


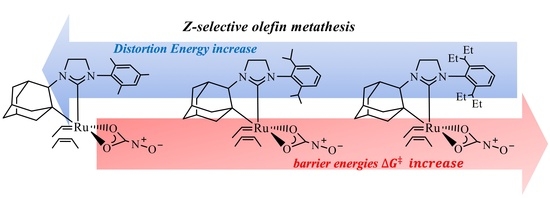
Role of Electronic and Steric Effects on Ruthenium Catalysts with Bulky NHC Ligands and Relationship with the Z-Selectivity in Olefin Metathesis

Abstract
:
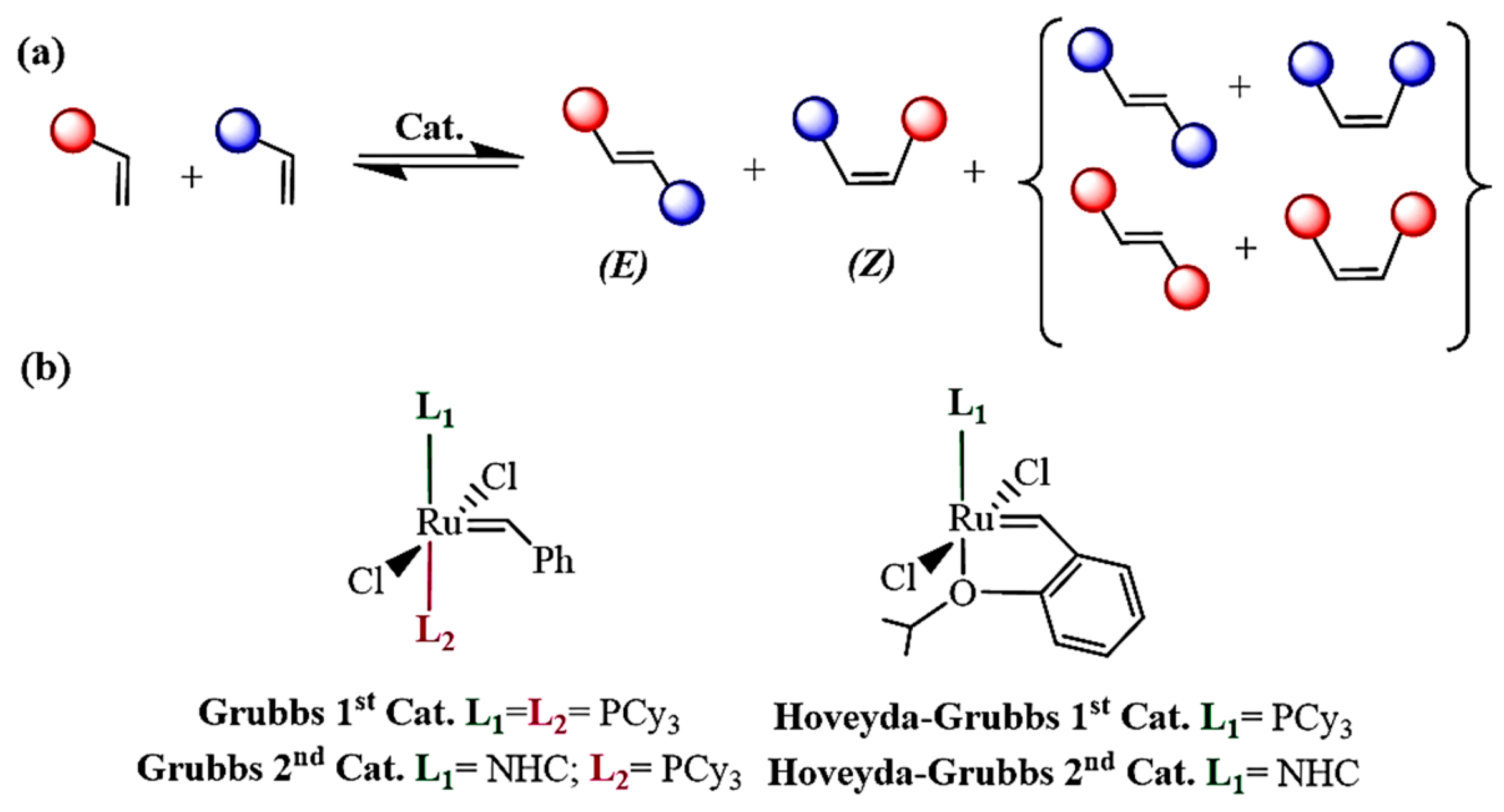
1. Introduction

2. Results
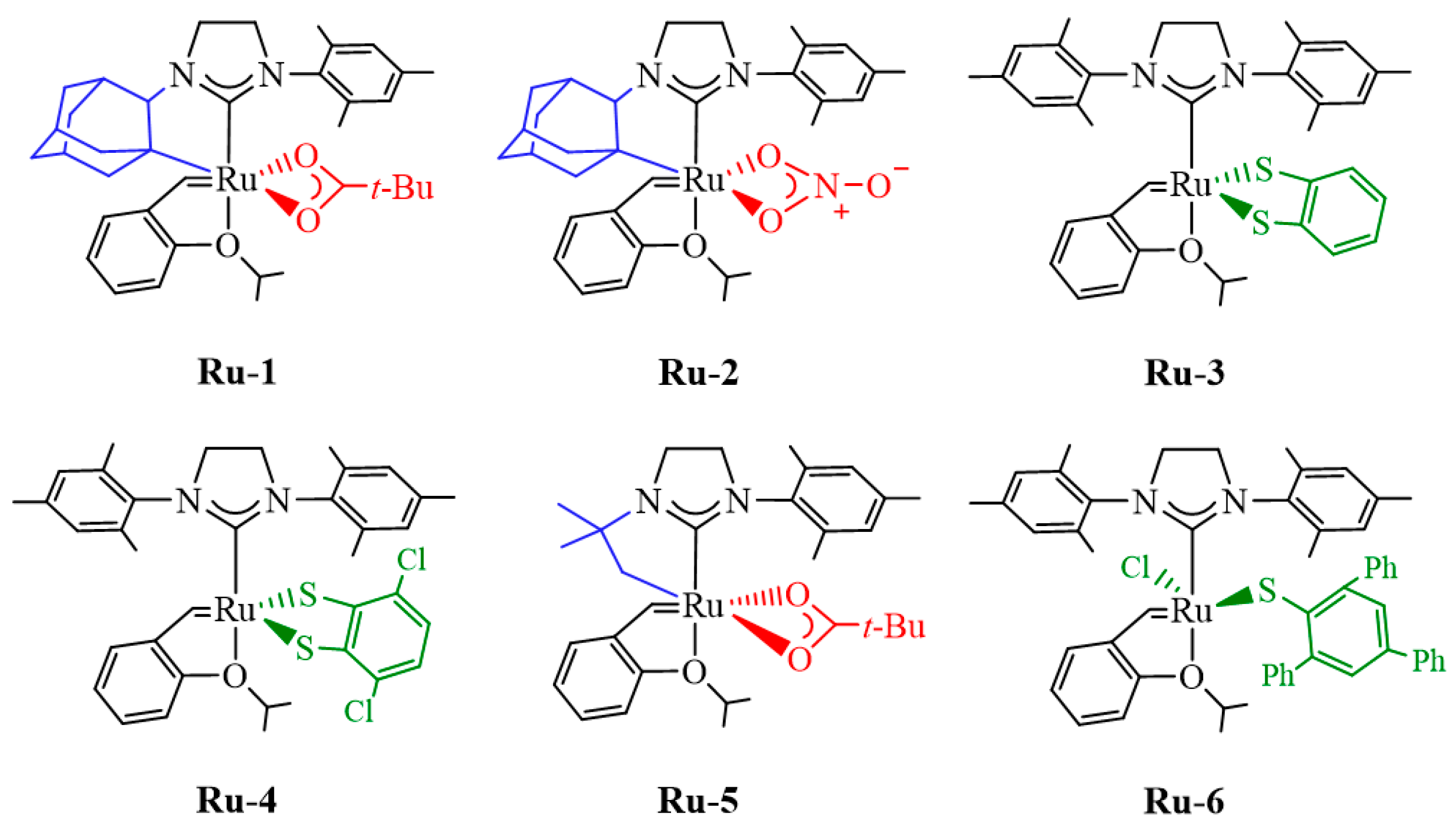
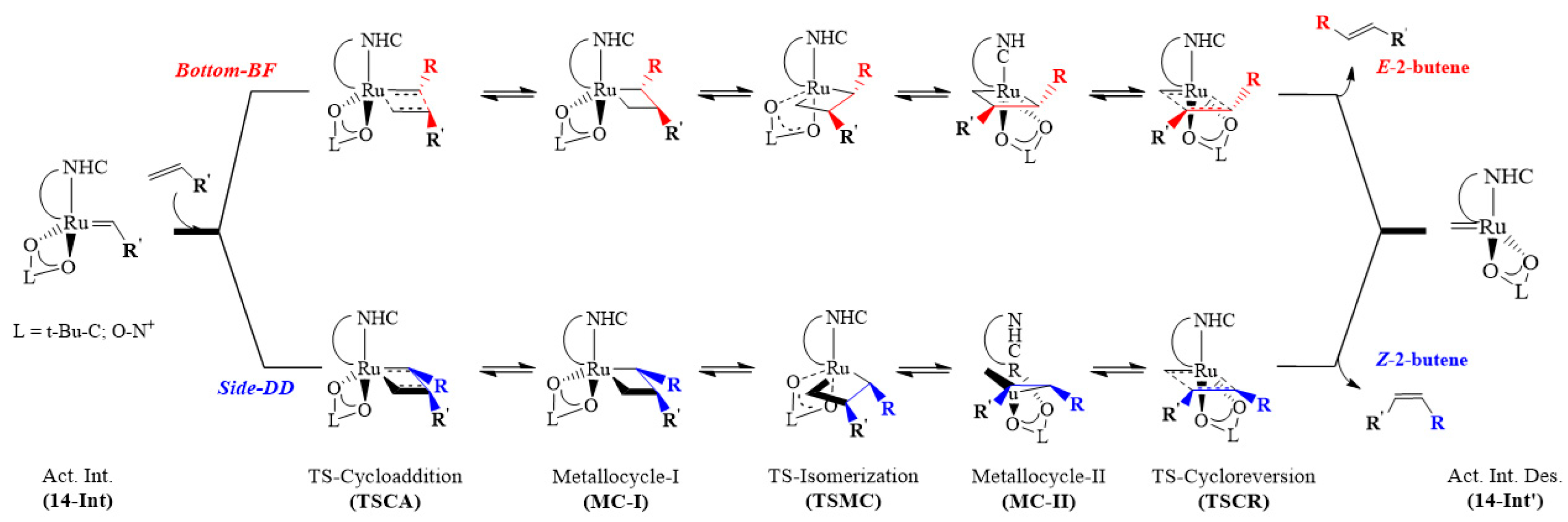
2.1. Bottom and Side Mechanism Employing the Keitz–Grubbs-Substituted, Ru-1 and Ru-2 Catalysts
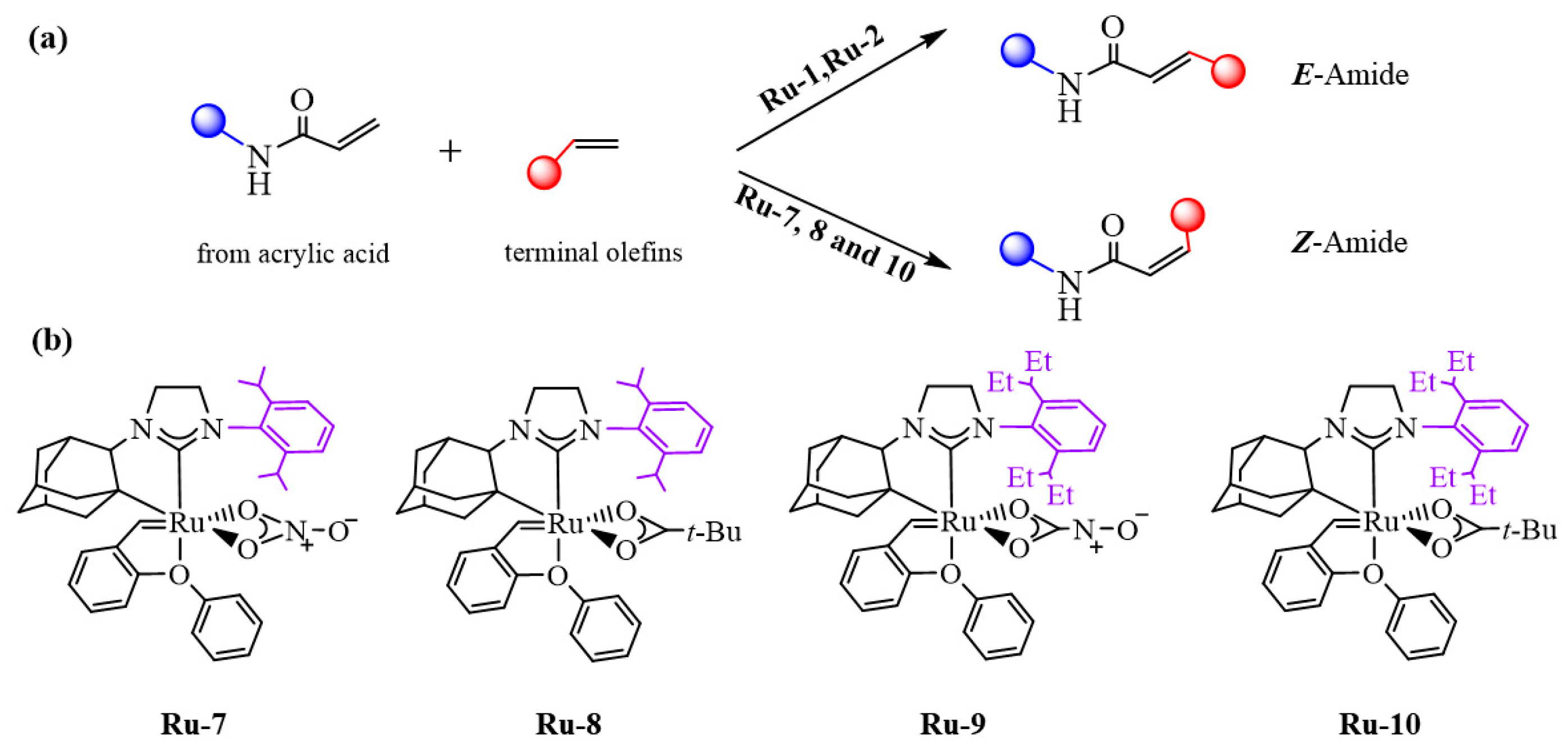
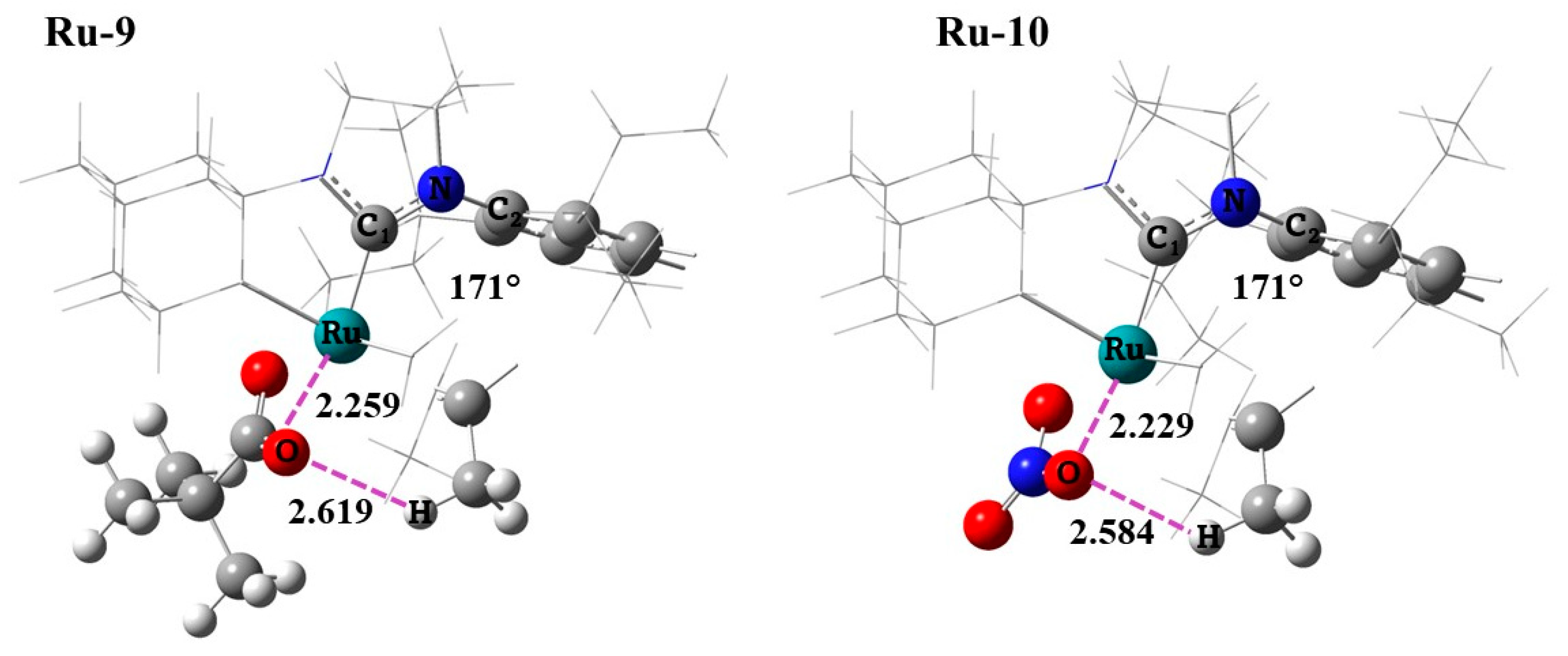
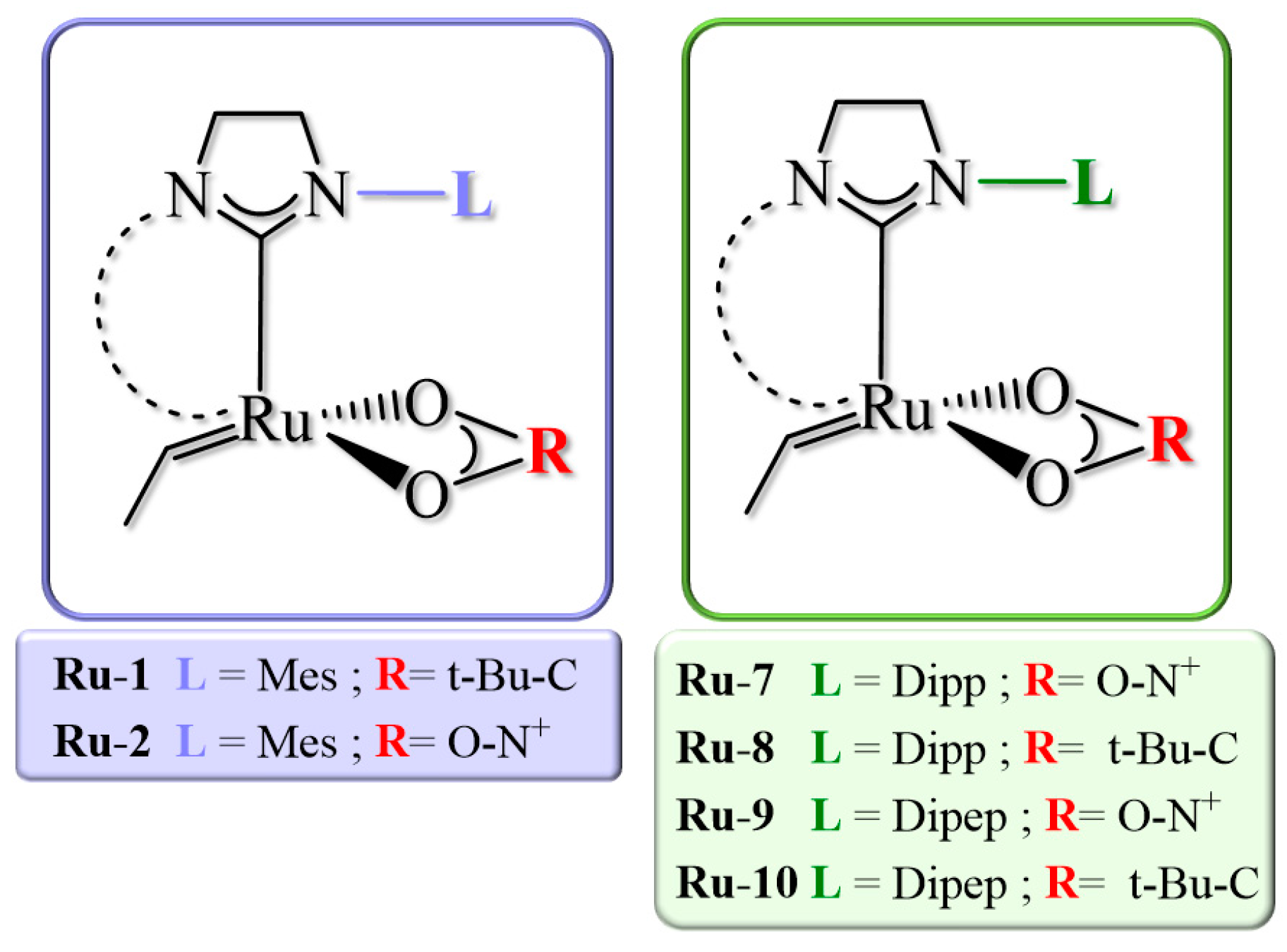
2.2. Bottom and Side DFT Mechanism Using Novel Ruthenium Catalysts, Ru-7 to Ru-10
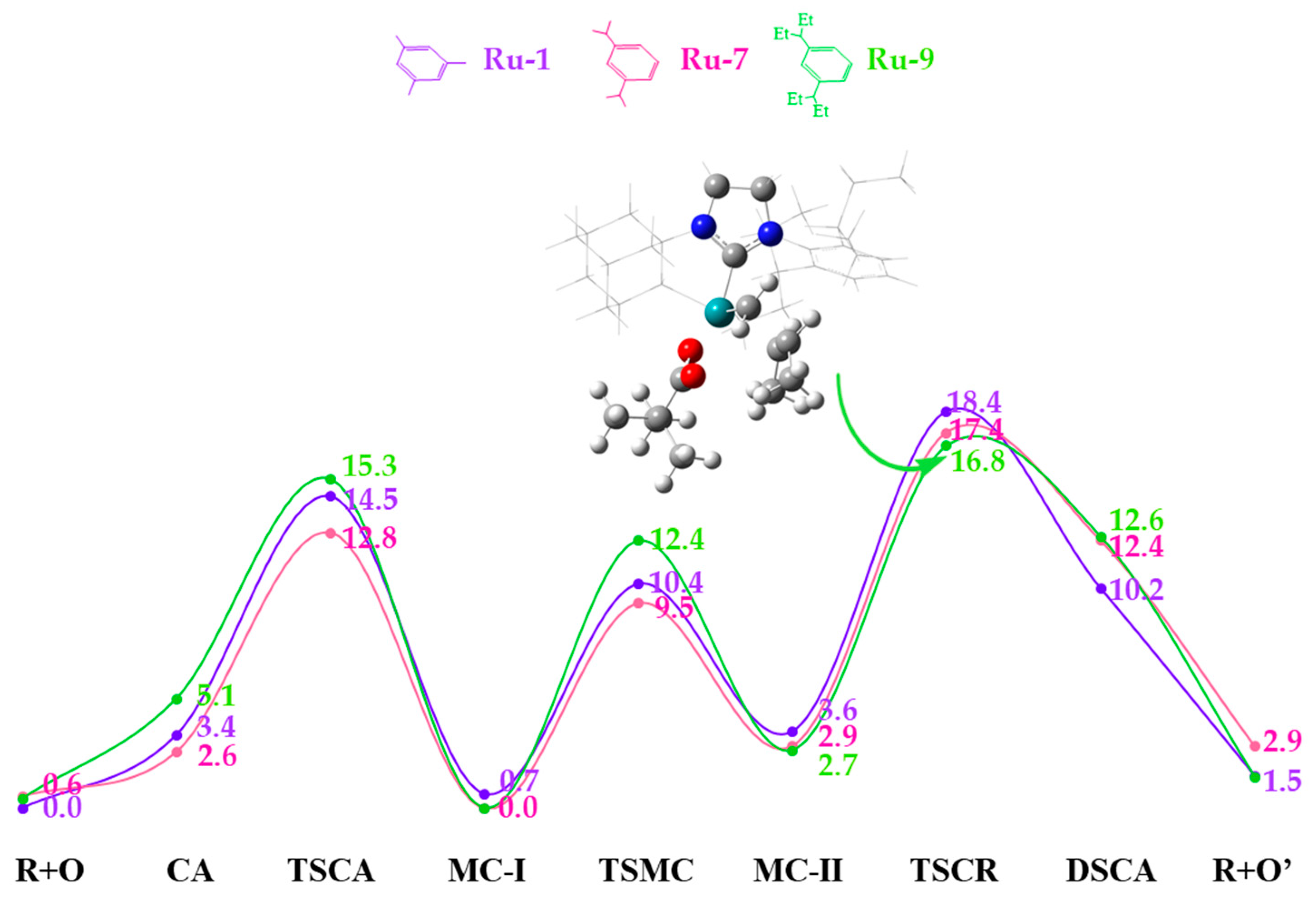
2.3. Side Mechanism and Analysis of the Electronic and Steric Role in Recent Z-Selective Catalysts (Ru-1, Ru-7, and Ru-9) Containing a Carboxylate as Anionic Group
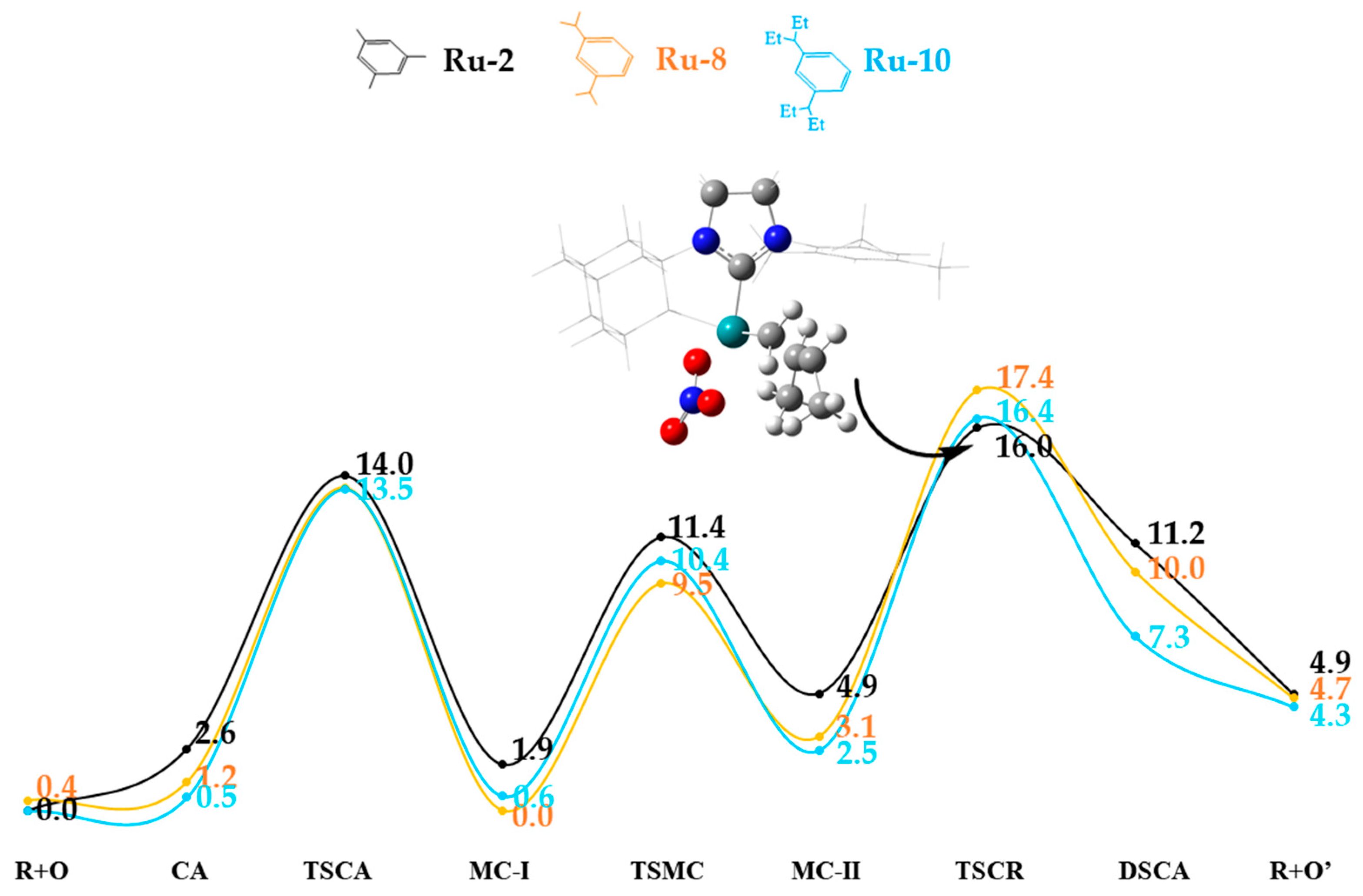
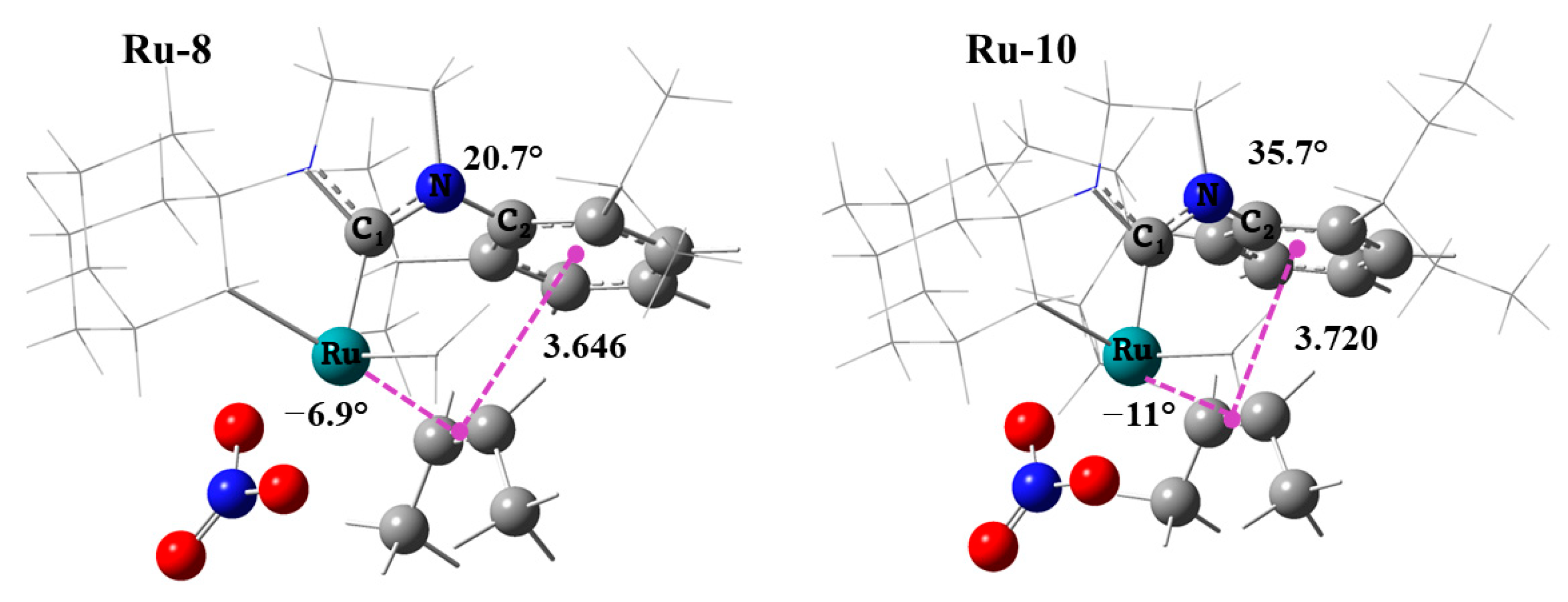
2.4. Side Mechanism and Analysis of the Electronic and Steric Role in Recent Z-Selective Catalyst (Ru-2, Ru-8, and Ru-10) with Nitrate as Anionic Group
3. Discussion
4. Materials and Methods
4.1. Models
4.2. Computational Details
4.3. Theoretical Background
4.3.1. Energy Decomposition Analysis (EDA)
4.3.2. Topographic Maps
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Turczel, G.; Kovács, E.; Merza, G.; Coish, P.; Anastas, P.T.; Tuba, R. Synthesis of Semiochemicals via Olefin Metathesis. ACS Sustain. Chem. Eng. 2019, 7, 33–48. [Google Scholar] [CrossRef]
- Eivgi, O.; Guidone, S.; Frenklah, A.; Kozuch, S.; Goldberg, I.; Lemcoff, N.G. Photoactivation of Ruthenium Phosphite Complexes for Olefin Metathesis. ACS Catal. 2018, 8, 6413–6418. [Google Scholar] [CrossRef]
- Biermann, U.; Bornscheuer, U.T.; Feussner, I.; Meier, M.A.R.; Metzger, J.O. Fatty Acids and Their Derivatives as Renewable Platform Molecules for the Chemical Industry. Angew. Chem.–Int. Ed. 2021, 60, 20144–20165. [Google Scholar] [CrossRef]
