
Development of an Environment-Friendly and Electrochemical Method for the Synthesis of an Oxadiazole Drug-Scaffold That Targets Poly(ADP-Ribose)Polymerase in Human Breast Cancer Cells
 , , , , , and
, , , , , and 
Abstract
:
1. Introduction
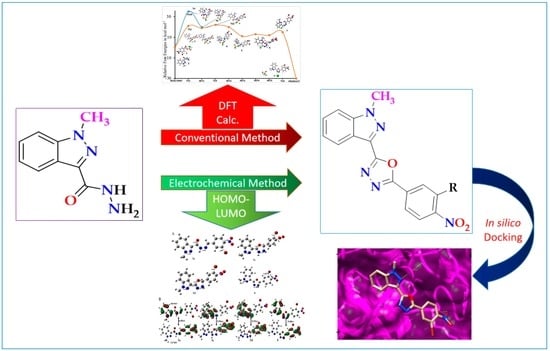
2. Results and Discussion
2.1. Chemical Synthesis of 1,3,4-Oxadiazoles via Conventional Methods
2.2. Synthesis of 1,3,4-Oxadiazoles via the Electrochemical Method
2.3. DFT Calculations
2.4. Efficacy of Oxadiazoles in Breast Cancer Cells
2.5. In Silico Analysis of the Interaction of Compound 8 with the PARP1 Catalytic Domain
3. Materials and Methods
3.1. General Procedure for Synthesizing New Oxadiazoles via the Conventional Method
3.2. General Procedure for Synthesizing New Oxadiazoles via the Electrochemical Method
3.3. The 2-(3-bromo-4-nitrophenyl)-5-(1-methyl-1H-indazol-3-yl)-1,3,4-oxadiazole (8)
3.4. The 2-(1-methyl-1H-indazol-3-yl)-5-(4-nitro-3-(pyridin-3-yl)phenyl)-1,3,4-oxadiazole (9a)
3.5. The 2-(1-methyl-1H-indazol-3-yl)-5-(4-nitro-3-(pyridin-4-yl)phenyl)-1,3,4-oxadiazole (9b)
3.6. The 2-(3-(6-chloro-5-methylpyridin-3-yl)-4-nitrophenyl)-5-(1-methyl-1H-indazol-3-yl)-1,3,4-oxadiazole (9c)
3.7. The 2-(3-(6-fluoro-5-methylpyridin-2-yl)-4-nitrophenyl)-5-(1-methyl-1H-indazol-3-yl)-1,3,4- oxadiazole (9d)
3.8. The 2-(4′-chloro-6-nitro-[1,1′-biphenyl]-3-yl)-5-(1-methyl-1H-indazol-3-yl)-1,3,4-oxadiazole (9e)
3.9. The 2-(3-(benzofuran-6-yl)-4-nitrophenyl)-5-(1-methyl-1H-indazol-3-yl)-1,3,4-oxadiazole (9f)
3.10. The 2-(3-cyclopentyl-4-nitrophenyl)-5-(1-methyl-1H-indazol-3-yl)-1,3,4-oxadiazole (9g)
3.11. The N-cyclopentyl-5′-(5-(1-methyl-1H-indazol-3-yl)-1,3,4-oxadiazol-2-yl)-2′-nitro-[1,1′-biphenyl]-4-carboxamide (9h)
3.12. The 2-(4′-bromo-6-nitro-[1,1′-biphenyl]-3-yl)-5-(1-methyl-1H-indazol-3-yl)-1,3,4-oxadiazole (9i)
3.13. The 5′-(5-(1-methyl-1H-indazol-3-yl)-1,3,4-oxadiazol-2-yl)-2′-nitro-[1,1′-biphenyl]-4-carbaldehyde (9j)
3.14. The 2-(1-methyl-1H-indazol-3-yl)-5-(6-nitro-[1,1′-biphenyl]-3-yl)-1,3,4-oxadiazole (9k)
3.15. Cell Viability Assay
3.16. Bioinformatic Studies for the Compound (8)
3.17. DFT Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Yuan, Y.; Lei, A. Is Electrosynthesis always Green and Advantageous Compared to Traditional Methods? Nat. Commun. 2020, 11, 802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jutand, A. Contribution of Electrochemistry to Organometallic Catalysis. Chem. Rev. 2008, 108, 2300–2347. [Google Scholar] [CrossRef] [PubMed]
- Handler, G. Preface. Commun. Asteroseismol. 2007, 150, 10. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Wang, X.; Tan, H.; Shi, W.; Zhang, X.; Wei, S.; Giesy, J.P.; Yu, H. Molecular Initiating Events of Bisphenols on Androgen Receptor-Mediated Pathways Provide Guidelines for In Silico Screening and Design of Substitute Compounds. Environ. Sci. Technol. Lett. 2019, 6, 205–210. [Google Scholar] [CrossRef]
- Ganesh, K.N.; Zhang, D.; Miller, S.J.; Rossen, K.; Chirik, P.J.; Kozlowski, M.C.; Zimmerman, J.B.; Brooks, B.W.; Savage, P.E.; Allen, D.T.; et al. Green Chemistry: A Framework for a Sustainable Future. Org. Process Res. Dev. 2021, 25, 1455–1459. [Google Scholar] [CrossRef]
