
Morpholine Radical in the Electrochemical Reaction with Quinoline N-Oxide

 , and
, and
Abstract
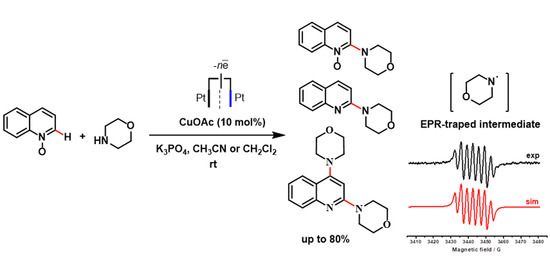
:
1. Introduction

2. Results
2.1. Electrochemical Synthesis
2.2. Voltammetry Data
2.3. EPR Study
2.4. Mechanistic Considerations
3. Materials and Methods
3.1. Materials
3.2. NMR Measurements
3.3. Mass Spectrometric Studies
3.4. EPR Experiments
3.5. Cyclic Voltammetry (CV)
3.6. Preparative Electrolysis
3.7. General Electrolysis Procedure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tzara, A.; Xanthopoulos, D.; Kourounakis, A.P. Morpholine as a Scaffold in Medicinal Chemistry: An Update on Synthetic Strategies. ChemMedChem 2020, 15, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Kőnig, B.; Sztanó, G.; Holczbauer, T.; Soós, T. Syntheses of 2- and 3-Substituted Morpholine Congeners via Ring Opening of 2-Tosyl-1,2-Oxazetidine. J. Org. Chem. 2023, 88, 6182–6191. [Google Scholar] [CrossRef]
- Kourounakis, A.P.; Xanthopoulos, D.; Tzara, A. Morpholine as a Privileged Structure: A Review on the Medicinal Chemistry and Pharmacological Activity of Morpholine Containing Bioactive Molecules. Med. Res. Rev. 2020, 40, 709–752. [Google Scholar] [CrossRef] [PubMed]
- Kumari, A.; Singh, R.K. Morpholine as Ubiquitous Pharmacophore in Medicinal Chemistry: Deep Insight into the Structure-Activity Relationship (SAR). Bioorg. Chem. 2020, 96, 103578. [Google Scholar] [CrossRef] [PubMed]
- Pal’chikov, V.A. Morpholines. Synthesis and Biological Activity. Russ. J. Org. Chem. 2013, 49, 787–814. [Google Scholar] [CrossRef]
- Palchykov, V.A.; Chebanov, V.A. Recent Progress in the Synthesis of Morpholines. Chem. Heterocycl. Compd. 2019, 55, 324–332. [Google Scholar] [CrossRef]
- Wallace, T.J.; Gritter, R.J.; Walsh, H.G. Free Radical Chemistry of Cyclic Ethers: P-Dioxane and Morpholine. Nature 1963, 198, 284–285. [Google Scholar] [CrossRef]
- Speckmeier, E.; Maier, T.C. ART—An Amino Radical Transfer Strategy for C(Sp2)–C(Sp3) Coupling Reactions, Enabled by Dual Photo/Nickel Catalysis. J. Am. Chem. Soc. 2022, 144, 9997–10005. [Google Scholar] [CrossRef] [PubMed]
- Vijayakrishna, K.; Manojkumar, K.; Haribabu, P.; GyanaRanjan, B.; Tilottama, B.; Agirre, A.; Meabe, L.; Mantione, D.; Porcarelli, L.; Leiza, J.R.; et al. Morpholine-based RAFT Agents for the Reversible Deactivation Radical Polymerization of Vinyl Acetate and N-vinylimidazole. Polym. Int. 2020, 69, 883–890. [Google Scholar] [CrossRef]
- Tereshchenko, A.D.; Myronchuk, J.S.; Leitchenko, L.D.; Knysh, I.V.; Tokmakova, G.O.; Litsis, O.O.; Tolmachev, A.; Liubchak, K.; Mykhailiuk, P. Synthesis of 3-Oxadiazolyl/Triazolyl Morpholines: Novel Scaffolds for Drug Discovery. Tetrahedron 2017, 73, 750–757. [Google Scholar] [CrossRef]
