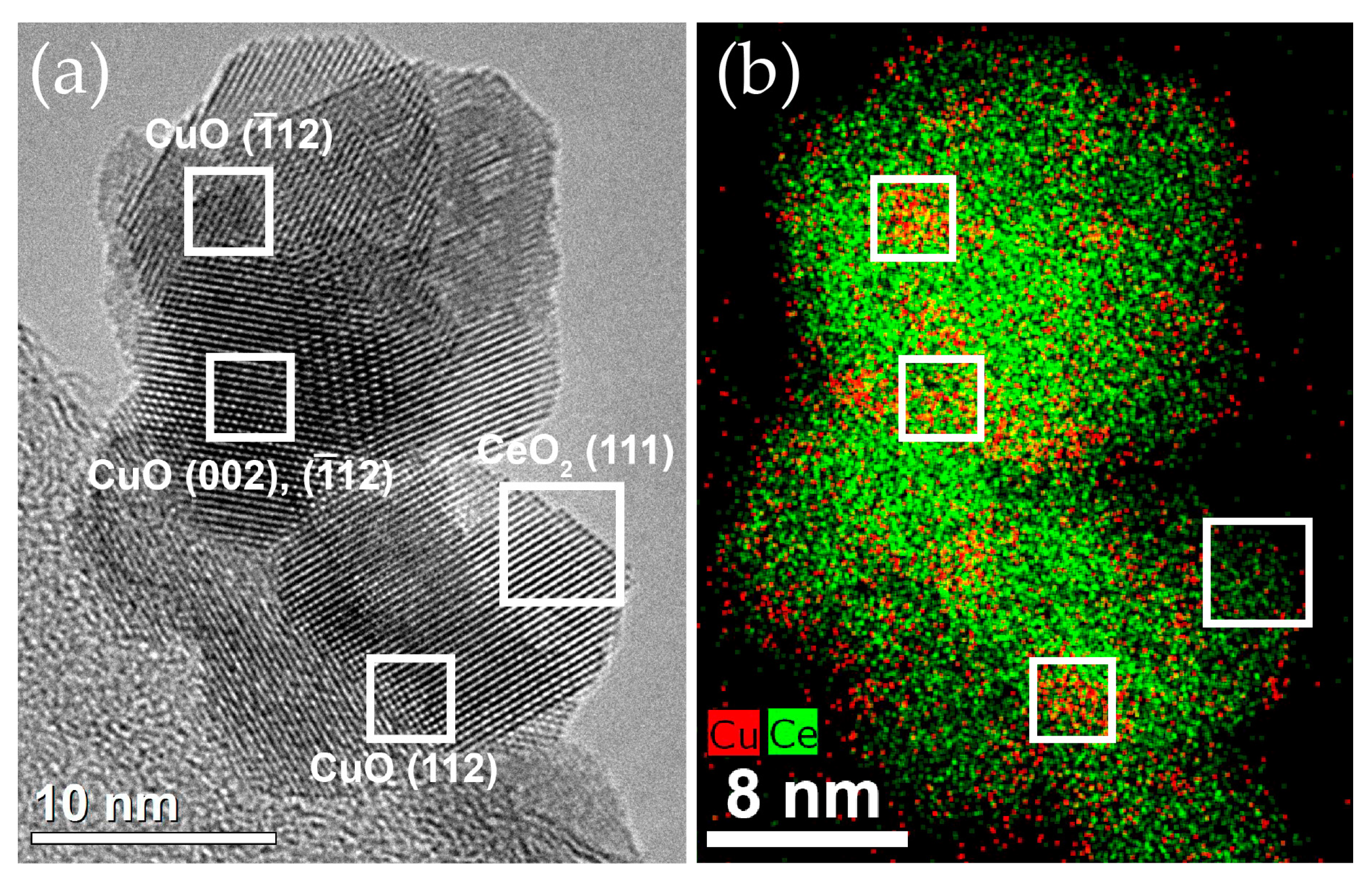
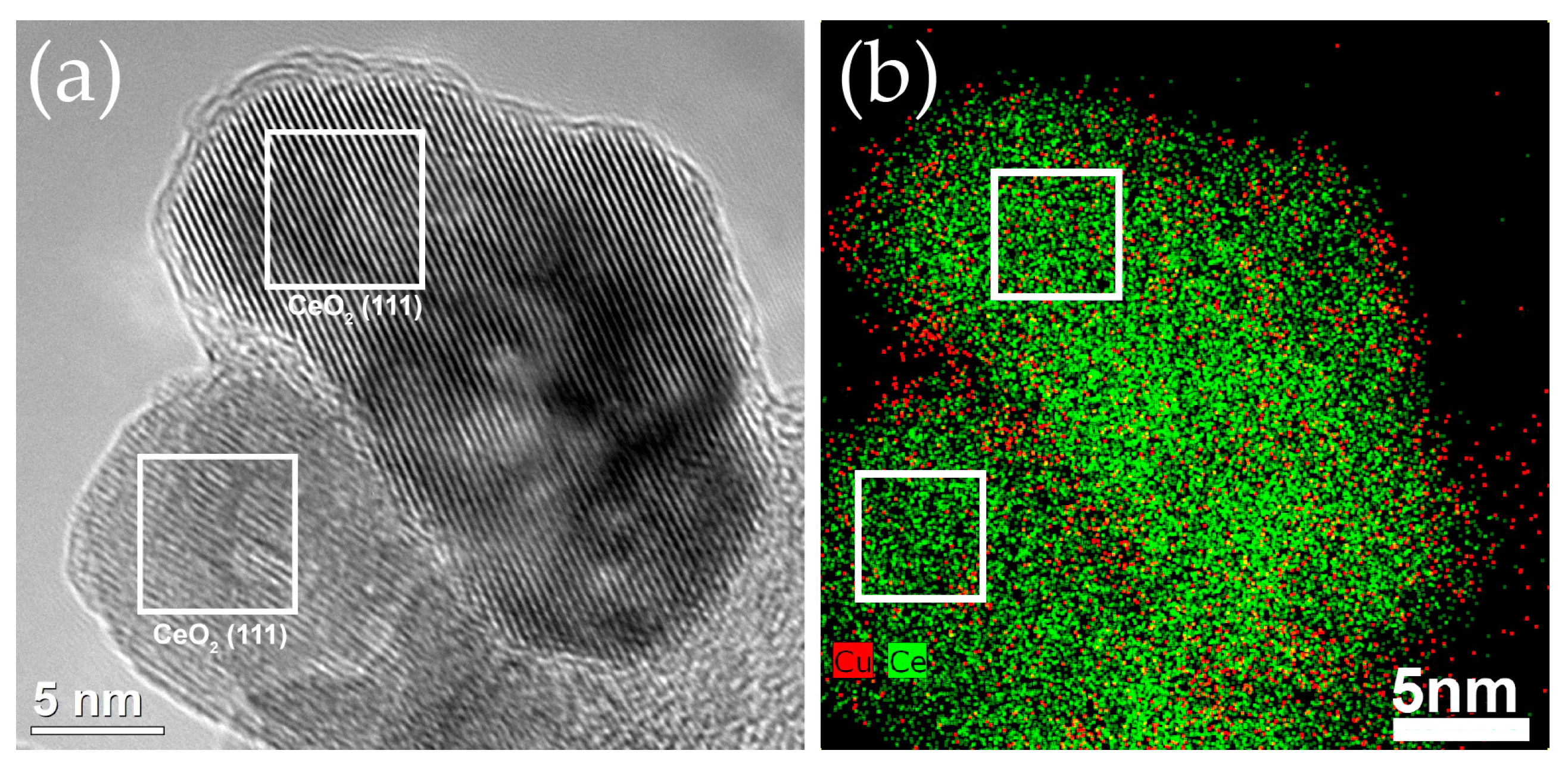
2.2.1. Distribution of Cu Supported on CeO2
2.2.2. Cerium Oxide Powders after Grinding for 30 min
2.2.3. Structure of Cu–CeO2 Composites
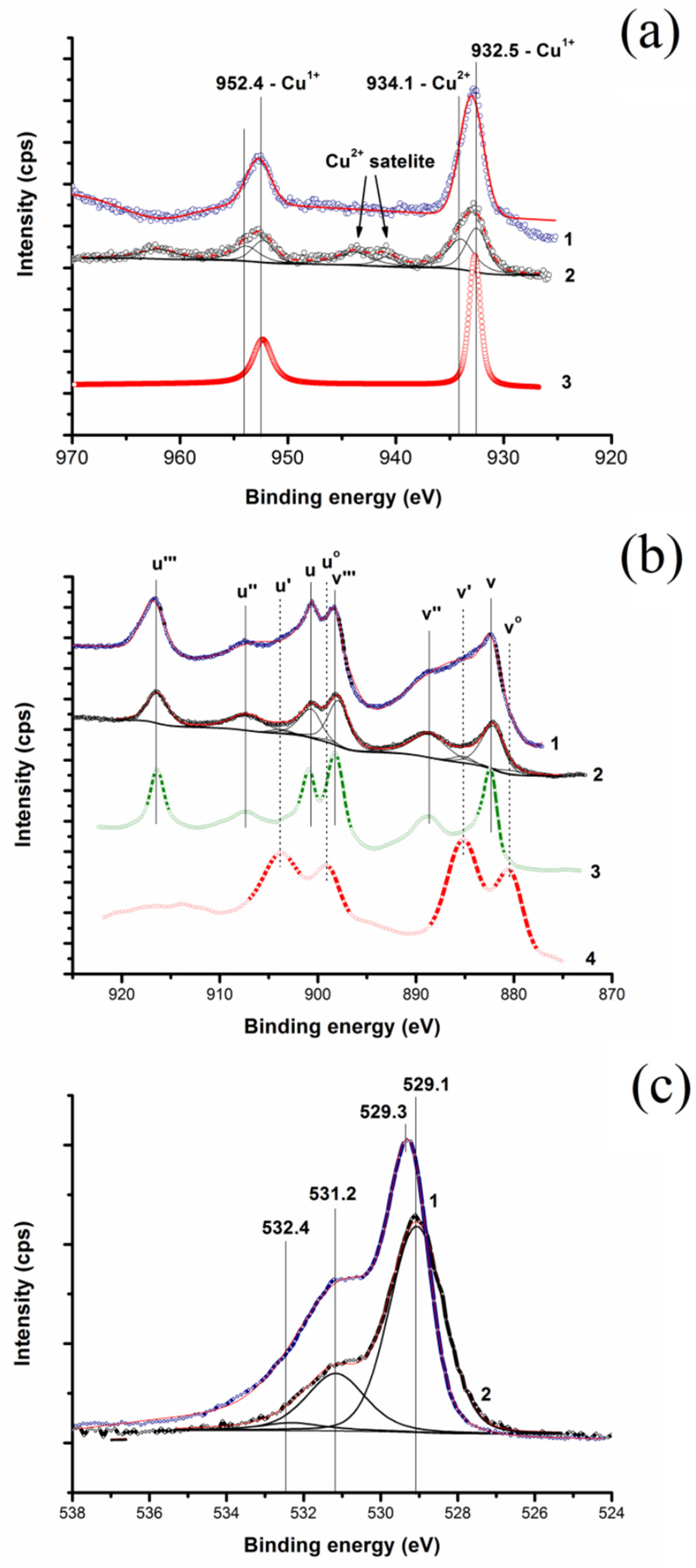
2.3. Chemical State of Cu–CeO2 Surface
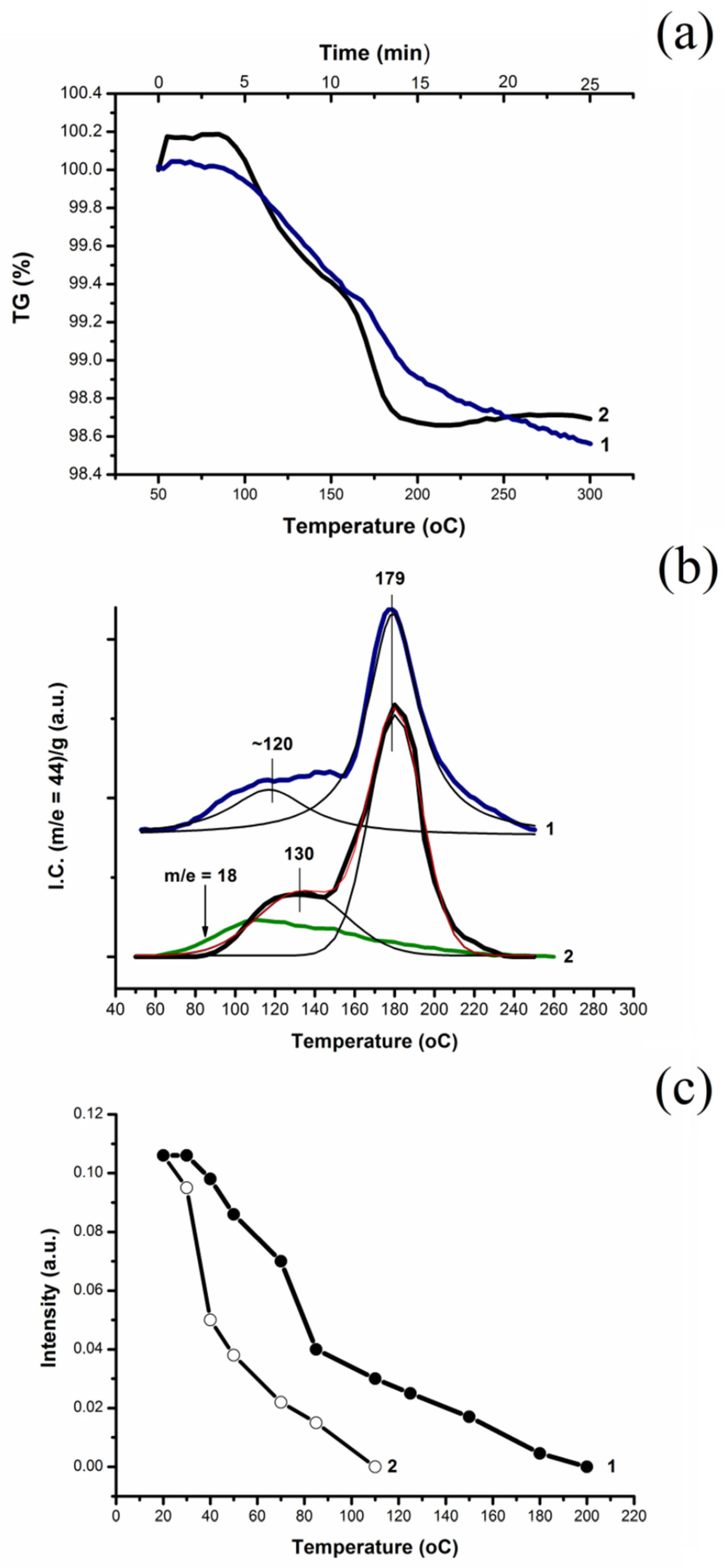
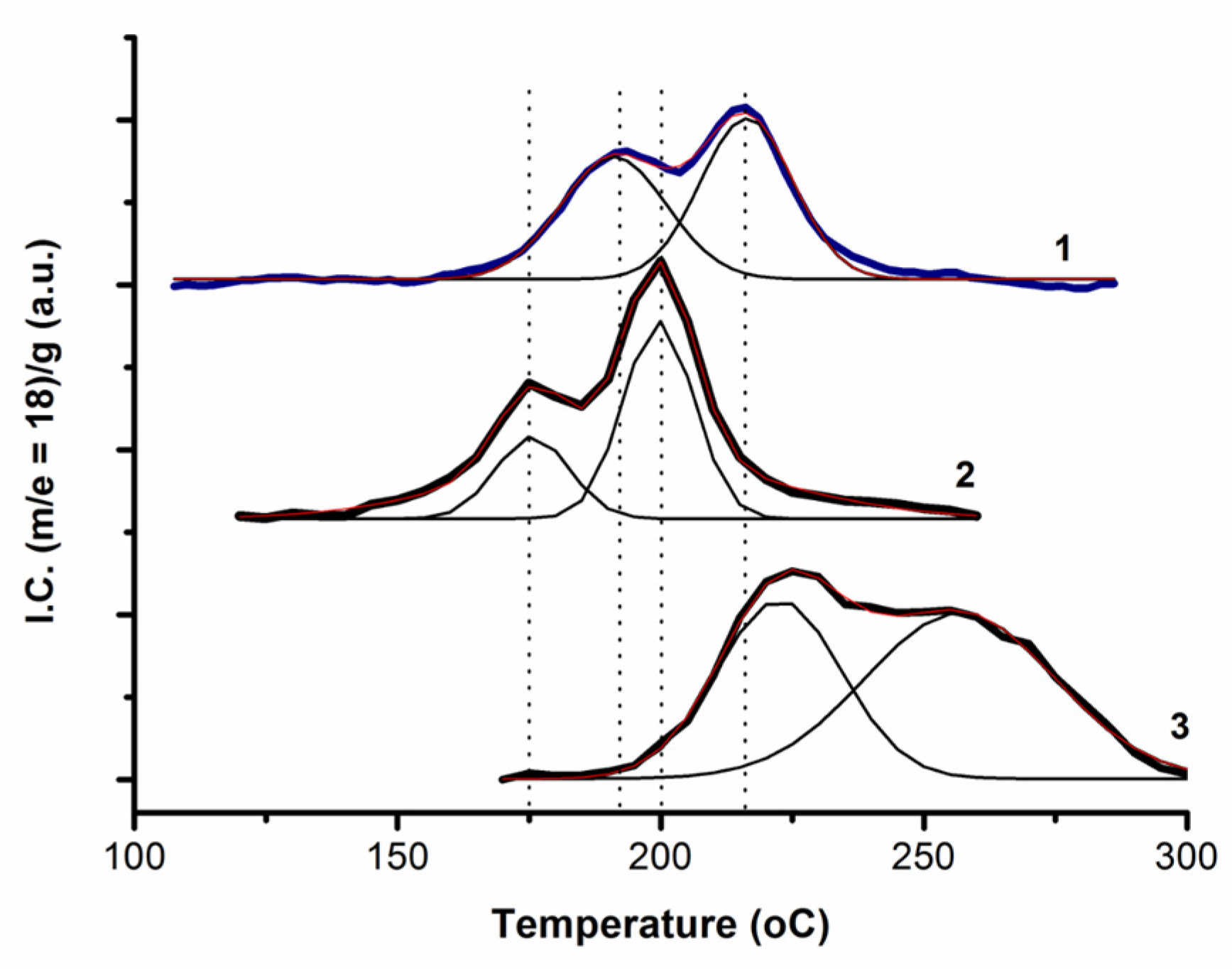
2.4. Oxidation of CO and H2 by Surface and Lattice Oxygen of Cu(D)–CeO2 and Cu(L)–CeO2
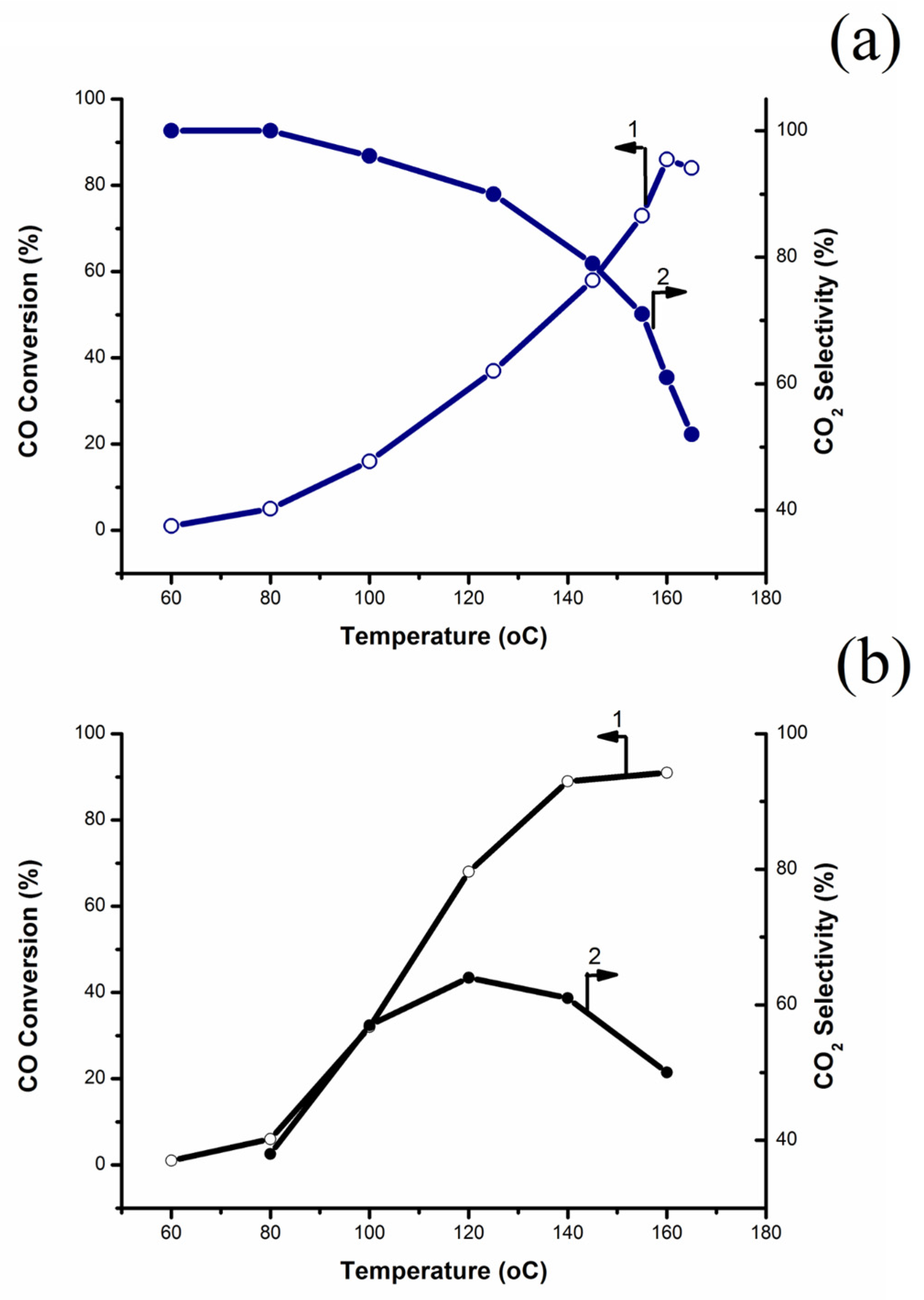
2.5. Catalytic Tests
3. Materials and Methods
3.1. Reagents
3.2. Preparation of Cu–CeO2 Catalysts
3.3. Catalytic Activity in CO-PROX Reaction
3.4. Catalyst Characterization
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Aboukaïs, A.; Skaf, M.; Hany, S.; Cousin, R.; Aouad, S.; Labaki, M.; Abi-Aad, E. A comparative study of Cu, Ag and Au doped CeO2 in the total oxidation of volatile organic compounds (VOCs). Mater. Chem. Phys. 2016, 177, 570–576. [Google Scholar] [CrossRef]
- Di Sarli, V.; Landi, G.; Di Benedetto, A.; Lisi, L. Synergy Between Ceria and Metals (Ag or Cu) in Catalytic Diesel Particulate Filters: Effect of the Metal Content and of the Preparation Method on the Regeneration Performance. Top. Catal. 2020, 64, 256–269. [Google Scholar] [CrossRef]
- Rico-Pérez, V.; Aneggi, E.; Trovarelli, A. The Effect of Sr Addition in Cu- and Fe-Modified CeO2 and ZrO2 Soot Combustion Catalysts. Catalysts 2017, 7, 28. [Google Scholar] [CrossRef]
- Lee, Y.-H.; Kim, H.-M.; Jeong, C.-H.; Jeong, D.-W. Effects of precipitants on the catalytic performance of Cu/CeO2catalysts for the water–gas shift reaction. Catal. Sci. Technol. 2021, 11, 6380–6389. [Google Scholar] [CrossRef]
- Zhang, Y.; Liang, L.; Chen, Z.; Wen, J.; Zhong, W.; Zou, S.; Fu, M.; Chen, L.; Ye, D. Highly efficient Cu/CeO2-hollow nanospheres catalyst for the reverse water-gas shift reaction: Investigation on the role of oxygen vacancies through in situ UV-Raman and DRIFTS. Appl. Surf. Sci. 2020, 516, 146035. [Google Scholar] [CrossRef]
