
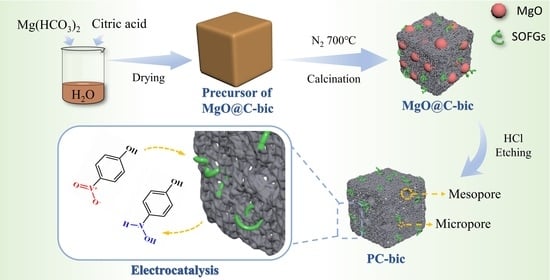
Templating Synthesis of Hierarchically Porous Carbon with Magnesium Salts for Electrocatalytic Reduction of 4-Nitrophenol

Abstract
:
1. Introduction
2. Results and Discussion
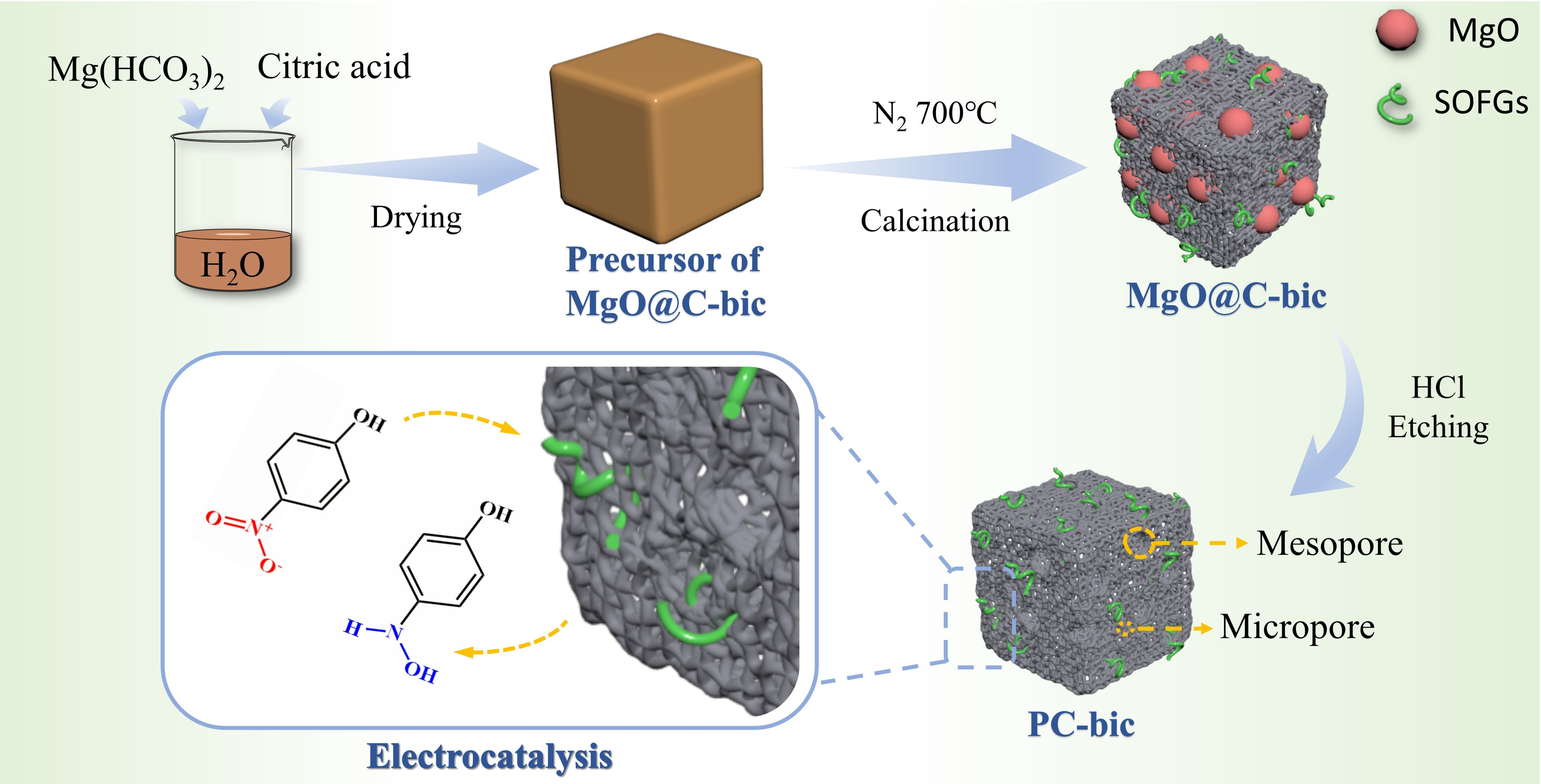
2.1. XRD Patterns
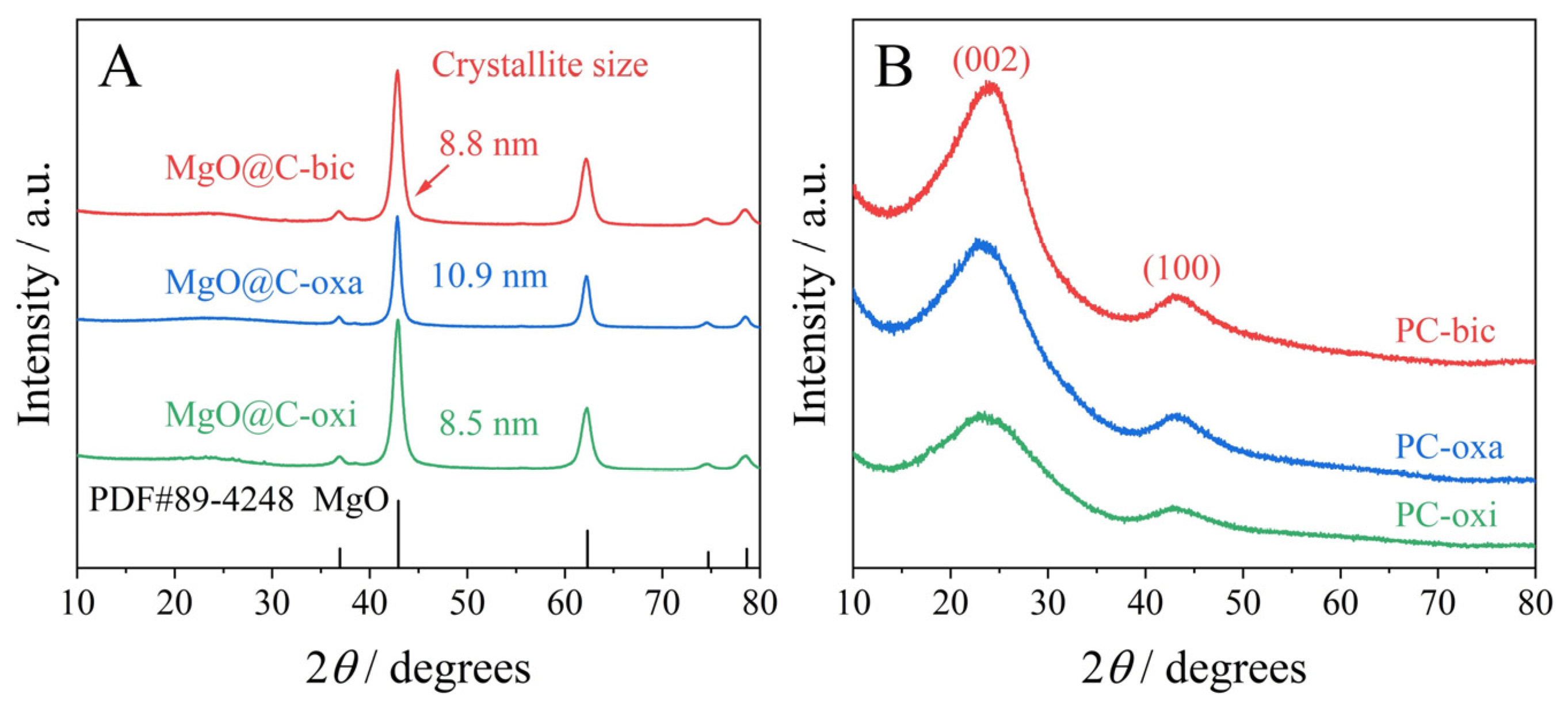
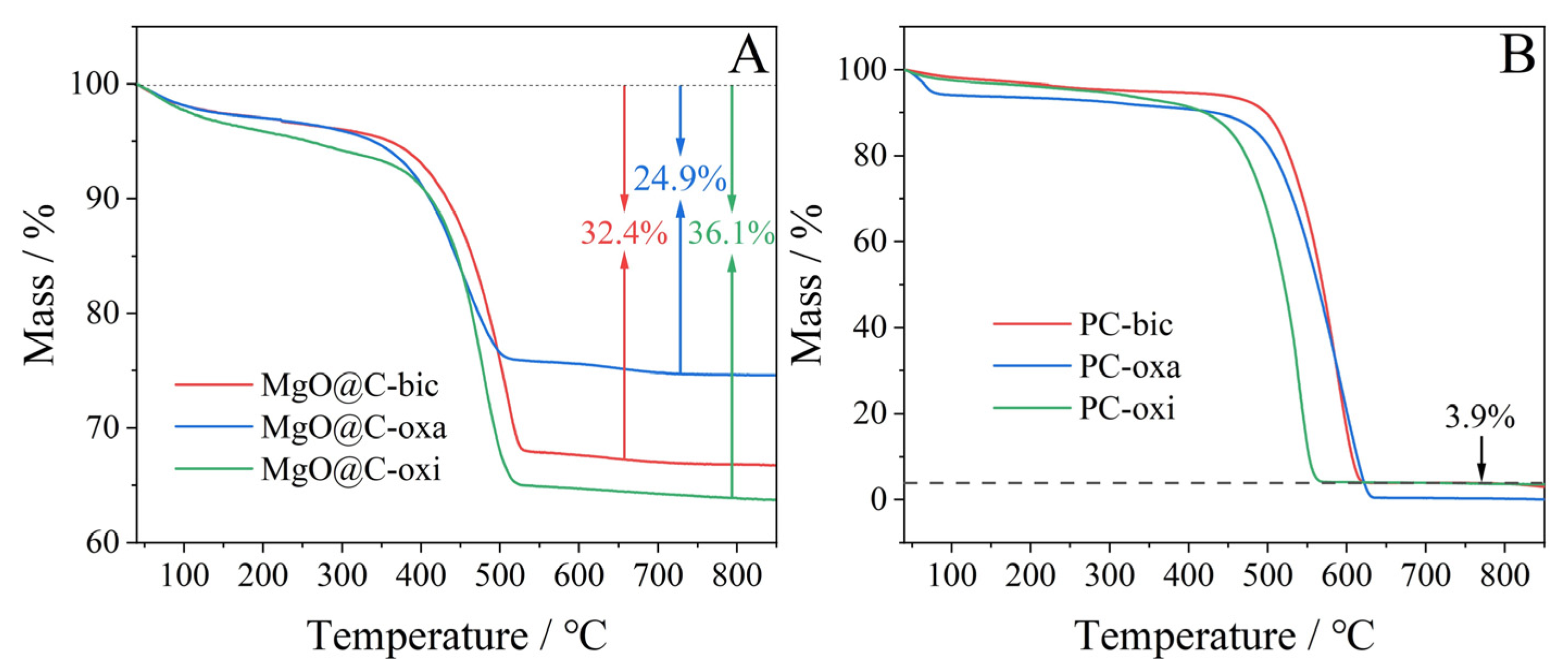
2.2. TGA-DSC Profiles
2.3. TEM Images