- Gao, X.; Wang, P.; Zeng, L.; Tang, S.; Lei, A. Cobalt(II)-Catalyzed Electrooxidative C–H Amination of Arenes with Alkylamines. J. Am. Chem. Soc. 2018, 140, 4195–4199. [Google Scholar] [CrossRef]
- Zhang, S.; Samanta, R.C.; Sauermann, N.; Ackermann, L. Nickel-Catalyzed Electrooxidative C−H Amination: Support for Nickel(IV). Chem. A Eur. J. 2018, 24, 19166–19170. [Google Scholar] [CrossRef]
- Wang, Z.; Han, M.-Y.; Li, P.; Wang, L. Copper-Catalyzed Deoxygenative C-2 Amination of Quinoline N-Oxides. Eur. J. Org. Chem. 2018, 2018, 5954–5960. [Google Scholar] [CrossRef]
- Thakur, D.G.; Sahoo, T.; Sen, C.; Rathod, N.; Ghosh, S.C. Palladium-Catalyzed Directed Aldehyde C–H Arylation of Quinoline-8-Carbaldehydes: Exploring the Reactivity Differences between Aryl (Pseudo) Halides. J. Org. Chem. 2022, 87, 16343–16350. [Google Scholar] [CrossRef] [PubMed]
- Haozhi, W.; Tian, L.; Jianwen, J.; Jieping, W. KI-Catalyzed Selective C(5)-Sulfenylation and C(5),C(7)-Disulfenylation of Unprotected 8-Aminoquinolines and the Indole C(2),C(3)-Disulfenylation. Chin. J. Org. Chem. 2022, 42, 3721. [Google Scholar] [CrossRef]
- Kumagai, Y.; Murakami, N.; Kamiyama, F.; Tanaka, R.; Yoshino, T.; Kojima, M.; Matsunaga, S. C–H γ,γ,γ-Trifluoroalkylation of Quinolines via Visible-Light-Induced Sequential Radical Additions. Org. Lett. 2019, 21, 3600–3605. [Google Scholar] [CrossRef]
- Xu, P.; Xu, H.-C. Electrochemical Deoxygenation of N-Heteroaromatic N-Oxides. Synlett 2019, 30, 1219–1221. [Google Scholar] [CrossRef]
- Fukazawa, Y.; Rubtsov, A.E.; Malkov, A.V. A Mild Method for Electrochemical Reduction of Heterocyclic N-Oxides. Eur. J. Org. Chem. 2020, 2020, 3317–3319. [Google Scholar] [CrossRef]
- Yu, X.; Yang, S.; Zhang, Y.; Guo, M.; Yamamoto, Y.; Bao, M. Intermolecular Amidation of Quinoline N-Oxides with Arylsulfonamides under Metal-Free Conditions. Org. Lett. 2017, 19, 6088–6091. [Google Scholar] [CrossRef] [PubMed]
- Nuzhdin, K.B.; Nesterov, S.V.; Tyurin, D.A.; Feldman, V.I.; Wei, L.; Lund, A. Structure of Radical Cations of Saturated Heterocyclic Compounds with Two Heteroatoms as Studied by Electron Paramagnetic Resonance, Electron−Nuclear Double Resonance, and Density Functional Theory Calculations. J. Phys. Chem. A 2005, 109, 6166–6173. [Google Scholar] [CrossRef]
- Hudson, A.; Hussain, H.A. Electron Spin Resonance of Aliphatic Nitroxides. Part II. The Cyclic Radicals from Pyrrolidine, Piperidine, Morpholine, and Hexamethyleneimine. J. Chem. Soc. B Phys. Org. 1968, 251, 251–253. [Google Scholar] [CrossRef]
- Sreenath, K.; Yuan, Z.; Macias-Contreras, M.; Ramachandran, V.; Clark, R.J.; Zhu, L. Dual Role of Acetate in Copper(II) Acetate Catalyzed Dehydrogenation of Chelating Aromatic Secondary Amines: A Kinetic Case Study of Copper-Catalyzed Oxidation Reactions. Eur. J. Inorg. Chem. 2016, 2016, 3728–3743. [Google Scholar] [CrossRef]
