
Kinetic Modeling of the Direct Dimethyl Ether (DME) Synthesis over Hybrid Multi-Site Catalysts

 ,
,  , , , , , and
, , , , , and
Abstract
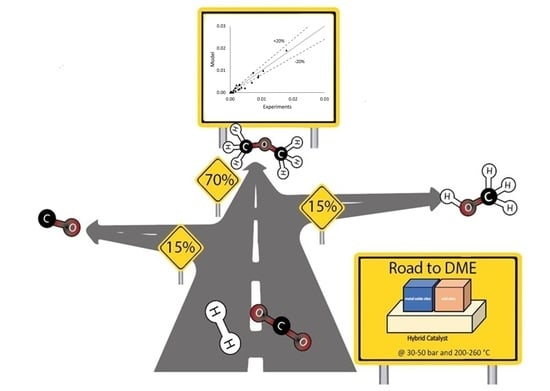
:
1. Introduction
2. Experimental Section
2.1. Catalyst Synthesis
2.2. Catalyst Testing
3. Kinetic Modeling
- Analysis of the implicated reactions and corresponding kinetic mechanisms and expressions.
- Estimation of missing parameters based on appropriate modeling of test conditions.
3.1. Reaction Kinetics Investigation
- CO2 hydrogenation to methanol (CHM):
- Methanol dehydration to DME (MDD):
- Reverse water–gas shift (rWGS):
3.1.1. CO2 Hydrogenation Mechanism
3.1.2. Methanol Dehydration Mechanism
3.1.3. rWGS Mechanism
3.2. Dual-Site Adsorption on the Hybrid Catalyst
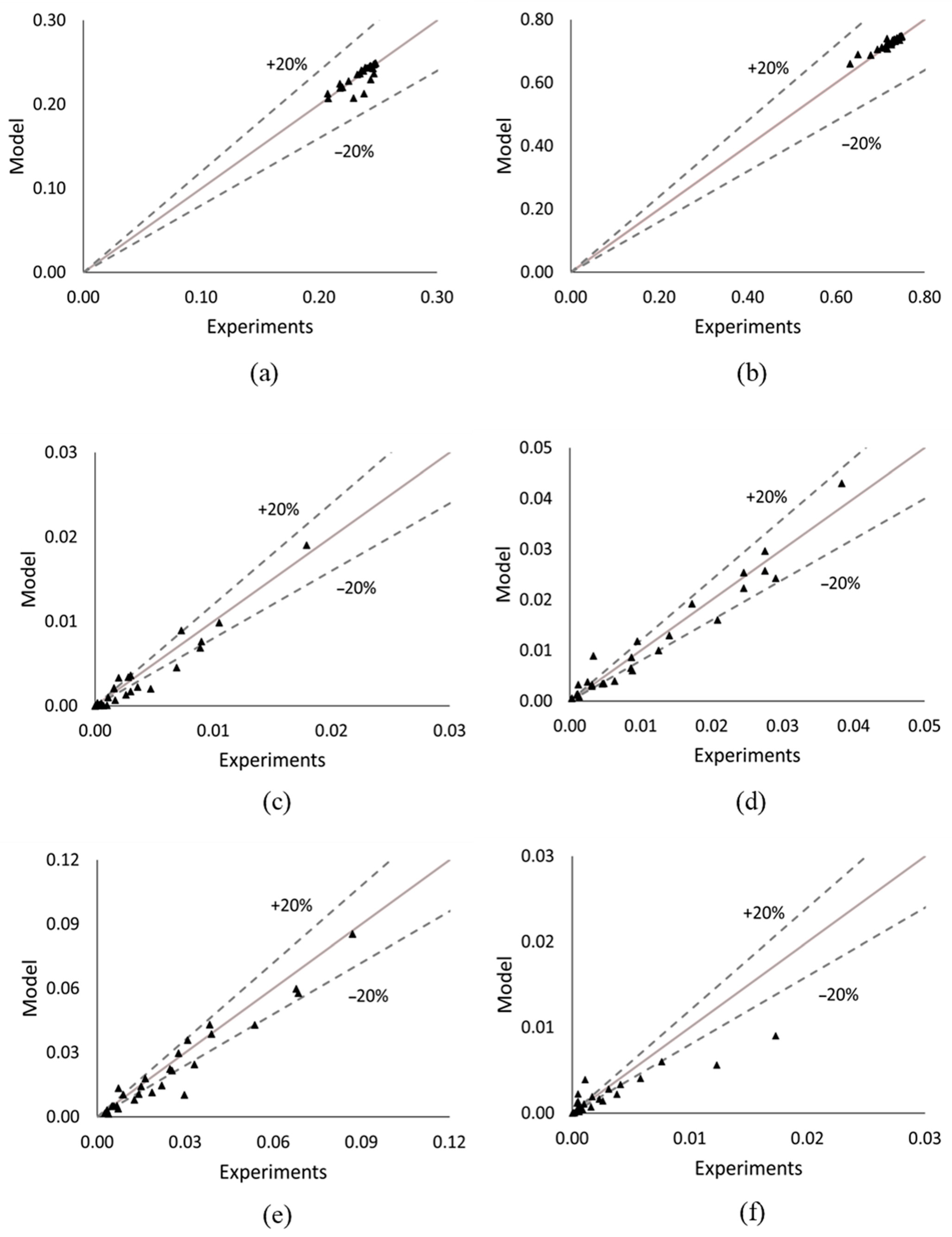
3.3. Parameter Estimation Method
4. Results and Discussion
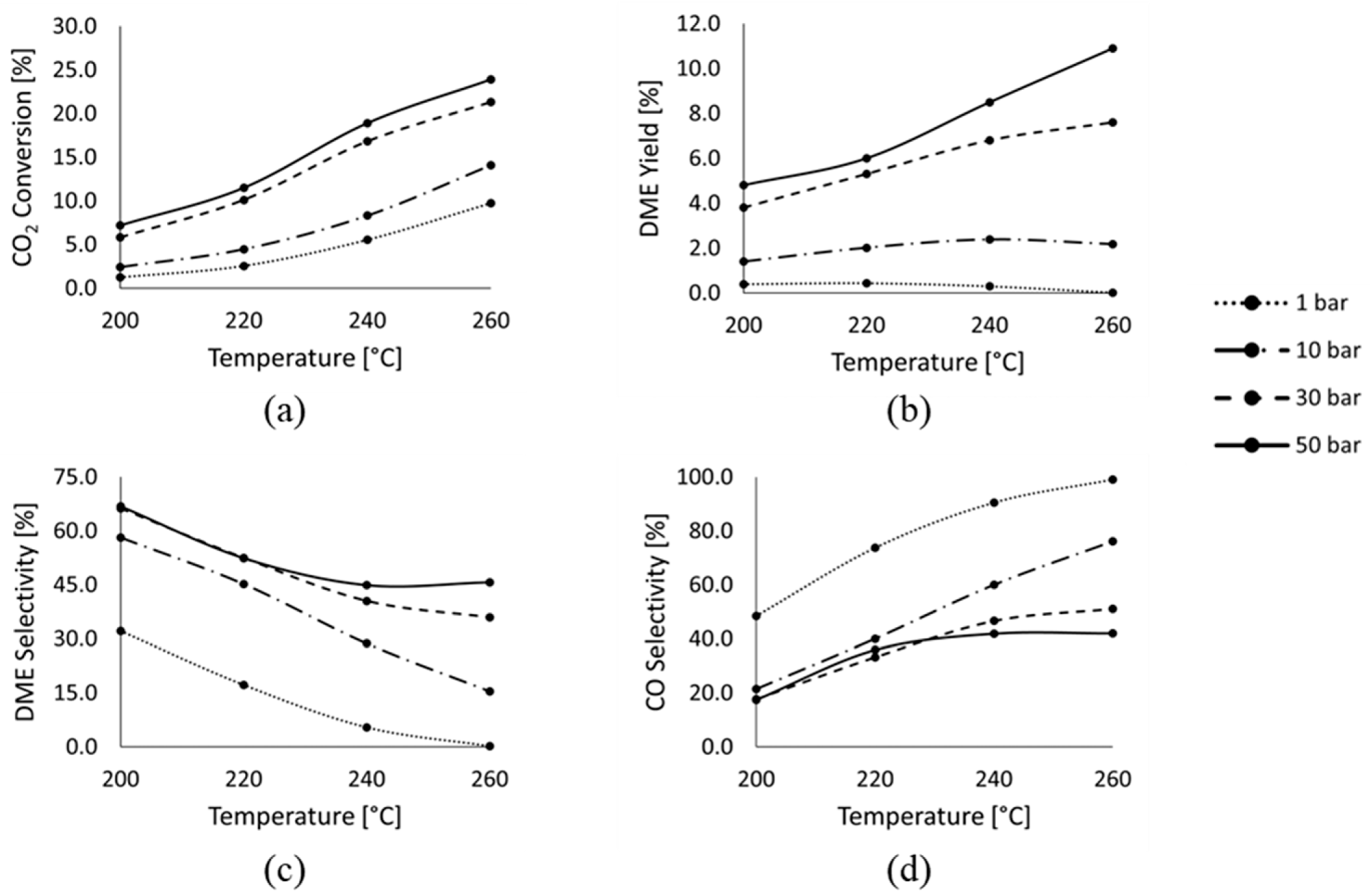
4.1. Experimental Results
4.2. Reaction Kinetics Investigation Results
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Fugacity Coefficient Evaluation

{kind=link}
{kind=link}
{kind=link}
| Compound | ||
|---|---|---|
| Water | 0.0279 | 0.0229 |
| Methanol | 0.0878 | 0.0560 |
| Dimethyl Ether | −0.01513 | 0 |
| 0.23 | - | - | - | - | - | - | - | - | |||||
| 0.21 | - | - | - | - | 0.96 | ||||||||
| 0.21 | |||||||||||||
| 0.20 | |||||||||||||
| 0.19 |
References
- Fleisch, T.H.; Basu, A.; Gradassi, M.J.; Masin, J.G. Dimethyl Ether: A Fuel for the 21st Century. In Studies in Surface Science and Catalysis; Elsevier: Amsterdam, The Netherlands, 1997; pp. 117–125. [Google Scholar]
- Matzen, M.; Demirel, Y. Methanol and Dimethyl Ether from Renewable Hydrogen and Carbon Dioxide: Alternative Fuels Production and Life-Cycle Assessment. J. Clean. Prod. 2016, 139, 1068–1077. [Google Scholar] [CrossRef]
- Makoś, P.; Słupek, E.; Sobczak, J.; Zabrocki, D.; Hupka, J.; Rogala, A. Dimethyl Ether (DME) as Potential Environmental Friendly Fuel. E3S Web Conf. 2019, 116, 00048. [Google Scholar] [CrossRef]
- Park, S.H.; Lee, C.S. Applicability of Dimethyl Ether (DME) in a Compression Ignition Engine as an Alternative Fuel. Energy Convers. Manag. 2014, 86, 848–863. [Google Scholar] [CrossRef]