- Ji, W.; Chen, X.; Li, Q.; Lin, K.; Deng, J.; Xing, X. Insights into CO Oxidation in Cu/CeO2 Catalysts: O2 Activation at the Dual-Interfacial Sites. Eur. J. Inorg. Chem. 2023, 26, e202200656. [Google Scholar] [CrossRef]
- Du, Z.; Liu, C.; Zhai, J.; Guo, X.; Xiong, Y.; Su, W.; He, G. A Review of Hydrogen Purification Technologies for Fuel Cell Vehicles. Catalysts 2021, 11, 393. [Google Scholar] [CrossRef]
- Wang, W.W.; Yu, W.Z.; Du, P.P.; Xu, H.; Jin, Z.; Si, R.; Ma, C.; Shi, S.; Jia, C.J.; Yan, C.H. Crystal plane effect of ceria on supported copper oxide cluster catalyst for CO oxidation: Importance of metal–support interaction. ACS Catal. 2017, 7, 1313–1329. [Google Scholar] [CrossRef]
- Konsolakis, M. The role of Copper–Ceria interactions in catalysis science: Recent theoretical and experimental advances. Appl. Catal. B Environ. 2016, 198, 49–66. [Google Scholar] [CrossRef]
- Liu, D.; Hu, F.; Yan, Y.; Ye, R.; Chen, X.; Han, B.; Lu, Z.-H.; Zhou, L.; Feng, G.; Zhang, R. Promotion of oxygen vacancies and metal-support interaction over 3DOM Au/CeO2 catalyst for CO oxidation. Appl. Surf. Sci. 2023, 629, 157438. [Google Scholar] [CrossRef]
- Kim, H.J.; Shin, D.; Jeong, H.; Jang, M.G.; Lee, H.; Han, J.W. Design of an Ultrastable and Highly Active Ceria Catalyst for CO Oxidation by Rare-Earth- and Transition-Metal Co-Doping. ACS Catal. 2020, 10, 14877–14886. [Google Scholar] [CrossRef]
- Vanpoucke, D.E.P.; Bultinck, P.; Cottenier, S.; Van Speybroeck, V.; Van Driessche, I. Aliovalent doping of CeO2: DFT study of oxidation state and vacancy effects. J. Mater. Chem. A 2014, 2, 13723–13737. [Google Scholar] [CrossRef]
- D’Angelo, A.M.; Wu, Z.; Overbury, S.H.; Chaffee, A.L. Cu enhanced surface defects and lattice mobility of Pr-CeO2 mixed oxides. J. Phys. Chem. C 2016, 120, 27996–28008. [Google Scholar] [CrossRef]