- Nienałtowski, T.; Krzesiński, P.; Baumert, M.E.; Skoczeń, A.; Suska-Kauf, E.; Pawłowska, J.; Kajetanowicz, A.; Grela, K. 4-Methyltetrahydropyran as a Convenient Alternative Solvent for Olefin Metathesis Reaction: Model Studies and Medicinal Chemistry Applications. ACS Sustain. Chem. Eng. 2020, 8, 18215–18223. [Google Scholar] [CrossRef]
- Gawin, R.; Czarnecka, P.; Grela, K. Ruthenium Catalysts Bearing Chelating Carboxylate Ligands: Application to Metathesis Reactions in Water. Tetrahedron 2010, 66, 1051–1056. [Google Scholar] [CrossRef]
- Herbert, M.B.; Grubbs, R.H. Z-Selective Cross Metathesis with Ruthenium Catalysts: Synthetic Applications and Mechanistic Implications. Angew. Chem.–Int. Ed. 2015, 54, 5018–5024. [Google Scholar] [CrossRef]
- Hughes, D.L. Highlights of the Recent U.S. Patent Literature: Focus on Metathesis. Org. Process Res. Dev. 2016, 20, 1008–1015. [Google Scholar] [CrossRef]
- Patrick Montgomery, T.; Johns, A.M.; Grubbs, R.H. Recent Advancements in Stereoselective Olefin Metathesis Using Ruthenium Catalysts. Catalysts 2017, 7, 87. [Google Scholar] [CrossRef]
- Paredes-Gil, K.; Sivasamy, R.; Mendizábal, F. A Mechanistic DFT Study of Z-Selective Ring-Opening Metathesis Polymerization by MAP Catalysts. Mol. Catal. 2022, 527, 112418. [Google Scholar] [CrossRef]
- Sousa-Silva, A.; Paredes-Gil, K.; De Matos, J.M.E.; Sá, É. Singlet Spin State Drives [V]-Carbene to Catalyze Olefin Metathesis: A Computational Analysis. Organometallics 2022, 41, 1295–1303. [Google Scholar] [CrossRef]
- Peryshkov, D.V.; Schrock, R.R.; Takase, M.K.; Müller, P.; Hoveyda, A.H. Z-Selective Olefin Metathesis Reactions Promoted by Tungsten Oxo Alkylidene Complexes. J. Am. Chem. Soc. 2011, 133, 20754–20757. [Google Scholar] [CrossRef] [PubMed]
- Ogba, O.M.; Warner, N.C.; O’Leary, D.J.; Grubbs, R.H. Recent Advances in Ruthenium-Based Olefin Metathesis. Chem. Soc. Rev. 2018, 47, 4510–4544. [Google Scholar] [CrossRef] [PubMed]