- Park, S.H.; Lee, C.S. Combustion Performance and Emission Reduction Characteristics of Automotive DME Engine System. Prog. Energy Combust. Sci. 2013, 39, 147–168. [Google Scholar] [CrossRef]
- Semelsberger, T.A.; Borup, R.L.; Greene, H.L. Dimethyl Ether (DME) as an Alternative Fuel. J. Power Sources 2006, 156, 497–511. [Google Scholar] [CrossRef]
- Khadzhiev, S.N.; Kolesnichenko, N.V.; Ezhova, N.N. Manufacturing of Lower Olefins from Natural Gas through Methanol and Its Derivatives (Review). Pet. Chem. 2008, 48, 325–334. [Google Scholar] [CrossRef]
- Cordero-Lanzac, T.; Martínez, C.; Aguayo, A.T.; Castaño, P.; Bilbao, J.; Corma, A. Activation of N-Pentane While Prolonging HZSM-5 Catalyst Lifetime during Its Combined Reaction with Methanol or Dimethyl Ether. Catal. Today 2022, 383, 320–329. [Google Scholar] [CrossRef]
- Pérez-Uriarte, P.; Ateka, A.; Aguayo, A.T.; Gayubo, A.G.; Bilbao, J. Kinetic Model for the Reaction of DME to Olefins over a HZSM-5 Zeolite Catalyst. Chem. Eng. J. 2016, 302, 801–810. [Google Scholar] [CrossRef]
- Catizzone, E.; Bonura, G.; Migliori, M.; Frusteri, F.; Giordano, G. CO2 Recycling to Dimethyl Ether: State-of-the-Art and Perspectives. Molecules 2017, 23, 31–58. [Google Scholar] [CrossRef]
- Bonura, G.; Cordaro, M.; Spadaro, L.; Cannilla, C.; Arena, F.; Frusteri, F. Hybrid Cu–ZnO–ZrO2/H-ZSM5 System for the Direct Synthesis of DME by CO2 Hydrogenation. Appl. Catal. B 2013, 140–141, 16–24. [Google Scholar] [CrossRef]
- Wawrzyńczak, D.; Majchrzak-Kuceba, I.; Pevida, C.; Bonura, G.; Nogueira, R.; De Falco, M. The Carbon Chain in Carbon Dioxide Industrial Utilization Technologies: A Case Study; Taylor & Francis: Abingdon, UK, 2023. [Google Scholar]
- Sankhe, S.; Krishna, S.V.M.; Juturu, R.M.; Subrahmanyam, C. Power-to-X (PtX) Technologies and Their Potential Role in the Transition towards a Fossil-Free Energy Future: A Review of EFuels Synthesis and Direct Air Capture (DAC) Technology. In Proceedings of the International Conference on Automotive Materials and Manufacturing AMM 2023, Pune, India, 1–3 June 2023; SAE Technical Paper 2023-28-1333. pp. 1–5. [Google Scholar] [CrossRef]
