
Mechanistic Insight into the Propane Oxidation Dehydrogenation by N2O over Cu-BEA Zeolite with Diverse Active Site Structures

Abstract
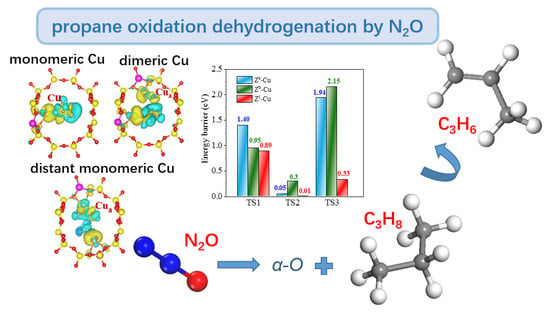
:
1. Introduction
2. Result and Discussion
2.1. N2O-ODHP Mechanism Simulation over Diverse ACTIVE Site
2.1.1. N2O-ODHP over Monomeric [Cu]+ Site of Route A
2.1.2. N2O-ODHP over Dimeric [Cu−Cu]2+ Site of Route B
2.1.3. N2O-ODHP over Distant [Cu]+—[Cu]+ Site of Route C
2.2. Microkinetic Modeling
2.3. Static Charge Difference PDOS and Analyses
3. N2O-ODHP Activity Measurement
4. Computational Modeling and Methodology
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pérez-Ramírez, J.; Gallardo-Llamas, A. Framework composition effects on the performance of steam-activated FeMFI zeolites in the N2O-mediated propane oxidative dehydrogenation to propylene. J. Phys. Chem. B 2005, 109, 20529–20538. [Google Scholar] [CrossRef] [PubMed]
- Kondratenko, E.; Cherian, M.; Baerns, M.; Su, D.; Schlogl, R.; Wang, X.; Wachs, I. Oxidative dehydrogenation of propane over V/MCM-41 catalysts: Comparison of O2 and N2O as oxidants. J. Catal. 2005, 234, 131–142. [Google Scholar] [CrossRef] [Green Version]
- Sanchezgalofre, O.; Segura, Y.; Perezramirez, J. Deactivation and regeneration of iron-containing MFI zeolites in propane oxidative dehydrogenation by N2O. J. Catal. 2007, 249, 123–133. [Google Scholar] [CrossRef]
- Wei, W.; Moulijn, J.A.; Mul, G. FAPO and Fe-TUD-1: Promising catalysts for N2O mediated selective oxidation of propane. J. Catal. 2009, 262, 1–8. [Google Scholar] [CrossRef]
- Kondratenko, E.V.; Brückner, A. On the nature and reactivity of active oxygen species formed from O2 and N2O on VOx/MCM-41 used for oxidative dehydrogenation of propane. J. Catal. 2010, 274, 111–116. [Google Scholar] [CrossRef]
- Orlyk, S.; Kyriienko, P.; Kapran, A.; Chedryk, V.; Balakin, D.; Gurgul, J.; Zimowska, M.; Millot, Y.; Dzwigaj, S. CO2-assisted dehydrogenation of propane to propene over Zn-BEA zeolites: Impact of acid–base characteristics on catalytic performance. Catalysts 2023, 13, 681. [Google Scholar] [CrossRef]
- Vogt, E.T.; Weckhuysen, B.M. Fluid catalytic cracking: Recent developments on the grand old lady of zeolite catalysis. Chem. Soc. Rev. 2015, 44, 7342–7370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.Q.; Bozzano, A.; Glover, B.; Fuglerud, T.; Kvisle, S. Recent advancements in ethylene and propylene production using the UOP/Hydro MTO process. Catal. Today 2005, 106, 103–107. [Google Scholar] [CrossRef]
- Bulánek, R.; Wichterlová, B.; Novoveská, K.; Kreibich, V. Oxidation of propane with oxygen and/or nitrous oxide over Fe-ZSM-5 with low iron concentrations. Appl. Catal. A 2004, 264, 13–22. [Google Scholar] [CrossRef]
- Novoveská, K.; Bulánek, R.; Wichterlová, B. Oxidation of propane with oxygen, nitrous oxide and oxygen/nitrous oxide mixture over Co- and Fe-zeolites. Catal. Today 2005, 100, 315–319. [Google Scholar] [CrossRef]
- Wu, G.; Hao, Y.; Zhang, N.; Guan, N.; Li, L.; Grünert, W. Oxidative dehydrogenation of propane with nitrous oxide over Fe–O–Al species occluded in ZSM-5: Reaction and deactivation mechanisms. Microporous Mesoporous Mater. 2014, 198, 82–91. [Google Scholar] [CrossRef]
- Jiang, X.; Sharma, L.; Fung, V.; Park, S.J.; Jones, C.W.; Sumpter, B.G.; Baltrusaitis, J.; Wu, Z. Oxidative dehydrogenation of propane to propylene with soft oxidants via heterogeneous catalysis. ACS Catal. 2021, 11, 2182–2234. [Google Scholar] [CrossRef]