- Anastas, P.; Eghbali, N. Green Chemistry: Principles and Practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef]
- Jiang, Y.; Xu, K.; Zeng, C. Use of Electrochemistry in the Synthesis of Heterocyclic Structures. Chem. Rev. 2017, 118, 4485–4540. [Google Scholar] [CrossRef]
- Ibrar, A.; Khan, A.; Ali, M.; Sarwar, R.; Mehsud, S.; Farooq, U.; Halimi, S.M.A.; Khan, I.; Al-Harrasi, A. Combined In Vitro and In Silico Studies for the Anticholinesterase Activity and Pharmacokinetics of Coumarinyl Thiazoles and Oxadiazoles. Front. Chem. 2018, 6, 61. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Qi, L.; Liu, H.; Lei, K.; Wang, X.; Liu, R. Synthesis of 1,3,4-Oxadiazoles Derivatives with Antidepressant Activity and Their Binding to the 5-HT1A Receptor. RSC Adv. 2020, 10, 30848–30857. [Google Scholar] [CrossRef]
- Vaidya, A.; Pathak, D.; Shah, K. 1,3,4-oxadiazole and Its Derivatives: A Review on Recent Progress in Anticancer Activities. Chem. Biol. Drug Des. 2020, 97, 572–591. [Google Scholar] [CrossRef]
- De Oliveira, C.S.; Lira, B.F.; Barbosa-Filho, J.M.; Lorenzo, J.G.F.; de Athayde-Filho, P.F. Synthetic Approaches and Pharmacological Activity of 1,3,4-Oxadiazoles: A Review of the Literature from 2000–2012. Molecules 2012, 17, 10192–10231. [Google Scholar] [CrossRef] [Green Version]
- Biernacki, K.; Daśko, M.; Ciupak, O.; Kubiński, K.; Rachon, J.; Demkowicz, S. Novel 1,2,4-Oxadiazole Derivatives in Drug Discovery. Pharmaceuticals 2020, 13, 111. [Google Scholar] [CrossRef]
- Rana, K.; Salahuddin; Sahu, J.K. Significance of 1,3,4-Oxadiazole Containing Compounds in New Drug Development. Curr. Drug Res. Rev. 2021, 13, 90–100. [Google Scholar] [CrossRef]
- Taher, A.T.; Georgey, H.H.; El-Subbagh, H.I. Novel 1,3,4-Heterodiazole Analogues: Synthesis and In-Vitro Antitumor Activity. Eur. J. Med. Chem. 2012, 47, 445–451. [Google Scholar] [CrossRef]
- Gu, W.; Jin, X.-Y.; Li, D.-D.; Wang, S.-F.; Tao, X.-B.; Chen, H. Design, Synthesis and In Vitro Anticancer Activity of Novel Quinoline and Oxadiazole Derivatives of Ursolic Acid. Bioorganic Med. Chem. Lett. 2017, 27, 4128–4132. [Google Scholar] [CrossRef]
- Castanet, A.-S.; Nafie, M.S.; Said, S.A.; Arafa, R.K. Discovery of PIM-1 Kinase Inhibitors Based on the 2,5-Disubstituted 1,3,4-Oxadiazole Scaffold against Prostate Cancer: Design, Synthesis, In Vitro and In Vivo Cytotoxicity Investigation. Eur. J. Med. Chem. 2023, 250, 115220. [Google Scholar] [CrossRef]
- Nieddu, V.; Pinna, G.; Marchesi, I.; Sanna, L.; Asproni, B.; Pinna, G.A.; Bagella, L.; Murineddu, G. Synthesis and Antineoplastic Evaluation of Novel Unsymmetrical 1,3,4-Oxadiazoles. J. Med. Chem. 2016, 59, 10451–10469. [Google Scholar] [CrossRef]
- Basappa; Sugahara, K.; Thimmaiah, K.N.; Bid, H.K.; Houghton, P.J.; Rangappa, K.S. Anti-tumor activity of a novel HS-mimetic-vascular endothelial growth factor binding small molecule. PLoS One 2012, 7, e39444. [Google Scholar] [CrossRef] [Green Version]
- Zoroddu, S.; Corona, P.; Sanna, L.; Borghi, F.; Bordoni, V.; Asproni, B.; Pinna, G.A.; Bagella, L.; Murineddu, G. Novel 1,3,4-Oxadiazole Chalcogen Analogues: Synthesis and Cytotoxic Activity. Eur. J. Med. Chem. 2022, 238, 114440. [Google Scholar] [CrossRef]