- Bleaney, B.; Bowers, K.D. Anomalous Paramagnetism of Copper Acetate. Proc. R. Soc. Lond. Ser. A Math. Phys. Sci. 1952, 214, 451–465. [Google Scholar] [CrossRef]
- Chavan, S. The Structural Basis for the Enhanced Oxygenase Activity of Copper Acetate Dimers Encapsulated in Zeolites. Top. Catal. 2000, 11, 359–367. [Google Scholar] [CrossRef]
- Gryaznova, T.V.; Kholin, K.V.; Nikanshina, E.O.; Khrizanforova, V.V.; Strekalova, S.O.; Fayzullin, R.R.; Budnikova, Y.H. Copper or Silver-Mediated Oxidative C(Sp2)–H/N–H Cross-Coupling of Phthalimide and Heterocyclic Arenes: Access to N-Arylphthalimides. Organometallics 2019, 38, 3617–3628. [Google Scholar] [CrossRef]
- Li, G.; Jia, C.; Sun, K. Copper-Catalyzed Intermolecular Dehydrogenative Amidation/Amination of Quinoline N-Oxides with Lactams/Cyclamines. Org. Lett. 2013, 15, 5198–5201. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Song, H.; Cui, X.; Pi, C.; Du, W.; Wu, Y. Sulfonylation of Quinoline N-Oxides with Aryl Sulfonyl Chlorides via Copper-Catalyzed C–H Bonds Activation. Org. Lett. 2013, 15, 1270–1273. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Jia, C.; Sun, K.; Lv, Y.; Zhao, F.; Zhou, K.; Wu, H. Copper(ii)-Catalyzed Electrophilic Amination of Quinoline N-Oxides with O-Benzoyl Hydroxylamines. Org. Biomol. Chem. 2015, 13, 3207–3210. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, F.; Tang, L.; Geng, H.; Yao, Z. Preparation of 4-aminoquinoline derivatives by palladium and copper synergistically catalyzed oxidative aromatization. CN115947684 A, 11 April 2023. [Google Scholar]
- Margolis, B.J.; Long, K.A.; Laird, D.L.T.; Ruble, J.C.; Pulley, S.R. Assembly of 4-Aminoquinolines via Palladium Catalysis: A Mild and Convenient Alternative to S N Ar Methodology. J. Org. Chem. 2007, 72, 2232–2235. [Google Scholar] [CrossRef]
- Strekowski, L.; Patterson, S.E.; Janda, L.; Wydra, R.L.; Harden, D.B.; Lipowska, M.; Cegla, M.T. Further studies on the cyclization of aromatic azomethines ortho-substituted with a trifluoromethyl group: Synthesis of 2,4-di- or 2,3,4-trisubstituted quinolines. J. Org. Chem. 1992, 57, 196–201. [Google Scholar] [CrossRef]
- Tapan, S.; Dinesh, T.; Baran, P.A.; Chandra, G.S. Copper/manganese oxide catalyzed regioselective amination of quinoline N-oxides: An example of synergistic cooperative catalysis. Tetrahedron Lett. 2021, 83, 153415. [Google Scholar] [CrossRef]
- Wong, S.M.; Choy, P.Y.; Zhao, Q.; Yuen, O.Y.; Yeung, C.C.; So, C.M.; Kwong, F.Y. Design of Benzimidazolyl Phosphines Bearing Alterable P,O or P,N-Coordination: Synthesis, Characterization, and Insights into Their Reactivity. Organometallics 2021, 40, 2265–2271. [Google Scholar] [CrossRef]