- Holgado, J.; Munuera, G.; Espinós, J.; González-Elipe, A. XPS study of oxidation processes of CeOx defective layers. Appl. Surf. Sci. 2000, 158, 164–171. [Google Scholar] [CrossRef]
- Zeng, S.; Zhang, W.; Śliwa, M.; Su, H. Comparative study of CeO2/CuO and CuO/CeO2 catalysts on catalytic performance for preferential CO oxidation. Int. J. Hydrogen Energy 2013, 38, 3597–3605. [Google Scholar] [CrossRef]
- Tang, X.; Zhang, B.; Li, Y.; Xu, Y.; Xin, Q.; Shen, W. CuO/CeO2 catalysts: Redox features and catalytic behaviors. Appl. Catal. A Gen. 2005, 288, 116–125. [Google Scholar] [CrossRef]
- Paier, J.; Penschke, C.; Sauer, J. Oxygen Defects and Surface Chemistry of Ceria: Quantum Chemical Studies Compared to Experiment. Chem. Rev. 2013, 113, 3949–3985. [Google Scholar] [CrossRef] [PubMed]
- Kullgren, J. Oxygen Vacancy Chemistry in Ceria. Digital Comprehensive Summaries of Uppsala. Ph.D. Dissertations, Faculty of Science and Technology, Acta Universitatis Upsaliensis, Uppsala, Sweden, 2012. [Google Scholar]
- Mars, P.; van Krevelen, D. Oxidations carried out by means of vanadium oxide catalysts. Chem. Eng. Sci. 1954, 3, 41–59. [Google Scholar] [CrossRef]
- Arango-Díaz, A.; Moretti, E.; Talon, A.; Storaro, L.; Lenarda, M.; Núñez, P.; Marrero-Jerez, J.; Jiménez-Jiménez, J.; Jiménez-López, A.; Rodríguez-Castellón, E. Preferential CO oxidation (CO-PROX) catalyzed by CuO supported on nanocrystalline CeO2 prepared by a freeze-drying method. Appl. Catal. A Gen. 2014, 477, 54–63. [Google Scholar] [CrossRef]
- Van Deelen, T.W.; Mejía, C.H.; De Jong, K.P. Control of metal-support interactions in heterogeneous catalysts to enhance activity and selectivity. Nat. Catal. 2019, 2, 955–970. [Google Scholar] [CrossRef]
- Luo, M.-F.; Song, Y.-P.; Lu, J.-Q.; Wang, X.-Y.; Pu, Z.-Y. Identification of CuO species in high surface area CuO-CeO2 catalyst and their catalytic activities for CO oxidation. J. Phys. Chem. C 2007, 111, 12686–12692. [Google Scholar] [CrossRef]