- Glomb, T.; Świątek, P. Antimicrobial Activity of 1,3,4-Oxadiazole Derivatives. Int. J. Mol. Sci. 2021, 22, 6979. [Google Scholar] [CrossRef]
- Yu, G.; Chen, S.; Guo, S.; Xu, B.; Wu, J. Trifluoromethylpyridine 1,3,4-Oxadiazole Derivatives: Emerging Scaffolds as Bacterial Agents. ACS Omega 2021, 6, 31093–31098. [Google Scholar] [CrossRef] [PubMed]
- Jian, W.; He, D.; Song, S. Synthesis, Biological Evaluation and Molecular Modeling Studies of New Oxadiazole-Stilbene Hybrids against Phytopathogenic Fungi. Sci. Rep. 2016, 6, 31045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szydłowski, M.; Prochorec-Sobieszek, M.; Szumera-Ciećkiewicz, A.; Derezińska, E.; Hoser, G.; Wasilewska, D.; Szymańska-Giemza, O.; Jabłońska, E.; Białopiotrowicz, E.; Sewastianik, T.; et al. Expression of PIM Kinases in Reed-Sternberg Cells Fosters Immune Privilege and Tumor Cell Survival in Hodgkin Lymphoma. Blood 2017, 130, 1418–1429. [Google Scholar] [CrossRef] [Green Version]
- Szydłowski, M.; Dębek, S.; Prochorec-Sobieszek, M.; Szołkowska, M.; Tomirotti, A.M.; Juszczyński, P.; Szumera-Ciećkiewicz, A. PIM Kinases Promote Survival and Immune Escape in Primary Mediastinal Large B-Cell Lymphoma through Modulation of JAK-STAT and NF-κB Activity. Am. J. Pathol. 2021, 191, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Mohan, C.D.; Anilkumar, N.C.; Rangappa, S.; Shanmugam, M.K.; Mishra, S.; Chinnathambi, A.; Alharbi, S.A.; Bhattacharjee, A.; Sethi, G.; Kumar, A.P.; et al. Novel 1,3,4-Oxadiazole Induces Anticancer Activity by Targeting NF-κB in Hepatocellular Carcinoma Cells. Front. Oncol. 2018, 8, 42. [Google Scholar] [CrossRef] [Green Version]
- Demény, M.A.; Virág, L. The PARP Enzyme Family and the Hallmarks of Cancer Part 1. Cell Intrinsic Hallmarks. Cancers 2021, 13, 2042. [Google Scholar] [CrossRef]
- Livraghi, L.; Garber, J.E. PARP Inhibitors in the Management of Breast Cancer: Current Data and Future Prospects. BMC Med. 2015, 13, 188. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Liu, C.; Chang, C.; Shen, C.; Yin, Y.; Yin, X.; Jiang, Z.; Zhao, Z.; Mu, M.; Cao, D.; et al. Comparative Safety and Tolerability of Approved PARP Inhibitors in Cancer: A Systematic Review and Network Meta-Analysis. Pharmacol. Res. 2021, 172, 105808. [Google Scholar] [CrossRef]
- Sethi, G.S.; Dharwal, V.; Naura, A.S. Poly(ADP-Ribose)Polymerase-1 in Lung Inflammatory Disorders: A Review. Front. Immunol. 2017, 8, 1172. [Google Scholar] [CrossRef] [Green Version]
- Javle, M.; Curtin, N.J. The Role of PARP in DNA Repair and Its Therapeutic Exploitation. Br. J. Cancer 2011, 105, 1114–1122. [Google Scholar] [CrossRef] [Green Version]
- Morales, J.; Li, L.; Fattah, F.J.; Dong, Y.; Bey, E.A.; Patel, M.; Gao, J.; Boothman, D.A. Review of Poly (ADP-Ribose) Polymerase (PARP) Mechanisms of Action and Rationale for Targeting in Cancer and Other Diseases. Crit. Rev. Eukaryot. Gene Expr. 2014, 24, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Sousa, F.G.; Matuo, R.; Soares, D.G.; Escargueil, A.E.; Henriques, J.A.P.; Larsen, A.K.; Saffi, J. PARPs and the DNA Damage Response. Carcinogenesis 2012, 33, 1433–1440. [Google Scholar] [CrossRef] [Green Version]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef]