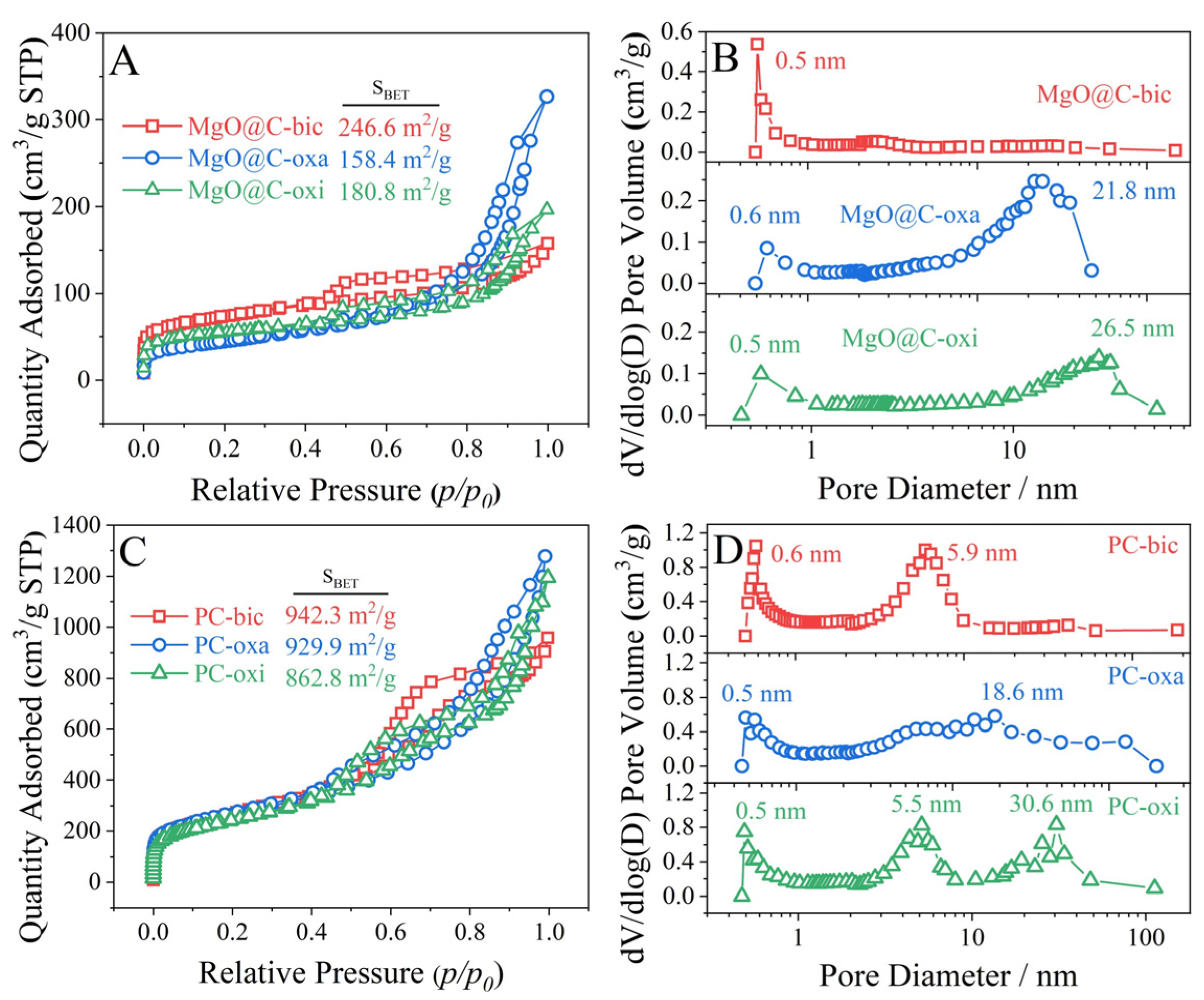
2.4. N2 Physisorption Isotherms
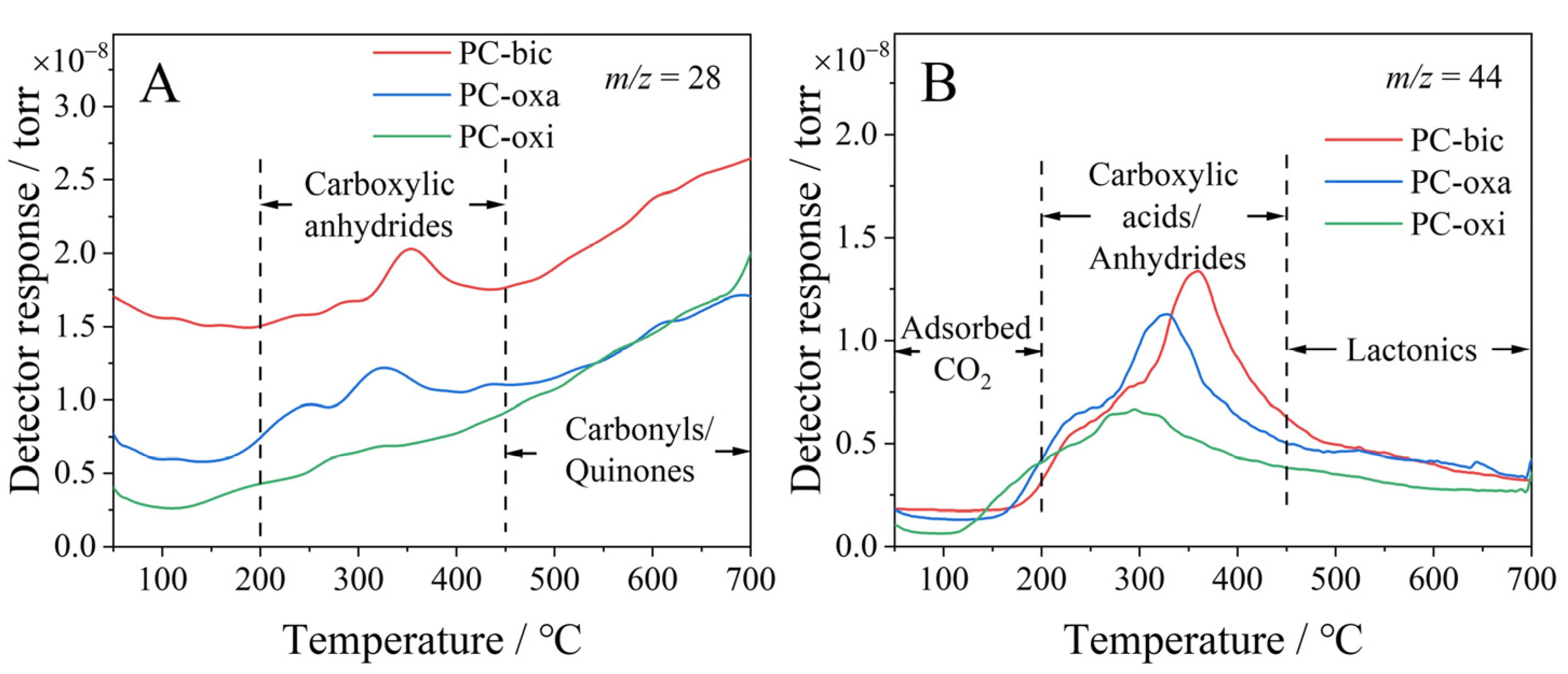
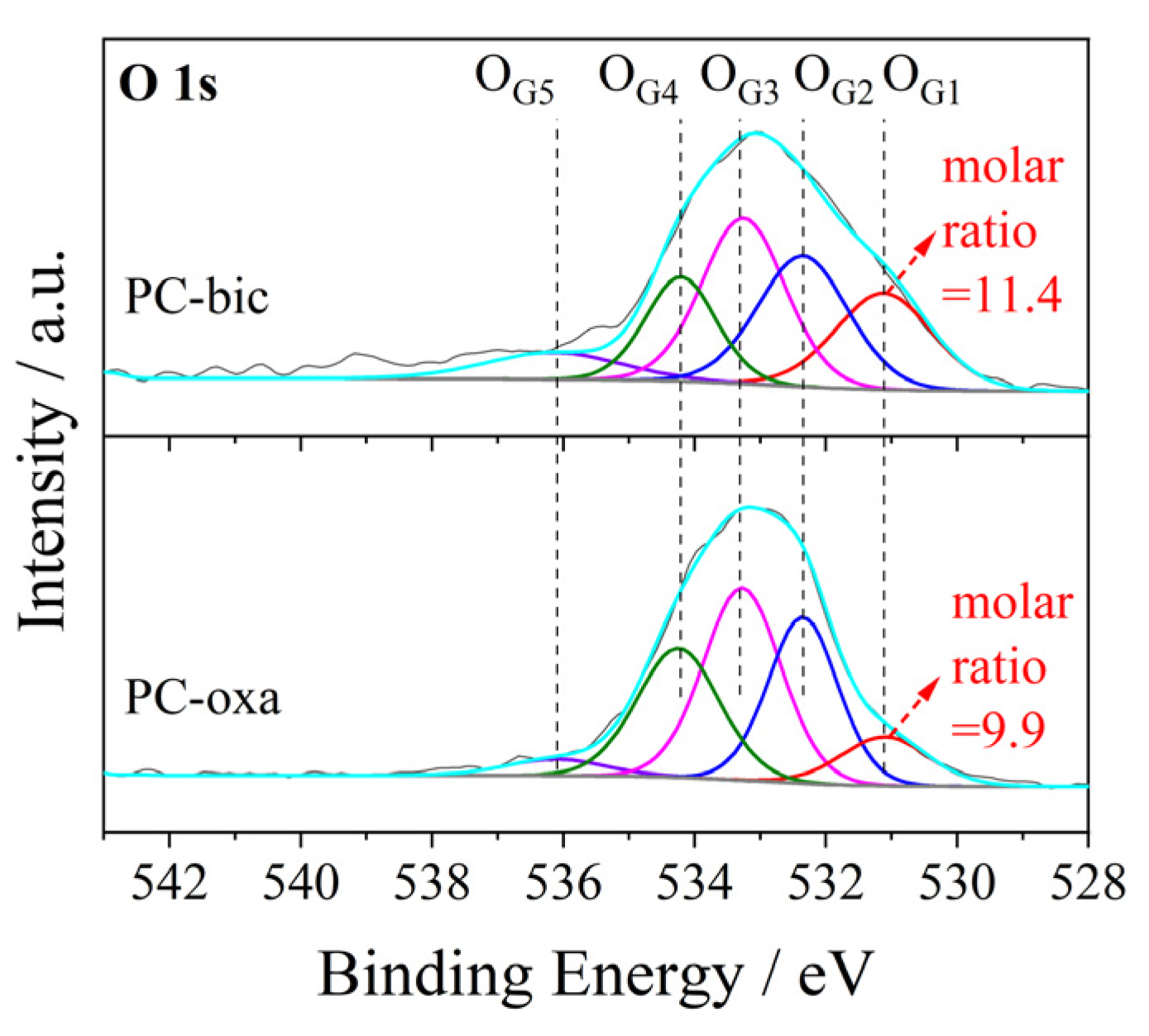
2.5. TPD and XPS
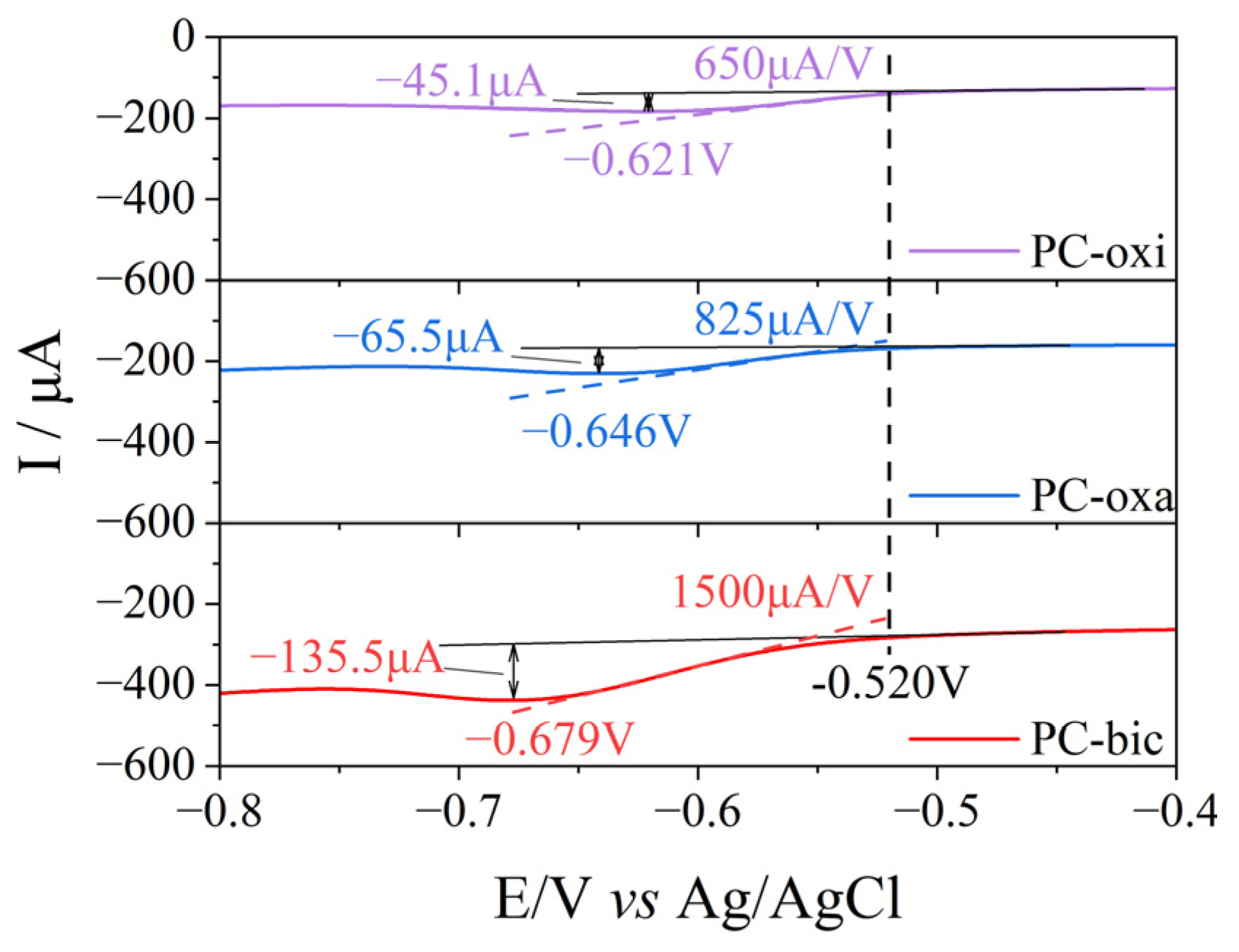
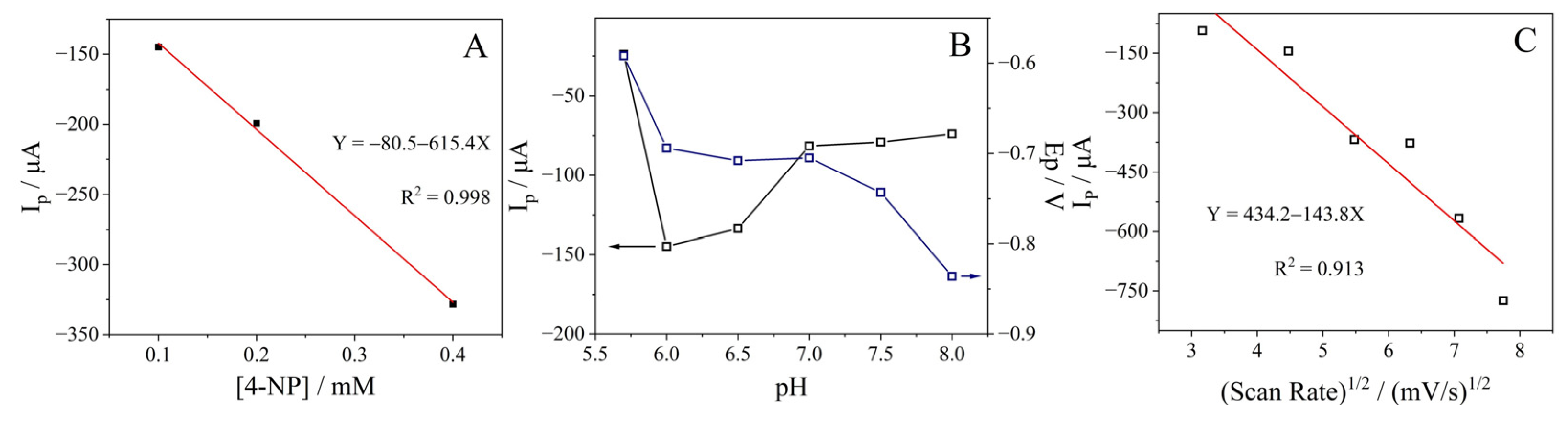
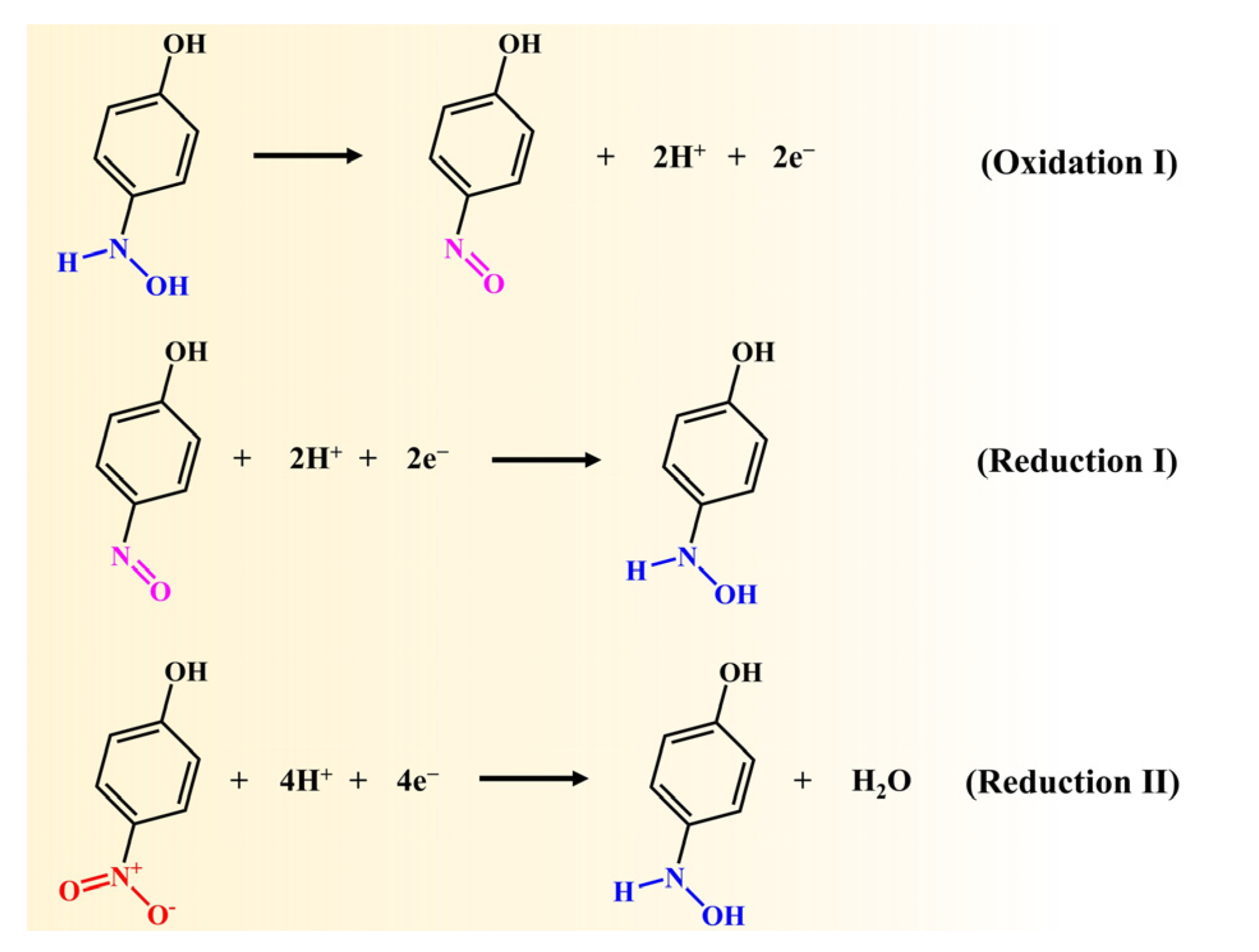
2.6. Electrocatalytic Reduction of 4-Nitrophenol
3. Experimental Section
3.1. Catalyst Preparation
3.2. Characterizations