- Cecilia, J.A.; Arango-Díaz, A.; Marrero-Jerez, J.; Núñez, P.; Moretti, E.; Storaro, L.; Rodríguez-Castellón, E. Catalytic Behaviour of CuO-CeO2 Systems Prepared by Different Synthetic Methodologies in the CO-PROX Reaction under CO2-H2O Feed Stream. Catalysts 2017, 7, 160. [Google Scholar] [CrossRef]
- Luong, N.T.; Okumura, H.; Yamasue, E.; Ishihara, K.N. Structure and catalytic behaviour of CuO–CeO2 prepared by high-energy ball milling. R. Soc. Open Sci. 2019, 6, 181861. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Yu, X.; Zhou, Y.; Miao, S.; Li, Y.; Kuld, S.; Sehested, J.; Liu, J.; Aoki, T.; Hong, S.; et al. Structure of the catalytically active copper–ceria interfacial perimeter. Nat. Catal. 2019, 2, 334–341. [Google Scholar] [CrossRef]
- Zhang, R.; Haddadin, T.; Rubiano, D.P.; Nair, H.; Polster, C.S.; Baertsch, C.D. Quantification of Reactive CO and H2 on CuOx-CeO2 during CO Preferential Oxidation by Reactive Titration and Steady State Isotopic Transient Kinetic Analysis. ACS Catal. 2011, 1, 519–525. [Google Scholar] [CrossRef]
- Gamarra, D.; Belver, C.; Fernández-García, M.; Martínez-Arias, A. Selective CO Oxidation in Excess H2 over Copper−Ceria Catalysts: Identification of Active Entities/Species. J. Am. Chem. Soc. 2007, 129, 12064–12065. [Google Scholar] [CrossRef] [PubMed]
- Borchers, C.; Martin, M.L.; Vorobjeva, G.A.; Morozova, O.S.; Firsova, A.A.; Leonov, A.V.; Kurmaev, E.Z.; Kukharenko, A.I.; Zhidkov, I.S.; Cholakh, S.O. Cu–CeO2 nanocomposites: Mechanochemical synthesis, physico-chemical properties, CO-PROX activity. J. Nanopart. Res. 2016, 18, 344. [Google Scholar] [CrossRef]
- Borchers, C.; Martin, M.L.; Vorobjeva, G.A.; Morozova, O.S.; Firsova, A.A.; Leonov, A.V.; Kurmaev, E.Z.; Kukharenko, A.I.; Zhidkov, I.S.; Cholakh, S.O. CuO-CeO2 nanocomposite catalysts produced by mechanochemical synthesis. AIP Adv. 2019, 9, 065115. [Google Scholar] [CrossRef]