- Zheng, F.; Zhang, Y.; Chen, S.; Weng, X.; Rao, Y.; Fang, H. Mechanism and Current Progress of Poly ADP-Ribose Polymerase (PARP) Inhibitors in the Treatment of Ovarian Cancer. Biomed. Pharmacother. 2020, 123, 109661. [Google Scholar] [CrossRef]
- De Gaetano, G.; Tonolli, M.C.; Bertoni, M.P.; Roncaglioni, M.C. Ditazole and Platelets. Pathophysiol. Haemost. Thromb. 1977, 6, 127–136. [Google Scholar] [CrossRef]
- Schelman, W.R.; Liu, G.; Wilding, G.; Morris, T.; Phung, D.; Dreicer, R. A Phase I Study of Zibotentan (ZD4054) in Patients with Metastatic, Castrate-Resistant Prostate Cancer. Investig. New Drugs 2009, 29, 118–125. [Google Scholar] [CrossRef]
- Luczynski, M.; Kudelko, A. Synthesis and Biological Activity of 1,3,4-Oxadiazoles Used in Medicine and Agriculture. Appl. Sci. 2022, 12, 3756. [Google Scholar] [CrossRef]
- Glomb, T.; Szymankiewicz, K.; Świątek, P. Anti-Cancer Activity of Derivatives of 1,3,4-Oxadiazole. Molecules 2018, 23, 3361. [Google Scholar] [CrossRef] [Green Version]
- Nayak, S.; Gaonkar, S.L.; Musad, E.A.; Dawsar, A.M.A. 1,3,4-Oxadiazole-Containing Hybrids as Potential Anticancer Agents: Recent Developments, Mechanism of Action and Structure-Activity Relationships. J. Saudi Chem. Soc. 2021, 25, 101284. [Google Scholar] [CrossRef]
- Benassi, A.; Doria, F.; Pirota, V. Groundbreaking Anticancer Activity of Highly Diversified Oxadiazole Scaffolds. Int. J. Mol. Sci. 2020, 21, 8692. [Google Scholar] [CrossRef]
- Ren, H.; Bakas, N.A.; Vamos, M.; Chaikuad, A.; Limpert, A.S.; Wimer, C.D.; Brun, S.N.; Lambert, L.J.; Tautz, L.; Celeridad, M.; et al. Design, Synthesis, and Characterization of an Orally Active Dual-Specific ULK1/2 Autophagy Inhibitor That Synergizes with the PARP Inhibitor Olaparib for the Treatment of Triple-Negative Breast Cancer. J. Med. Chem. 2020, 63, 14609–14625. [Google Scholar] [CrossRef] [PubMed]
- Voronkov, A.; Holsworth, D.D.; Waaler, J.; Wilson, S.R.; Ekblad, B.; Perdreau-Dahl, H.; Dinh, H.; Drewes, G.; Hopf, C.; Morth, J.P.; et al. Structural Basis and SAR for G007-LK, a Lead Stage 1,2,4-Triazole Based Specific Tankyrase 1/2 Inhibitor. J. Med. Chem. 2013, 56, 3012–3023. [Google Scholar] [CrossRef] [PubMed]
- Puthiyapurayil, P.; Poojary, B.; Chikkanna, C.; Buridipad, S.K. Design, Synthesis and Biological Evaluation of a Novel Series of 1,3,4-Oxadiazole Bearing N-Methyl-4-(Trifluoromethyl)Phenyl Pyrazole Moiety as Cytotoxic Agents. Eur. J. Med. Chem. 2012, 53, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Strzelecka, M.; Glomb, T.; Drąg-Zalesińska, M.; Kulbacka, J.; Szewczyk, A.; Saczko, J.; Kasperkiewicz-Wasilewska, P.; Rembiałkowska, N.; Wojtkowiak, K.; Jezierska, A.; et al. Synthesis, Anticancer Activity and Molecular Docking Studies of Novel N-Mannich Bases of 1,3,4-Oxadiazole Based on 4,6-Dimethylpyridine Scaffold. Int. J. Mol. Sci. 2022, 23, 11173. [Google Scholar] [CrossRef]