- Keitz, B.K.; Endo, K.; Herbert, M.B.; Grubbs, R.H. Z -Selective Homodimerization of Terminal Olefins with a Ruthenium Metathesis Catalyst. J. Am. Chem. Soc. 2011, 133, 9686–9688. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Yu, X.; Zhang, H.; Wu, S.; Guo, W.; Wang, J. Synthesis and Evaluation of Ruthenium 2-Alkyl-6-Mercaptophenolate Catalysts for Olefin Metathesis. Appl. Organomet. Chem. 2019, 33, 4939. [Google Scholar] [CrossRef]
- Dawood, K.M.; Nomura, K. Recent Developments in Z-Selective Olefin Metathesis Reactions by Molybdenum, Tungsten, Ruthenium, and Vanadium Catalysts. Adv. Synth. Catal. 2021, 363, 1970–1997. [Google Scholar] [CrossRef]
- Grzesiński, Ł.; Milewski, M.; Nadirova, M.; Kajetanowicz, A.; Grela, K. Unexpected Latency of Z-Stereoretentive Ruthenium Olefin Metathesis Catalysts Bearing Unsymmetrical N-Heterocyclic Carbene or Cyclic(Alkyl)(Amino)Carbene Ligands. Organometallics 2022, 428. [Google Scholar] [CrossRef]
- Keitz, B.K.; Endo, K.; Patel, P.R.; Herbert, M.B.; Grubbs, R.H. Improved Ruthenium Catalysts for Z-Selective Olefin Metathesis. J. Am. Chem. Soc. 2012, 134, 693–699. [Google Scholar] [CrossRef]
- Khan, R.K.M.; Torker, S.; Hoveyda, A.H. Readily Accessible and Easily Modifiable Ru-Based Catalysts for Efficient and Z-Selective Ring-Opening Metathesis Polymerization and Ring-Opening/Cross- Metathesis. J. Am. Chem. Soc. 2013, 135, 10258–10261. [Google Scholar] [CrossRef]
- Koh, M.J.; Khan, R.K.M.; Torker, S.; Yu, M.; Mikus, M.S.; Hoveyda, A.H. High-Value Alcohols and Higher-Oxidation-State Compounds by Catalytic Z-Selective Cross-Metathesis. Nature 2015, 517, 181–186. [Google Scholar] [CrossRef]
- Rosebrugh, L.E.; Marx, V.M.; Keitz, B.K.; Grubbs, R.H. Synthesis of Highly Cis, Syndiotactic Polymers via Ring-Opening Metathesis Polymerization Using Ruthenium Metathesis Catalysts. J. Am. Chem. Soc. 2013, 135, 10032–10035. [Google Scholar] [CrossRef]
- Smit, W.; Ekeli, J.B.; Occhipinti, G.; Woźniak, B.; Törnroos, K.W.; Jensen, V.R. Z-Selective Monothiolate Ruthenium Indenylidene Olefin Metathesis Catalysts. Organometallics 2020, 39, 397–407. [Google Scholar] [CrossRef]