- Morozova, O.S.; Firsova, A.A.; Tyulenin, Y.P.; Vorobieva, G.A.; Leonov, A.V. Mechanochemical Synthesis as an Alternative Effective Technique for the Preparation of the Composite Catalysts. Kinet. Catal. 2020, 61, 824–831. [Google Scholar] [CrossRef]
- Shelekhov, E.V.; Sviridova, T.A. Programs for X-ray analysis of polycrystals. Met. Sci. Heat Treat. 2000, 42, 309–313. [Google Scholar] [CrossRef]
- Zhai, Y.; Zhao, J.; Di, X.; Di, S.; Wang, B.; Yue, Y.; Sheng, G.; Lai, H.; Guo, L.; Wang, H.; et al. Carbon-supported perovskite-like CsCuCl3 nanoparticles: A highly active and cost-effective heterogeneous catalyst for the hydrochlorination of acetylene to vinyl chloride. Catal. Sci. Technol. 2018, 8, 2901–2908. [Google Scholar] [CrossRef]
- Sun, H.; Zelekew, O.A.; Chen, X.; Guo, Y.; Kuo, D.-H.; Lu, Q.; Lin, J. A noble bimetal oxysulfide CuVOS catalyst for highly efficient catalytic reduction of 4-nitrophenol and organic dyes. RSC Adv. 2019, 9, 31828–31839. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Peng, F.; Li, J.; Liang, X.; Chen, B. Morphology-Dependent Properties of Cu/CeO2 Catalysts for the Water-Gas Shift Reaction. Catalysts 2017, 7, 48. [Google Scholar] [CrossRef]
- Park, J.-Y.; Lim, K.-A.; Ramsier, R.D.; Kang, Y.-C. Spectroscopic and Morphological Investigation of Copper Oxide Thin Films Prepared by Magnetron Sputtering at Various Oxygen Ratios. Bull. Korean Chem. Soc. 2011, 32, 3395–3399. [Google Scholar] [CrossRef]
- Svintsitskiy, D.; Stadnichenko, A.; Demidov, D.; Koscheev, S.; Boronin, A. Investigation of oxygen states and reactivities on a nanostructured cupric oxide surface. Appl. Surf. Sci. 2011, 257, 8542–8549. [Google Scholar] [CrossRef]
- Sun, S.; Mao, D.; Yu, J.; Yang, Z.; Lua, G.; Ma, Z. Low-temperature CO oxidation on CuO/CeO2 catalysts: The significant effect of copper precursor and calcination temperature. Catal. Sci. Technol. 2015, 5, 3166. [Google Scholar]
- Park, P.W.; Ledford, J.S. Effect of Crystallinity on the Photoreduction of Cerium Oxide: A Study of CeO2 and Ce/Al2O3 Catalysts. Langmuir 1996, 12, 1794–1799. [Google Scholar] [CrossRef]