- Sun, J.; Ren, S.-Z.; Lu, X.-Y.; Li, J.-J.; Shen, F.-Q.; Xu, C.; Zhu, H.-L. Discovery of a Series of 1,3,4-Oxadiazole-2(3H)-Thione Derivatives Containing Piperazine Skeleton as Potential FAK Inhibitors. Bioorganic Med. Chem. 2017, 25, 2593–2600. [Google Scholar] [CrossRef]
- Zhang, S.; Luo, Y.; He, L.-Q.; Liu, Z.-J.; Jiang, A.-Q.; Yang, Y.-H.; Zhu, H.-L. Synthesis, Biological Evaluation, and Molecular Docking Studies of Novel 1,3,4-Oxadiazole Derivatives Possessing Benzotriazole Moiety as FAK Inhibitors with Anticancer Activity. Bioorganic Med. Chem. 2013, 21, 3723–3729. [Google Scholar] [CrossRef]
- Hamdy, R.; Ziedan, N.I.; Ali, S.; Bordoni, C.; El-Sadek, M.; Lashin, E.; Brancale, A.; Jones, A.T.; Westwell, A.D. Synthesis and Evaluation of 5-(1 H-Indol-3-Yl)-N-Aryl-1,3,4-Oxadiazol-2-Amines as Bcl-2 Inhibitory Anticancer Agents. Bioorganic Med. Chem. Lett. 2017, 27, 1037–1040. [Google Scholar] [CrossRef]
- Sun, J.; Zhu, H.; Yang, Z.-M.; Zhu, H.-L. Synthesis, Molecular Modeling and Biological Evaluation of 2-Aminomethyl-5-(Quinolin-2-Yl)-1,3,4-Oxadiazole-2(3H)-Thione Quinolone Derivatives as Novel Anticancer Agent. Eur. J. Med. Chem. 2013, 60, 23–28. [Google Scholar] [CrossRef]
- Basappa, B.; Jung, Y.Y.; Ravish, A.; Xi, Z.; Swamynayaka, A.; Madegowda, M.; Pandey, V.; Lobie, P.E.; Sethi, G.; Ahn, K.S. Methyl-Thiol-Bridged Oxadiazole and Triazole Heterocycles as Inhibitors of NF-κB in Chronic Myelogenous Leukemia Cells. Biomedicines 2023, 11, 1662. [Google Scholar] [CrossRef]
- Sebastian, A.; Pandey, V.; Mohan, C.D.; Chia, Y.T.; Rangappa, S.; Mathai, J.; Baburajeev, C.P.; Paricharak, S.; Mervin, L.H.; Bulusu, K.C.; et al. Novel Adamantanyl-Based Thiadiazolyl Pyrazoles Targeting EGFR in Triple-Negative Breast Cancer. ACS Omega 2016, 1, 1412–1424. [Google Scholar] [CrossRef] [Green Version]
- Dukanya; Shanmugam, M.K.; Rangappa, S.; Metri, P.K.; Mohan, S.; Basappa; Rangappa, K.S. Exploring the Newer Oxadiazoles as Real Inhibitors of Human SIRT2 in Hepatocellular Cancer Cells. Bioorganic Med. Chem. Lett. 2020, 30, 127330. [Google Scholar] [CrossRef]
- Malojirao, V.H.; Girimanchanaika, S.S.; Shanmugam, M.K.; Sherapura, A.; Dukanya; Metri, P.K.; Vigneshwaran, V.; Chinnathambi, A.; Alharbi, S.A.; Rangappa, S.; et al. Novel 1,3,4-Oxadiazole Targets STAT3 Signaling to Induce Antitumor Effect in Lung Cancer. Biomedicines 2020, 8, 368. [Google Scholar] [CrossRef]
- Vishwanath, D.; Girimanchanaika, S.S.; Dukanya, D.; Rangappa, S.; Yang, J.-R.; Pandey, V.; Lobie, P.E.; Basappa, B. Design and Activity of Novel Oxadiazole Based Compounds That Target Poly(ADP-Ribose) Polymerase. Molecules 2022, 27, 703. [Google Scholar] [CrossRef]
- Ma, H.-Y.; Zha, Z.-G.; Zhang, Z.-L.; Meng, L.; Wang, Z.-Y. Electrosynthesis of Oxadiazoles from Benzoylhydrazines. Chin. Chem. Lett. 2013, 24, 780–782. [Google Scholar] [CrossRef]
- Haines, B.E.; Steussy, C.N.; Stauffacher, C.V.; Wiest, O. Molecular Modeling of the Reaction Pathway and Hydride Transfer Reactions of HMG-CoA Reductase. Biochemistry 2012, 51, 7983–7995. [Google Scholar] [CrossRef] [Green Version]
- Lu, F.; Gong, F.; Li, L.; Zhang, K.; Li, Z.; Zhang, X.; Yin, Y.; Wang, Y.; Gao, Z.; Zhang, H.; et al. Electrochemical Synthesis of 2,5-Disubstituted 1,3,4-Oxadiazoles from α-Keto Acids and Acylhydrazines under Mild Conditions. Eur. J. Org. Chem. 2020, 2020, 3257–3260. [Google Scholar] [CrossRef]