- Catizzone, E.; Freda, C.; Braccio, G.; Frusteri, F.; Bonura, G. Dimethyl Ether as Circular Hydrogen Carrier: Catalytic Aspects of Hydrogenation/Dehydrogenation Steps. J. Energy Chem. 2021, 58, 55–77. [Google Scholar] [CrossRef]
- Azizi, Z.; Rezaeimanesh, M.; Tohidian, T.; Rahimpour, M.R. Dimethyl Ether: A Review of Technologies and Production Challenges. Chem. Eng. Process. Process Intensif. 2014, 82, 150–172. [Google Scholar] [CrossRef]
- Mondal, U.; Yadav, G.D. Perspective of Dimethyl Ether as Fuel: Part I. Catalysis. J. CO2 Util. 2019, 32, 299–320. [Google Scholar] [CrossRef]
- Bonura, G.; Migliori, M.; Frusteri, L.; Cannilla, C.; Catizzone, E.; Giordano, G.; Frusteri, F. Acidity Control of Zeolite Functionality on Activity and Stability of Hybrid Catalysts during DME Production via CO2 Hydrogenation. J. CO2 Util. 2018, 24, 398–406. [Google Scholar] [CrossRef]
- Tokay, K.C.; Dogu, T.; Dogu, G. Dimethyl Ether Synthesis over Alumina Based Catalysts. Chem. Eng. J. 2012, 184, 278–285. [Google Scholar] [CrossRef]
- Akhoondi, A.; Osman, A.I.; Alizadeh Eslami, A. Direct Catalytic Production of Dimethyl Ether from CO and CO2: A Review. Synth. Sinter. 2021, 1, 105–125. [Google Scholar] [CrossRef]
- Chen, W.H.; Hsu, C.L.; Wang, X.D. Thermodynamic Approach and Comparison of Two-Step and Single Step DME (Dimethyl Ether) Syntheses with Carbon Dioxide Utilization. Energy 2016, 109, 326–340. [Google Scholar] [CrossRef]
- Banivaheb, S.; Pitter, S.; Delgado, K.H.; Rubin, M.; Sauer, J.; Dittmeyer, R. Recent Progress in Direct DME Synthesis and Potential of Bifunctional Catalysts. Chem. Ing. Tech. 2022, 94, 240–255. [Google Scholar] [CrossRef]
- Bonura, G.; Cannilla, C.; Frusteri, L.; Catizzone, E.; Todaro, S.; Migliori, M.; Giordano, G.; Frusteri, F. Interaction Effects between CuO-ZnO-ZrO2 Methanol Phase and Zeolite Surface Affecting Stability of Hybrid Systems during One-Step CO2 Hydrogenation to DME. Catal. Today 2020, 345, 175–182. [Google Scholar] [CrossRef]