- Hardacre, C.; Roe, G.M.; Lambert, R.M. Structure, composition and thermal properties of cerium oxide films on platinum {111}. Surf. Sci. 1995, 326, 1–10. [Google Scholar] [CrossRef]
- Cruz, A.R.M.; Assaf, E.M.; Gomes, J.F.; Assaf, J.M. Active copper species of co-precipitated copper-ceria catalysts in the CO-PROX reaction: An in situ XANES and DRIFTS study. Catal. Today 2021, 381, 42–49. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, L.; van Hoof, A.J.; Hensen, E.J. On the surface-dependent oxidation of Cu2O during CO oxidation: Cu2+ is more active than Cu+. Appl. Catal. A Gen. 2020, 602, 117712. [Google Scholar] [CrossRef]
- Vayssilov, G.N.; Mihaylov, M.; Petkov, P.S.; Hadjiivanov, K.I.; Neyman, K.M. Reassignment of the Vibrational Spectra of Carbonates, Formates, and Related Surface Species on Ceria: A Combined Density Functional and Infrared Spectroscopy Investigation. J. Phys. Chem. C 2011, 115, 23435–23454. [Google Scholar] [CrossRef]
- Mariño, F.; Descorme, C.; Duprez, D. Supported base metal catalysts for the preferential oxidation of carbon monoxide in the presence of excess hydrogen (PROX). Appl. Catal. B Environ. 2005, 58, 175–183. [Google Scholar] [CrossRef]
- Gamarra, D.; Martínez-Arias, A. Preferential oxidation of CO in rich H2 over CuO/CeO2: Operando-DRIFTS analysis of deactivating effect of CO2 and H2O. J. Catal. 2009, 263, 189–195. [Google Scholar] [CrossRef]
- James, T.E.; Hemmingson, S.L.; Ito, T.; Campbell, C.T. Energetics of Cu Adsorption and Adhesion onto Reduced CeO2(111) Surfaces by Calorimetry. J. Phys. Chem. C 2015, 119, 17209–17217. [Google Scholar] [CrossRef]
- Yu, W.Z.; Wang, W.W.; Li, S.Q.; Fu, X.P.; Wang, X.; Wu, K.; Si, R.; Ma, C.; Jia, C.J.; Yan, C.H. Construction of active site in a sintered capper-ceria nanorod catalyst. J. Am. Chem. Soc. 2019, 141, 17548–17557. [Google Scholar] [CrossRef] [PubMed]