3.3. Electrochemical Reductions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Fang, X.; Li, W.; Chen, X.; Wu, Z.; Zhang, Z.; Zou, Y. Controlling the microstructure of biomass-derived porous carbon to assemble structural absorber for broadening bandwidth. Carbon 2022, 198, 70–79. [Google Scholar] [CrossRef]
- Yong, X.; Yin, Q.; Sha, P.; Zhang, Q.; Pang, B.; Yu, J.; Dong, H.; Yu, L.; Dong, L. Walnut shell-derived porous carbon with MgSO4 modification for high-performance capacitive deionization. Diam. Relat. Mater. 2023, 136, 110063. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, R.; Chen, W.; Song, K.; Tian, Y.; Li, J.; Shi, G. Microstructure and electrochemical properties of porous carbon derived from biomass. Int. J. Electrochem. Sci. 2023, 18, 100190. [Google Scholar] [CrossRef]
- Peng, L.; Peng, H.; Li, W.; Zhao, D. Monomicellar assembly to synthesize structured and functional mesoporous carbonaceous nanomaterials. Nat. Protoc. 2023, 18, 1155–1178. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Yang, W.; Xu, C.; Che, S.; Li, Y.; Liu, H.; Chen, N.; Zhang, G.; Liu, H.; Wu, N.; et al. Nitrogen-Doped porous carbon embedded Sn/SnO nanoparticles as high-performance lithium-ion battery anode. Electrochim. Acta 2022, 428, 140898. [Google Scholar]