- Ren, S.; Shoemaker, W.R.; Wang, X.; Shang, Z.; Klinghoffer, N.; Li, S.; Yu, M.; He, X.; White, T.A.; Liang, X. Highly Active and Selective Cu-ZnO Based Catalyst for Methanol and Dimethyl Ether Synthesis via CO2 Hydrogenation. Fuel 2019, 239, 1125–1133. [Google Scholar] [CrossRef]
- Abu-Dahrieh, J.; Rooney, D.; Goguet, A.; Saih, Y. Activity and Deactivation Studies for Direct Dimethyl Ether Synthesis Using CuO–ZnO–Al2O3 with NH4ZSM-5, HZSM-5 or γ-Al2O3. Chem. Eng. J. 2012, 203, 201–211. [Google Scholar] [CrossRef]
- Frusteri, L.; Bonura, G.; Cannilla, C.; Todaro, S.; Giordano, G.; Migliori, M.; Frusteri, F. Promoting Direct CO2 Conversion to DME over Zeolite-Based Hybrid Catalysts. Pet. Chem. 2020, 60, 508–515. [Google Scholar] [CrossRef]
- Ateka, A.; Rodriguez-Vega, P.; Ereña, J.; Aguayo, A.T.; Bilbao, J. Kinetic Modeling and Reactor Design of the Direct Synthesis of Dimethyl Ether for CO2 Valorization. A Review. Fuel 2022, 327, 125148. [Google Scholar] [CrossRef]
- Hadipour, A.; Sohrabi, M. Synthesis of some bifunctional catalysts and determination of kinetic parameters for direct conversion of syngas to dimethyl ether. Chem. Eng. J. 2008, 137, 294–301. [Google Scholar] [CrossRef]
- Frusteri, F.; Cordaro, M.; Cannilla, C.; Bonura, G. Multifunctionality of Cu–ZnO–ZrO2/H-ZSM5 Catalysts for the One-Step CO2-to-DME Hydrogenation Reaction. Appl. Catal. B 2015, 162, 57–65. [Google Scholar] [CrossRef]
- Delgado Otalvaro, N.; Bilir, P.G.; Herrera Delgado, K.; Pitter, S.; Sauer, J. Kinetics of the Direct DME Synthesis: State of the Art and Comprehensive Comparison of Semi-Mechanistic, Data-Based and Hybrid Modeling Approaches. Catalysts 2022, 12, 347. [Google Scholar] [CrossRef]
- Naik, S.P.; Ryu, T.; Bui, V.; Miller, J.D.; Drinnan, N.B.; Zmierczak, W. Synthesis of DME from CO2/H2 Gas Mixture. Chem. Eng. J. 2011, 167, 362–368. [Google Scholar] [CrossRef]
- De Falco, M.; Capocelli, M.; Centi, G. Dimethyl Ether Production from CO2 Rich Feedstocks in a One-Step Process: Thermodynamic Evaluation and Reactor Simulation. Chem. Eng. J. 2016, 294, 400–409. [Google Scholar] [CrossRef]
- Studt, F.; Behrens, M.; Kunkes, E.L.; Thomas, N.; Zander, S.; Tarasov, A.; Schumann, J.; Frei, E.; Varley, J.B.; Abild-Pedersen, F.; et al. The Mechanism of CO and CO2 Hydrogenation to Methanol over Cu-Based Catalysts. ChemCatChem 2015, 7, 1105–1111. [Google Scholar] [CrossRef]