- Davó-Quiñonero, A.; López-Rodríguez, S.; Chaparro-Garnica, C.; Martín-García, I.; Bailón-García, E.; Lozano-Castelló, D.; Bueno-López, A.; García-Melchor, M. Investigations of the Effect of H2 in CO Oxidation over Ceria Catalysts. Catalysts 2021, 11, 1556. [Google Scholar] [CrossRef]
- Puigdollers, A.R.; Schlexer, P.; Tosoni, S.; Pacchioni, G. Increasing Oxide Reducibility: The Role of Metal/Oxide Interfaces in the Formation of Oxygen Vacancies. ACS Catal. 2017, 7, 6493–6513. [Google Scholar] [CrossRef]
- Kappis, K.; Papavasiliou, J. Influence of the Hydrothermal Parameters on the Physicochemical Characteristics of Cu−Ce Oxide Nanostructures. Chemcatchem 2019, 11, 4765–4776. [Google Scholar] [CrossRef]
- Moretti, E.; Lenarda, M.; Riello, P.; Storaro, L.; Talon, A.; Frattini, R.; Reyes-Carmona, A.; Jiménez-López, A.; Rodríguez-Castellón, E. Influence of synthesis parameters on the performance of CeO2–CuO and CeO2–ZrO2–CuO systems in the catalytic oxidation of CO in excess of hydrogen. Appl. Catal. B Environ. 2013, 129, 556–565. [Google Scholar] [CrossRef]
- Lu, J.; Wang, J.; Zou, Q.; He, D.; Zhang, L.; Xu, Z.; He, S.; Luo, Y. Unravelling the Nature of the Active Species as well as the Doping Effect over Cu/Ce-Based Catalyst for Carbon Monoxide Preferential Oxidation. ACS Catal. 2019, 9, 2177–2195. [Google Scholar] [CrossRef]
- Martínez-Munuera, J.; Giménez-Mañogil, J.; Yeste, M.; Hungría, A.; Cauqui, M.; García-García, A.; Calvino, J. New findings regarding the role of copper entity particle size on the performance of Cu/ceria-based catalysts in the CO-PROX reaction. Appl. Surf. Sci. 2021, 575, 151717. [Google Scholar] [CrossRef]
- Zhigach, A.N.; Kuskov, M.L.; Leipunskii, I.O.; Stoenko, N.I.; Storozhev, V.B. Levitation-jet method for obtaining ultra-fine metal powders, alloys, metal compounds by Gene-Miller technique: History, state of art, perspective. Bull. Russ. Acad. Sci. Energetic 2012, 3, 80–96. [Google Scholar]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