- Xu, Y.; Wong, J.J.; Samkian, A.E.; Ko, J.H.; Chen, S.; Houk, K.N.; Grubbs, R.H. Efficient Z-Selective Olefin-Acrylamide Cross-Metathesis Enabled by Sterically Demanding Cyclometalated Ruthenium Catalysts. J. Am. Chem. Soc. 2020, 142, 20987–20993. [Google Scholar] [CrossRef]
- Ritter, T.; Hejl, A.; Wenzel, A.G.; Funk, T.W.; Grubbs, R.H. A Standard System of Characterization for Olefin Metathesis Catalysts. Organometallics 2006, 25, 5740–5745. [Google Scholar] [CrossRef]
- Rosebrugh, L.E.; Herbert, M.B.; Marx, V.M.; Keitz, B.K.; Grubbs, R.H. Highly Active Ruthenium Metathesis Catalysts Exhibiting Unprecedented Activity and Z-Selectivity. J. Am. Chem. Soc. 2013, 135, 1276–1279. [Google Scholar] [CrossRef] [PubMed]
- Dumas, A.; Tarrieu, R.; Vives, T.; Roisnel, T.; Dorcet, V.; Baslé, O.; Mauduit, M. A Versatile and Highly Z -Selective Olefin Metathesis Ruthenium Catalyst Based on a Readily Accessible N -Heterocyclic Carbene. ACS Catal. 2018, 8, 3257–3262. [Google Scholar] [CrossRef]
- Małecki, P.; Gajda, K.; Gajda, R.; Woźniak, K.; Trzaskowski, B.; Kajetanowicz, A.; Grela, K. Specialized Ruthenium Olefin Metathesis Catalysts Bearing Bulky Unsymmetrical NHC Ligands: Computations, Synthesis, and Application. ACS Catal. 2019, 9, 587–598. [Google Scholar] [CrossRef]
- Nelson, J.W.; Grundy, L.M.; Dang, Y.; Wang, Z.X.; Wang, X. Mechanism of Z -Selective Olefin Metathesis Catalyzed by a Ruthenium Monothiolate Carbene Complex: A DFT Study. Organometallics 2014, 33, 4290–4294. [Google Scholar] [CrossRef]
- Occhipinti, G.; Koudriavtsev, V.; Törnroos, K.W.; Jensen, V.R. Theory-Assisted Development of a Robust and Z-Selective Olefin Metathesis Catalyst. Dalton Trans. 2014, 43, 11106–11117. [Google Scholar] [CrossRef]
- Nuñez-Zarur, F.; Solans-Monfort, X.; Rodríguez-Santiago, L.; Sodupe, M. Differences in the Activation Processes of Phosphine-Containing and Grubbs-Hoveyda-Type Alkene Metathesis Catalysts. Organometallics 2012, 31, 4203–4215. [Google Scholar] [CrossRef]