- Dolman, S.J.; Gosselin, F.; O’Shea, P.D.; Davies, I.W. Superior Reactivity of Thiosemicarbazides in the Synthesis of 2-Amino-1,3,4-Oxadiazoles. J. Org. Chem. 2006, 71, 9548–9551. [Google Scholar] [CrossRef]
- Baburajeev, C.P.; Dhananjaya Mohan, C.; Ananda, H.; Rangappa, S.; Fuchs, J.E.; Jagadish, S.; Sivaraman Siveen, K.; Chinnathambi, A.; Ali Alharbi, S.; Zayed, M.E.; et al. Development of Novel Triazolo-Thiadiazoles from Heterogeneous "Green" Catalysis as Protein Tyrosine Phosphatase 1B Inhibitors. Sci. Rep. 2015, 5, 14195. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Sharma, L.K.; Saraswat, A.; Siddiqui, I.R.; Kehri, H.K.; Singh, R.K.P. Electrosynthesis and Screening of Novel 1,3,4-Oxadiazoles as Potent and Selective Antifungal Agents. RSC Adv. 2013, 3, 4237. [Google Scholar] [CrossRef]
- Basappa, B.; Chumadathil Pookunoth, B.; Shinduvalli Kempasiddegowda, M.; Knchugarakoppal Subbegowda, R.; Lobie, P.E.; Pandey, V. Novel Biphenyl Amines Inhibit Oestrogen Receptor (ER)-α in ER-Positive Mammary Carcinoma Cells. Molecules 2021, 26, 783. [Google Scholar] [CrossRef]
- Huey, R.; Morris, G.M.; Olson, A.J.; Goodsell, D.S. A Semiempirical Free Energy Force Field with Charge–Based Desolvation. J. Comput. Chem. 2007, 28, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, N.B.; Mohan, C.D.; Basappa, S.; Pandey, V.; Rangappa, S.; Bharathkumar, H.; Kumar, A.P.; Lobie, P.E.; Rangappa, K.S. An azaspirane derivative suppresses growth and induces apoptosis of ER-positive and ER-negative breast cancer cells through the modulation of JAK2/STAT3 signaling pathway. Int. J. Oncol. 2016, 49, 1221–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bharathkumar, H.; Mohan, C.D.; Ananda, H.; Fuchs, J.E.; Li, F.; Rangappa, S.; Surender, M.; Bulusu, K.C.; Girish, K.S.; Sethi, G.; et al. Microwave-assisted synthesis, characterization and cytotoxic studies of novel estrogen receptor α ligands towards human breast cancer cells. Bioorg. Med. Chem. Lett. 2015, 25, 1804–1807. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, V.; Chevalier, F.; Imberty, A.; Leeflang, B.R.; Basappa; Sugahara, K.; Kamerling, J.P. Conformational studies on five octasaccharides isolated from chondroitin sulfate using NMR spectroscopy and molecular modeling. Biochemistry 2007, 46, 1167–1175. [Google Scholar] [CrossRef]
- Basappa; Rangappa, K.S.; Sugahara, K. Roles of glycosaminoglycans and glycanmimetics in tumor progression and metastasis. Glycoconj. J. 2014, 31, 461–467. [Google Scholar] [CrossRef]
- Kumar, C.A.; Jayarama, S.; Basappa; Salimath, B.P.; Rangappa, K.S. Pro-apoptotic activity of imidazole derivatives mediated by up-regulation of Bax and activation of CAD in Ehrlich Ascites Tumor cells. Investig. New Drugs 2007, 25, 343–350. [Google Scholar] [CrossRef]
- BIOVIA Dassault Systèmes. Discovery Studio Visualizer; 21.1.0.20298; Dassault Systèmes: San Diego, CA, USA, 2020. [Google Scholar]
- Schrödinger, L.L.C.; DeLano, W. PyMOL. 2020. Available online: http://www.pymol.org/pymol (accessed on 15 February 2023).