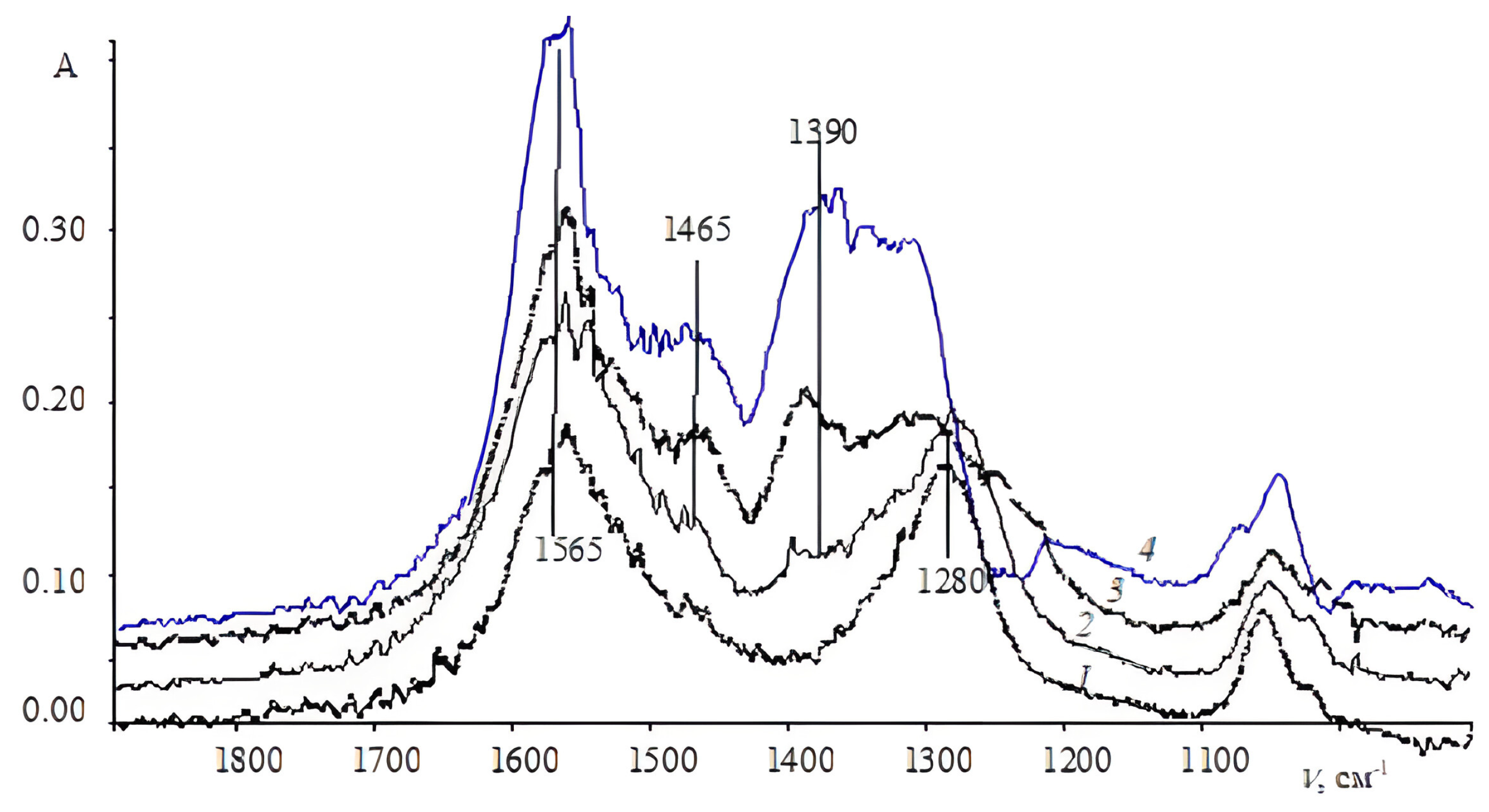{kind=link}
| No | Sample | Lattice Constant, a (nm) | Crystallite Size (nm) 1 | Specific Surface Area (m2g−1) |
|---|---|---|---|---|
| 1 | CeO2 original | 0.5411 | 10–12 | 89.0 |
| 2 | CeO2 milled for 30 min | 0.5413 | ~13 | 46.5 |
| 3 | 8 mass% Cu(D)–CeO2 milled for 30 min | 0.5414 | 9–12 | 39.0 |
| 4 | 8 mass% Cu(L)–CeO2 milled for 30 min | 0.5410 | 10–13 | 50.5 |
| Sample | Ratio of the Main Components on the Catalyst Surface | |||
|---|---|---|---|---|
| Cu/Ce * | Cu+/Cu2+ * | O/Ce * | Os/Ol ** | |
| Cu(D)–CeO2 | 0.47 | 3.80 | 5.13 | 65.6 |
| Cu(L)–CeO2 | 0.46 | 0.54 | 3.42 | 25.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhigalina, O.M.; Morozova, O.S.; Khmelenin, D.N.; Firsova, A.A.; Silchenkova, O.V.; Vorobieva, G.A.; Bukhtiyarov, A.V.; Cherkovskiy, E.N.; Basu, V.G. Effect of Copper Particle Size on the Surface Structure and Catalytic Activity of Cu–CeO2 Nanocomposites Prepared by Mechanochemical Synthesis in the Preferential CO Oxidation in a H2-Rich Stream (CO-PROX). Catalysts 2024, 14, 222. https://doi.org/10.3390/catal14040222
Zhigalina OM, Morozova OS, Khmelenin DN, Firsova AA, Silchenkova OV, Vorobieva GA, Bukhtiyarov AV, Cherkovskiy EN, Basu VG. Effect of Copper Particle Size on the Surface Structure and Catalytic Activity of Cu–CeO2 Nanocomposites Prepared by Mechanochemical Synthesis in the Preferential CO Oxidation in a H2-Rich Stream (CO-PROX). Catalysts. 2024; 14(4):222. https://doi.org/10.3390/catal14040222
Chicago/Turabian StyleZhigalina, Olga M., Olga S. Morozova, Dmitry N. Khmelenin, Alla A. Firsova, Olga V. Silchenkova, Galina A. Vorobieva, Andrey V. Bukhtiyarov, Evgeny N. Cherkovskiy, and Victoria G. Basu. 2024. "Effect of Copper Particle Size on the Surface Structure and Catalytic Activity of Cu–CeO2 Nanocomposites Prepared by Mechanochemical Synthesis in the Preferential CO Oxidation in a H2-Rich Stream (CO-PROX)" Catalysts 14, no. 4: 222. https://doi.org/10.3390/catal14040222