- Paredes-Gil, K.; Jaque, P. Theoretical Characterization of First- and Second-Generation Grubbs Catalysts in Styrene Cross-Metathesis Reactions: Insights from Conceptual DFT. Catal. Sci. Technol. 2016, 6, 755–766. [Google Scholar] [CrossRef]
- Herbert, M.B.; Suslick, B.A.; Liu, P.; Zou, L.; Dornan, P.K.; Houk, K.N.; Grubbs, R.H. Cyclometalated Z-Selective Ruthenium Metathesis Catalysts with Modified N-Chelating Groups. Organometallics 2015, 34, 2858–2869. [Google Scholar] [CrossRef]
- Liu, P.; Xu, X.; Dong, X.; Keitz, B.K.; Herbert, M.B.; Grubbs, R.H.; Houk, K.N. Z-Selectivity in Olefin Metathesis with Chelated Ru Catalysts: Computational Studies of Mechanism and Selectivity. J. Am. Chem. Soc. 2012, 134, 1464–1467. [Google Scholar] [CrossRef] [PubMed]
- Patra, S.G.; Das, N.K. Recent Advancement on the Mechanism of Olefin Metathesis by Grubbs Catalysts: A Computational Perspective. Polyhedron 2021, 200, 115096. [Google Scholar] [CrossRef]
- Dang, Y.; Wang, Z.X.; Wang, X. Does the Ruthenium Nitrato Catalyst Work Differently in Z -Selective Olefin Metathesis? A Dft Study. Organometallics 2012, 31, 8654–8657. [Google Scholar] [CrossRef]
- Sandford, M.S.; Love, J.A.; Grubbs, R.H. Mechanism and Activity of Ruthenium Olefin Metathesis Catalysts. J. Am. Chem. Soc. 2001, 123, 6543–6554. [Google Scholar] [CrossRef]
- Suresh, C.H.; Koga, N. Orbital Interactions in the Ruthenium Olefin Metathesis Catalysts. Organometallics 2004, 23, 76–80. [Google Scholar] [CrossRef]
- Montgomery, T.P.; Grandner, J.M.; Houk, K.N.; Grubbs, R.H. Synthesis and Evaluation of Sterically Demanding Ruthenium Dithiolate Catalysts for Stereoretentive Olefin Metathesis. Organometallics 2017, 36, 3940–3953. [Google Scholar] [CrossRef]
- Martínez, J.P.; Trzaskowski, B. Olefin Metathesis Catalyzed by a Hoveyda-Grubbs-like Complex Chelated to Bis(2-Mercaptoimidazolyl) Methane: A Predictive DFT Study. J. Phys. Chem. A 2022, 126, 720–732. [Google Scholar] [CrossRef]