- Cao, A.; Wang, Z.; Li, H.; Elnabawy, A.O.; Nørskov, J.K. New Insights on CO and CO2 Hydrogenation for Methanol Synthesis: The Key Role of Adsorbate-Adsorbate Interactions on Cu and the Highly Active MgO-Cu Interface. J. Catal. 2021, 400, 325–331. [Google Scholar] [CrossRef]
- Campbell, C.T. Finding the Rate-Determining Step in a Mechanism: Comparing DeDonder Relations with the “Degree of Rate Control”. J. Catal. 2001, 204, 520–524. [Google Scholar] [CrossRef]
- Lu, W.Z.; Teng, L.H.; Xiao, W. De Simulation and Experiment Study of Dimethyl Ether Synthesis from Syngas in a Fluidized-Bed Reactor. Chem. Eng. Sci. 2004, 59, 5455–5464. [Google Scholar] [CrossRef]
- Li, Z.; Li, N.; Wang, N.; Zhou, B.; Yin, P.; Song, B.; Yu, J.; Yang, Y. Mechanism Investigations on Water Gas Shift Reaction over Cu(111), Cu(100), and Cu(211) Surfaces. ACS Omega 2022, 7, 3514–3521. [Google Scholar] [CrossRef] [PubMed]
- Bonura, G.; Todaro, S.; Frusteri, L.; Majchrzak-Kucęba, I.; Wawrzyńczak, D.; Pászti, Z.; Tálas, E.; Tompos, A.; Ferenc, L.; Solt, H.; et al. Inside the Reaction Mechanism of Direct CO2 Conversion to DME over Zeolite-Based Hybrid Catalysts. Appl. Catal. B 2021, 294, 120255. [Google Scholar] [CrossRef]
- Motagamwala, A.H.; Dumesic, J.A. Microkinetic Modeling: A Tool for Rational Catalyst Design. Chem. Rev. 2021, 121, 1049–1076. [Google Scholar] [CrossRef]
- Saravanan, K.; Ham, H.; Tsubaki, N.; Bae, J.W. Recent progress for direct synthesis of dimethyl ether from syngas on the heterogeneous bifunctional hybrid catalysts. Appl. Catal. B Environ. 2017, 217, 494–522. [Google Scholar] [CrossRef]
- Din, I.U.; Shaharun, M.S.; Alotaibi, M.A.; Alharthi, A.I.; Naeem, A. Recent Developments on Heterogeneous Catalytic CO2 Reduction to Methanol. J. CO2 Util. 2019, 34, 20–33. [Google Scholar] [CrossRef]
- Guil-López, R.; Mota, N.; Llorente, J.; Millán, E.; Pawelec, B.; Fierro, J.L.G.; Navarro, R.M. Methanol Synthesis from CO2: A Review of the Latest Developments in Heterogeneous Catalysis. Materials 2019, 12, 3902. [Google Scholar] [CrossRef]