- Patet, R.E.; Koehle, M.; Lobo, R.F.; Caratzoulas, S.; Vlachos, D.G. General acid-type catalysis in the dehydrative aromatization of furans to aromatics in H-[Al]-BEA, H-[Fe]-BEA, H-[Ga]-BEA, and H-[B]-BEA zeolites. J. Phys. Chem. C 2017, 121, 13666–13679. [Google Scholar] [CrossRef]
- Chalupka, K.; Thomas, C.; Millot, Y.; Averseng, F.; Dzwigaj, S. Mononuclear pseudo-tetrahedral V species of VSiBEA zeolite as the active sites of the selective oxidative dehydrogenation of propane. J. Catal. 2013, 305, 46–55. [Google Scholar] [CrossRef]
- Mauvezin, M.; Delahay, G.; Kißlich, F.; Coq, B.; Kieger, S. Catalytic reduction of N2O by NH3 in presence of oxygen using Fe-exchanged zeolites. Catal. Lett. 1999, 62, 41–44. [Google Scholar] [CrossRef]
- Sobalik, Z.; Sazama, P.; Dedecek, J.; Wichterlová, B. Critical evaluation of the role of the distribution of Al atoms in the framework for the activity of metallo-zeolites in redox N2O/NOx reactions. Appl. Catal. A 2014, 474, 178–185. [Google Scholar] [CrossRef]
- Liu, N.; Zhang, R.; Chen, B.; Li, Y.; Li, Y. Comparative study on the direct decomposition of nitrous oxide over M (Fe, Co, Cu)–BEA zeolites. J. Catal. 2012, 294, 99–112. [Google Scholar] [CrossRef]
- Xu, R.; Liu, N.; Dai, C.; Li, Y.; Zhang, J.; Wu, B.; Yu, G.; Chen, B. H2O-built proton transfer bridge enhances continuous methane oxidation to methanol over Cu-BEA zeolite. Angew. Chem. 2021, 60, 16634–16640. [Google Scholar] [CrossRef]
- Widmann, D.; Behm, R.J. Dynamic surface composition in a Mars-van Krevelen type reaction: CO oxidation on Au/TiO2. J. Catal. 2018, 357, 263–273. [Google Scholar] [CrossRef]
- Wang, C.; Gu, X.-K.; Yan, H.; Lin, Y.; Li, J.; Liu, D.; Li, W.-X.; Lu, J. Water-mediated Mars–Van Krevelen mechanism for CO oxidation on ceria-supported single-atom Pt1 catalyst. ACS Catal. 2016, 7, 887–891. [Google Scholar] [CrossRef]
- Jan, F.; Lian, Z.; Zhi, S.; Yang, M.; Si, C.; Li, B. Revealing the role of HBr in propane dehydrogenation on CeO2(111) via DFT-based microkinetic simulation. Phys. Chem. Chem. Phys. 2022, 24, 9718–9726. [Google Scholar] [CrossRef] [PubMed]
- Suo, W.; Sun, S.; Liu, N.; Li, X.; Wang, Y. The adsorption and dissociation of N2O on CuO(111) surface: The effect of surface structures. Surf. Sci. 2020, 696, 121596. [Google Scholar] [CrossRef]
- Li, Y.; Liu, N.; Dai, C.; Xu, R.; Yu, G.; Wang, N.; Zhang, J.; Chen, B. Synergistic Effect of neighboring Fe and Cu cation sites boosts FenCum-BEA activity for the continuous direct oxidation of Methane to Methanol. Catalysts 2021, 11, 1444. [Google Scholar] [CrossRef]
- Jianwen, Z.; Zhifeng, H.; Ziqian, Y.; Meijuan, L.; Fei, C.; Qiang, S. The influence of alkaline earth elements on electronic properties of α-Si3N4 via DFT calculation. J. Wuhan Univ. Technol. Mater. Sci. Ed. 2020, 35, 863–871. [Google Scholar]
- Zhou, Z.; Qin, B.; Li, S.; Sun, Y. A DFT-based microkinetic study on methanol synthesis from CO2 hydrogenation over the In2O3 catalyst. Phys. Chem. Chem. Phys. 2021, 23, 1888–1895. [Google Scholar] [CrossRef] [PubMed]
- Montejo-Valencia, B.D.; Curet-Arana, M.C. Periodic DFT study of the opening of fructose and glucose rings and the further conversion of fructose to trioses catalyzed by M-BEA (M = Sn, Ti, Zr, or Hf). J. Phys. Chem. C 2019, 123, 3532–3540. [Google Scholar] [CrossRef]
- Mahyuddin, M.H.; Staykov, A.; Shiota, Y.; Yoshizawa, K. Direct conversion of methane to methanol by metal-exchanged ZSM5 zeolite (Metal = Fe, Co, Ni, Cu). ACS Catal. 2016, 6, 8321–8331. [Google Scholar] [CrossRef]