- Poater, A.; Solans-Monfort, X.; Clot, E.; Copéret, C.; Eisenstein, O. Understanding D0-Olefin Metathesis Catalysts: Which Metal, Which Ligands? J. Am. Chem. Soc. 2007, 129, 8207–8216. [Google Scholar] [CrossRef]
- Zhao, F.; Li, Y.; Houk, K.N.; Lu, Q.; Liu, F. Computational Elucidation on the Conformational Control of Selectivity in Intramolecular Ring-Closing Metathesis vs Intermolecular Homometathesis. J. Org. Chem. 2023, 88, 8512–8521. [Google Scholar] [CrossRef]
- Falivene, L.; Credendino, R.; Poater, A.; Petta, A.; Serra, L.; Oliva, R.; Scarano, V.; Cavallo, L. SambVca 2. A Web Tool for Analyzing Catalytic Pockets with Topographic Steric Maps. Organometallics 2016, 35, 2286–2293. [Google Scholar] [CrossRef]
- Nuñez-Zarur, F.; Poater, J.; Rodríguez-Santiago, L.; Solans-Monfort, X.; Solà, M.; Sodupe, M. On the Electronic Structure of Second Generation Hoveyda-Grubbs Alkene Metathesis Precursors. Comput. Theor. Chem. 2012, 996, 57–67. [Google Scholar] [CrossRef]
- Paredes-Gil, K.; Mendizábal, F.; Jaque, P. Further Understanding of the Ru-Centered [2+2] Cycloreversion/Cycloaddition Involved into the Interconversion of Ruthenacyclobutane Using the Grubbs Catalysts from a Reaction Force Analysis. J. Mol. Model 2019, 25, 305. [Google Scholar] [CrossRef] [PubMed]
- Paredes-Gil, K.; Solans-Monfort, X.; Rodriguez-Santiago, L.; Sodupe, M.; Jaque, P. DFT Study on the Relative Stabilities of Substituted Ruthenacyclobutane Intermediates Involved in Olefin Cross-Metathesis Reactions and Their Interconversion Pathways. Organometallics 2014, 33, 6065–6075. [Google Scholar] [CrossRef]
- Ziegler, T.; Autschbach, J. Theoretical Methods of Potential Use for Studies of Inorganic Reaction Mechanisms. Chem. Rev. 2005, 105, 2695–2722. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Bickelhaupt, F.M.; Houk, K.N. Das Distortion/Interaction-Activation-Strain-Modell Zur Analyse von Reaktionsgeschwindigkeiten. Angew. Chem. 2017, 129, 10204–10221. [Google Scholar] [CrossRef]