- Blaszkowski, S.R.; van Santen, R.A. The Mechanism of Dimethyl Ether Formation from Methanol Catalyzed by Zeolitic Protons. J. Am. Chem. Soc. 1996, 118, 5152–5153. [Google Scholar] [CrossRef]
- Jones, A.J.; Iglesia, E. Kinetic, Spectroscopic, and Theoretical Assessment of Associative and Dissociative Methanol Dehydration Routes in Zeolites. Angew. Chem. 2014, 126, 12373–12377. [Google Scholar] [CrossRef]
- Ghorbanpour, A.; Rimer, J.D.; Grabow, L.C. Computational Assessment of the Dominant Factors Governing the Mechanism of Methanol Dehydration over H-ZSM-5 with Heterogeneous Aluminum Distribution. ACS Catal. 2016, 6, 2287–2298. [Google Scholar] [CrossRef]
- Tang, Q.-L.; Liu, Z.-P. Identification of the Active Cu Phase in the Water−Gas Shift Reaction over Cu/ZrO2 from First Principles. J. Phys. Chem. C 2010, 114, 8423–8430. [Google Scholar] [CrossRef]
- Madon, R.J.; Braden, D.; Kandoi, S.; Nagel, P.; Mavrikakis, M.; Dumesic, J.A. Microkinetic Analysis and Mechanism of the Water Gas Shift Reaction over Copper Catalysts. J. Catal. 2011, 281, 1–11. [Google Scholar] [CrossRef]
- Lacerda de Oliveira Campos, B.; Herrera Delgado, K.; Wild, S.; Studt, F.; Pitter, S.; Sauer, J. Surface Reaction Kinetics of the Methanol Synthesis and the Water Gas Shift Reaction on Cu/ZnO/Al2O3. React. Chem. Eng. 2021, 6, 868–887. [Google Scholar] [CrossRef]
- Behloul, C.R.; Commenge, J.-M.; Castel, C. Simulation of Reactors under Different Thermal Regimes and Study of the Internal Diffusional Limitation in a Fixed-Bed Reactor for the Direct Synthesis of Dimethyl Ether from a CO2-Rich Input Mixture and H2. Ind. Eng. Chem. Res. 2021, 60, 1602–1623. [Google Scholar] [CrossRef]
- Agarwal, A.K.; Brisk, M.L. Sequential Experimental Design for Precise Parameter Estimation. 1. Use of Reparameterization. Ind. Eng. Chem. Process Des. Dev. 1985, 24, 203–207. [Google Scholar] [CrossRef]
- Pritchard, D.J.; Bacon, D.W. Statistical Assessment of Chemical Kinetic Models. Chem. Eng. Sci. 1975, 30, 567–574. [Google Scholar] [CrossRef]