- Liu, N.; Zhang, R.; Li, Y.; Chen, B. Local electric field effect of TMI (Fe, Co, Cu)-BEA on N2O direct dissociation. J. Phys. Chem. C 2014, 118, 10944–10956. [Google Scholar] [CrossRef]
- Konsolakis, M. Recent advances on nitrous oxide (N2O) decomposition over non-noble-metal oxide catalysts: Catalytic performance, mechanistic considerations, and surface chemistry aspects. ACS Catal. 2015, 5, 6397–6421. [Google Scholar] [CrossRef]
- Ikuno, T.; Grundner, S.; Jentys, A.; Li, G.; Pidko, E.A.; Fulton, J.L.; Sanchez-Sanchez, M.; Lercher, J.A. Formation of Active Cu-oxo Clusters for Methane Oxidation in Cu-Exchanged Mordenite. J. Phys. Chem. C 2019, 123, 8759–8769. [Google Scholar] [CrossRef]
- Liu, N.; Yuan, X.; Zhang, R.; Xu, R.; Li, Y. Mechanistic insight into selective catalytic combustion of acrylonitrile (C2H3CN): NCO formation and its further transformation towards N2. Phys. Chem. Chem. Phys. 2017, 19, 7971–7979. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Yuan, X.; Zhang, R.; Li, Y.; Chen, B. Mechanistic insight into selective catalytic combustion of HCN over Cu-BEA: Influence of different active center structures. Phys. Chem. Chem. Phys. 2017, 19, 23960–23970. [Google Scholar] [CrossRef] [PubMed]
- Dixit, M.; Baruah, R.; Parikh, D.; Sharma, S.; Bhargav, A. Autothermal reforming of methane on rhodium catalysts: Microkinetic analysis for model reduction. Comput. Chem. Eng. 2016, 89, 149–157. [Google Scholar] [CrossRef] [Green Version]
- Ke, C.; Lin, Z. Density Functional Theory Based Micro- and Macro-Kinetic Studies of Ni-Catalyzed Methanol Steam Reforming. Catalysts 2020, 10, 349. [Google Scholar] [CrossRef] [Green Version]
- Gokhale, A.A.; Kandoi, S.; Greeley, J.P.; Mavrikakis, M.; Dumesic, J.A. Molecular-level descriptions of surface chemistry in kinetic models using density functional theory. Chem. Eng. Sci. 2004, 59, 4679–4691. [Google Scholar] [CrossRef]
- Cai, Q.-X.; Wang, J.-G.; Wang, Y.-G.; Mei, D. Mechanistic insights into the structure-dependent selectivity of catalytic furfural conversion on platinum catalysts. AIChE J. 2015, 61, 3812–3824. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Step | Elementary Steps | Reaction Rate Equations |
|---|---|---|
| R1 | Z−Cu−N2O(g) Z−Cu−N2O | r1 = k1PN2Oθv − k-1θN2O |
| R2 | Z−Cu−N2O → Z−Cu−O+N2(g) | r2 = k2θN2O |
| R3 | Z−Cu−O+C3H8(g) Z−Cu−O−C3H8 | r3 = k3PC3H8θO − k-3θO-C3H8 |
| R4 | Z−Cu−O−C3H8 Z−Cu−OH−C3H7 | r4 = k4θO-C3H8 − k-4θC3H7-OH |
| R5 | Z−Cu−OH−C3H7 Z−Cu−H2O−C3H6 | r5 = k5θC3H7-OH − k-5θC3H6-H2O |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, R.; Liu, N.; Dai, C.; Xu, R.; Yu, G.; Wang, N.; Chen, B. Mechanistic Insight into the Propane Oxidation Dehydrogenation by N2O over Cu-BEA Zeolite with Diverse Active Site Structures. Catalysts 2023, 13, 1212. https://doi.org/10.3390/catal13081212
Wu R, Liu N, Dai C, Xu R, Yu G, Wang N, Chen B. Mechanistic Insight into the Propane Oxidation Dehydrogenation by N2O over Cu-BEA Zeolite with Diverse Active Site Structures. Catalysts. 2023; 13(8):1212. https://doi.org/10.3390/catal13081212
Chicago/Turabian StyleWu, Ruiqi, Ning Liu, Chengna Dai, Ruinian Xu, Gangqiang Yu, Ning Wang, and Biaohua Chen. 2023. "Mechanistic Insight into the Propane Oxidation Dehydrogenation by N2O over Cu-BEA Zeolite with Diverse Active Site Structures" Catalysts 13, no. 8: 1212. https://doi.org/10.3390/catal13081212