- Pablo Martínez, J.; Solà, M.; Poater, A. Predictive Catalysis in Olefin Metathesis with Ru-Based Catalysts with Annulated C60 Fullerenes in the N-Heterocyclic Carbenes. Chem.–A Eur. J. 2021, 27, 18074–18083. [Google Scholar] [CrossRef]
- Arnedo, L.; Chauvin, R.; Poater, A. Olefin Metathesis with Ru-Based Catalysts Exchanging the Typical N-Heterocyclic Carbenes by a Phosphine-Phosphonium Ylide. Catalysts 2017, 7, 85. [Google Scholar] [CrossRef]
- Cruz, T.R.; Masson, G.H.C.; Amorim, K.A.E.; Machado, A.E.H.; Goi, B.E.; Carvalho-Jr, V.P. Ru/Pd Complex and Its Monometallic Fragments as Catalysts for Norbornene Polymerization via ROMP and Addition. Catalysts 2022, 12, 1111. [Google Scholar] [CrossRef]
- Gómez-Suárez, A.; Nelson, D.J.; Nolan, S.P. Quantifying and Understanding the Steric Properties of N-Heterocyclic Carbenes. Chem. Commun. 2017, 53, 2650–2660. [Google Scholar] [CrossRef] [PubMed]
- Troiano, R.; Costabile, C.; Grisi, F. Alternating Ring-Opening Metathesis Polymerization Promoted by Ruthenium Catalysts Bearing Unsymmetrical NHC Ligands. Catalysts 2023, 13, 34. [Google Scholar] [CrossRef]
- Romero, M.; Macchione, M.A.; Mattea, F.; Strumia, M. The Role of Polymers in Analytical Medical Applications. A Review. Microchem. J. 2020, 159, 105366. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | Mechanism | ΔE≠ | ||
|---|---|---|---|---|
Ru-1 | Bottom-BF TSCR | 8.7 | 50.9 | −42.1 |
| Side-DD TSCR | −5.7 | 41.5 | −47.1 | |
Ru-2 | Bottom-BF TSCA | 3.5 | 48.9 | −45.3 |
| Side-DD TSCR | −6.2 | 41.2 | −47.5 |
| Catalyst | CA | TSCA | MC-l | TSMC | MC-ll | TSCR | DSCA |
|---|---|---|---|---|---|---|---|
| Ru-7 | 3.5 | 31.0 | 20.9 | 23.9 | 21.8 | 33.3 | 17.0 |
| Ru-8 | 0.6 | 28.6 | 17.7 | 27.4 | 17.0 | 27.6 | 12.0 |
| Ru-9 | 4.3 | 32.8 | 23.2 | 26.2 | 22.8 | 33.3 | 17.0 |
| Ru-10 | 1.7 | 33.4 | 19.9 | 23.5 | 18.7 | 29.3 | 13.2 |
| Catalyst | CA | TSCA | MC-l | TSMC | MC-ll | TSCR | DSCA |
|---|---|---|---|---|---|---|---|
| Ru-7 | 2.6 | 12.8 | 0.0 | 9.5 | 2.9 | 17.4 | 12.4 |
| Ru-8 | 1.2 | 13.5 | 0.0 | 9.5 | 3.1 | 17.4 | 10.0 |