- Espie, D.M.; Macchietto, S. Nonlinear Transformations for Parameter Estimation. Ind. Eng. Chem. Res. 1988, 27, 2175–2179. [Google Scholar] [CrossRef]
- Ereña, J.; Sierra, I.; Aguayo, A.T.; Ateka, A.; Olazar, M.; Bilbao, J. Kinetic Modelling of Dimethyl Ether Synthesis from (H2 + CO2) by Considering Catalyst Deactivation. Chem. Eng. J. 2011, 174, 660–667. [Google Scholar] [CrossRef]
- Ahmad, K.; Upadhyayula, S. Greenhouse Gas CO2 Hydrogenation to Fuels: A Thermodynamic Analysis. Environ. Prog. Sustain. Energy 2019, 38, 98–111. [Google Scholar] [CrossRef]
- Marrelli, L. Termodinamica Degli Equilibri Di Fasi Fluide; Edizioni Efesto: Rome, Italy, 2017. [Google Scholar]
- De Santis, R. Introduzione al Calcolo Degli Equilibri Di Fasi Fluide; ESA: Paris, France, 1991. [Google Scholar]
- Tsonopoulos, C. An Empirical Correlation of Second Virial Coefficients. AIChE J. 1974, 20, 263–272. [Google Scholar] [CrossRef]
- Chueh, P.L.; Prausnitz, J.M. Vapor-Liquid Equilibria at High Pressures. Vapor-Phase Fugacity Coefficients in Nonpolar and Quantum-Gas Mixtures. Ind. Eng. Chem. Fundam. 1967, 6, 492–498. [Google Scholar] [CrossRef]


| Parameter | Temperature Dependence |
|---|---|
| Parameter | Unit |
Pre-Exponential Factor () |
Energetic Term () |
|---|---|---|---|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Ambrosio, A.; Bertino, A.; Todaro, S.; Santoro, M.; Cannilla, C.; Frusteri, F.; Bonura, G.; Mazzeo, L.; Piemonte, V. Kinetic Modeling of the Direct Dimethyl Ether (DME) Synthesis over Hybrid Multi-Site Catalysts. Catalysts 2024, 14, 61. https://doi.org/10.3390/catal14010061
D’Ambrosio A, Bertino A, Todaro S, Santoro M, Cannilla C, Frusteri F, Bonura G, Mazzeo L, Piemonte V. Kinetic Modeling of the Direct Dimethyl Ether (DME) Synthesis over Hybrid Multi-Site Catalysts. Catalysts. 2024; 14(1):61. https://doi.org/10.3390/catal14010061
Chicago/Turabian StyleD’Ambrosio, Antonio, Alice Bertino, Serena Todaro, Mariarita Santoro, Catia Cannilla, Francesco Frusteri, Giuseppe Bonura, Leone Mazzeo, and Vincenzo Piemonte. 2024. "Kinetic Modeling of the Direct Dimethyl Ether (DME) Synthesis over Hybrid Multi-Site Catalysts" Catalysts 14, no. 1: 61. https://doi.org/10.3390/catal14010061