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Boronic Acids (R) | Structure |
|---|---|
| Pyridin-3-Ylboronic Acid |  |
| Pyridin-4-Ylboronic Acid | |
| (6-Chloro-5-Methylpyridin-3-Yl)Boronic Acid | |
| (2-Fluoro-3-Methylpyridin-4-Yl)Boronic Acid | |
| (4-Chlorophenyl)Boronic Acid | |
| Benzofuran-2-Ylboronic Acid | |
| Cyclopentylboronic Acid | |
| (3-(Cyclopentylcarbamoyl)Phenyl)Boronic Acid | |
| (4-Bromophenyl)Boronic Acid | |
| (4-Formylphenyl)Boronic Acid | |
| Phenylboronic Acid |
| Compounds | IC50 (µM) | Compounds | IC50 (µM) |
|---|---|---|---|
 8 | 1.57 |  9f | 84.70 |
 9a | 36.36 |  9g | >100 |
 9b | >100 |  9h | 51.23 |
 9c | 62.07 |  9i | 76.84 |
 9d | >100 |  9j | 46.85 |
 9e | 64.89 |  9k | 44.40 |
| Entry | Binding Energy (kcal/mol) | Entry | Binding Energy (kcal/mol) |
|---|---|---|---|
| 8 | −7.29 | 9f | −8.67 |
| 9a | −7.43 | 9g | −7.75 |
| 9b | −9.28 | 9h | −8.44 |
| 9c | −7.62 | 9i | −8.11 |
| 9d | −8 | 9j | −7.94 |
| 9e | −7.86 | 9k | −7.52 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parameshwaraiah, S.M.; Xi, Z.; Ravish, A.; Mohan, A.; Shankarnaik, V.; Dukanya, D.; Basappa, S.; Preetham, H.D.; Periyasamy, G.; Gaonkar, S.L.; et al. Development of an Environment-Friendly and Electrochemical Method for the Synthesis of an Oxadiazole Drug-Scaffold That Targets Poly(ADP-Ribose)Polymerase in Human Breast Cancer Cells. Catalysts 2023, 13, 1185. https://doi.org/10.3390/catal13081185
Parameshwaraiah SM, Xi Z, Ravish A, Mohan A, Shankarnaik V, Dukanya D, Basappa S, Preetham HD, Periyasamy G, Gaonkar SL, et al. Development of an Environment-Friendly and Electrochemical Method for the Synthesis of an Oxadiazole Drug-Scaffold That Targets Poly(ADP-Ribose)Polymerase in Human Breast Cancer Cells. Catalysts. 2023; 13(8):1185. https://doi.org/10.3390/catal13081185
Chicago/Turabian StyleParameshwaraiah, Sindhu M., Zhang Xi, Akshay Ravish, Arunkumar Mohan, Vanishree Shankarnaik, Dukanya Dukanya, Shreeja Basappa, Habbanakuppe D. Preetham, Ganga Periyasamy, Santhosh L. Gaonkar, and et al. 2023. "Development of an Environment-Friendly and Electrochemical Method for the Synthesis of an Oxadiazole Drug-Scaffold That Targets Poly(ADP-Ribose)Polymerase in Human Breast Cancer Cells" Catalysts 13, no. 8: 1185. https://doi.org/10.3390/catal13081185