| Ru-9 | 5.4 | 15.3 | 0.0 | 12.4 | 1.7 | 16.8 | 12.6 |
| Ru-10 | 0.5 | 13.5 | 0.6 | 10.4 | 2.5 | 16.4 | 7.3 |
| Catalyst | |
|---|---|
| Ru-7 | 15.9 |
| Ru-8 | 11.2 |
| Ru-9 | 16.6 |
| Ru-10 | 19.3 |
| Catalyst | ΔE≠ | ||||
|---|---|---|---|---|---|
| Ru-1 | −5.7 | 41.5 | 20.1 | 21.4 | −47.1 |
| Ru-7 | −6.9 | 40.7 | 19.1 | 21.6 | −47.6 |
| Ru-9 | −6.6 | 44.3 | 20.9 | 23.4 | −50.9 |
| Catalyst | ΔE≠ | ||||
|---|---|---|---|---|---|
| Ru-2 | −6.2 | 41.2 | 20.0 | 21.2 | −47.5 |
| Ru-8 | −7.5 | 38.6 | 18.4 | 20.2 | −46.1 |
| Ru-10 | −13.3 | 40.0 | 21.2 | 18.8 | −54.5 |
| ≠ | ≠ | ||||
|---|---|---|---|---|---|
| Ru-1: Mes | 18.4 | 21.4 | Ru-2: Mes | 16.0 | 21.2 |
| Ru-7: Dipp | 17.4 | 21.6 | Ru-8: Dipp | 17.0 | 20.2 |
| Ru-9: Dipep | 16.8 | 23.4 | Ru-10: Dipep | 16.4 | 18.8 |
| Catalysts | ||||||
|---|---|---|---|---|---|---|
| Ru-1 | −0.497 | 0.509 | −0.187 | 0.032 | 0.078 | 0.065 |
| Ru-2 | −0.640 | 0.537 | −0.119 | 0.011 | 0.108 | 0.103 |
| Ru-7 | −0.487 | 0.541 | −0.192 | 0.017 | 0.059 | 0.062 |
| Ru-8 | −0.617 | 0.614 | −0.164 | 0.010 | 0.081 | 0.076 |
| Ru-9 | −0.503 | 0.548 | −0.193 | 0.017 | 0.071 | 0.060 |
| Ru-10 | −0.605 | 0.574 | −0.179 | 0.022 | 0.027 | 0.161 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diaz-González, V.; Paredes-Gil, K. Role of Electronic and Steric Effects on Ruthenium Catalysts with Bulky NHC Ligands and Relationship with the Z-Selectivity in Olefin Metathesis. Catalysts 2023, 13, 1305. https://doi.org/10.3390/catal13091305
Diaz-González V, Paredes-Gil K. Role of Electronic and Steric Effects on Ruthenium Catalysts with Bulky NHC Ligands and Relationship with the Z-Selectivity in Olefin Metathesis. Catalysts. 2023; 13(9):1305. https://doi.org/10.3390/catal13091305
Chicago/Turabian StyleDiaz-González, Valentina, and Katherine Paredes-Gil. 2023. "Role of Electronic and Steric Effects on Ruthenium Catalysts with Bulky NHC Ligands and Relationship with the Z-Selectivity in Olefin Metathesis" Catalysts 13, no. 9: 1305. https://doi.org/10.3390/catal13091305