- Kondratev, V.B.; Malenkov, M.Y.; Kaabak, L.V.; Golikov, A.G.; Golovkov, V.F. Method for the Production of N-hydroxy morpholine. Russia Patent RU2757983C1, 25 October 2021. [Google Scholar]
- Inami, A.; Nishii, Y.; Hirano, K.; Miura, M. Rhodium-Catalyzed Isoquinoline Synthesis Using Vinyl Selenone as Oxidizing Acetylene Surrogate. Org. Lett. 2023, 25, 3206–3209. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Ratio Morpholine: N-Oxide | Catalyst | Solvent | Base | Number of Electrons (Faradays) | Potential, V | Product (Yield, %) |
|---|---|---|---|---|---|---|---|
| 1 | 1.2:1 | Cu(OAc)2 | CH3CN | - | 2 | 1.80 | 5 (54) |
| 2 | 1.2:1 | Cu(OAc)2 | CH3CN | - | 3 | 2.00 | 6 (31) |
| 3 | 2.4:1 | Cu(OAc)2 | CH3CN | - | 4 | 2.20 | 4 (48) |
| 4 | 1.2:1 | Cu(OAc)2 | CH3CN | K3PO4 | 2 | 1.49 | 5 (80) |
| 5 | 1.2:1 | Cu(OAc)2 | CH3CN | K3PO4 | 3 | 1.80 | 6 (67) |
| 6 | 3:1 | Cu(OAc)2 | CH3CN | K3PO4 | 4 | 1.90 | 4 (52) |
| 7 * | 3:1 | Cu(OAc)2 | CH2Cl2 | K3PO4 | 4 | 0.30 | 3 (64) |
| 8 | 1.2:1 | AgOAc | CH3CN | - | 2 | 1.84 | 5 (32) |
| 9 | 1.2:1 | AgOAc | CH3CN | - | 3 | 2.08 | 6 (24) |
| 10 | 3:1 | AgOAc | CH3CN | - | 4 | 2.10 | 4 (46) |
| Entry | 1st Peak (vs. Fc/Fc+) | 2nd Peak (vs. Fc/Fc+) | 3rd Peak (vs. Fc/Fc+) |
|---|---|---|---|
| Quinoline N-ox | 1.10 V | - | - |
| Cu(OAc)2 | 1.41 V | 1.87 V | - |
| Morpholine | 0.85 V | - | - |
| Quinoline N-ox + Cu(OAc)2 | 1.24 V | - | - |
| Morpholine + Cu(OAc)2 | 0.74 V | 1.32 V | 1.83 V |
| Quinoline N-ox + Morpholine + Cu(OAc)2 | 0.74 V | 1.29 V | - |
| Entry | 1st Peak (vs. Fc/Fc+) | 2nd Peak (vs. Fc/Fc+) | 3rd Peak (vs. Fc/Fc+) |
|---|---|---|---|
| Quinoline N-ox | 1.18 V | - | - |
| Cu(OAc)2 | - | - | - |
| Morpholine | 0.89 V | - | - |
| Quinoline N-ox + Cu(OAc)2 | 1.16 V | 1.81 V | - |
| Morpholine + Cu(OAc)2 | 0.85 V | 1.55 V | - |
| Quinoline N-ox + Morpholine + Cu(OAc)2 | 0.84 V | 1.29 V | 1.63 V |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dolengovski, E.L.; Gryaznova, T.V.; Sinyashin, O.G.; Gavrilova, E.L.; Kholin, K.V.; Budnikova, Y.H. Morpholine Radical in the Electrochemical Reaction with Quinoline N-Oxide. Catalysts 2023, 13, 1279. https://doi.org/10.3390/catal13091279
Dolengovski EL, Gryaznova TV, Sinyashin OG, Gavrilova EL, Kholin KV, Budnikova YH. Morpholine Radical in the Electrochemical Reaction with Quinoline N-Oxide. Catalysts. 2023; 13(9):1279. https://doi.org/10.3390/catal13091279
Chicago/Turabian StyleDolengovski, Egor L., Tatyana V. Gryaznova, Oleg G. Sinyashin, Elena L. Gavrilova, Kirill V. Kholin, and Yulia H. Budnikova. 2023. "Morpholine Radical in the Electrochemical Reaction with Quinoline N-Oxide" Catalysts 13, no. 9: 1279. https://doi.org/10.3390/catal13091279