- Cheng, X.; Tang, C.; Yan, C.; Du, J.; Chen, A.; Liu, X.; Jewell, L.; Zhang, Q. Preparation of porous carbon spheres and their application as anode materials for lithium-ion batteries: A review. Mater. Today Nano 2023, 22, 100321. [Google Scholar] [CrossRef]
- Ou, J.; Deng, H.; Li, B.; Li, K.; Li, M. High content of nitrogen doped porous carbon prepared by one-step calcination for enviable rate lithium ion batteries. Diam. Relat. Mater. 2023, 133, 109696. [Google Scholar] [CrossRef]
- Koketsu, T.; Ma, J.; Morgan, B.J.; Body, M.; Legein, C.; Goddard, P.; Borkiewicz, O.J.; Strasser, P.; Dambournet, D. Exploiting cationic vacancies for increased energy densities in dual-ion batteries. Energy Storage Mater. 2020, 25, 154–163. [Google Scholar] [CrossRef]
- Deng, X.; Zhao, B.; Sha, Y.; Zhu, Y.; Xu, X.; Shao, Z. Three Strongly Coupled Allotropes in a Functionalized Porous All-Carbon Nanocomposite as a Superior Anode for Lithium-Ion Batteries. Chemelectrochem 2016, 3, 698–703. [Google Scholar] [CrossRef]
- Mo, F.; Wu, X. MgO template-assisted synthesis of hierarchical porous carbon with high content heteroatoms for supercapacitor. J. Energy Storage 2022, 54, 105287. [Google Scholar] [CrossRef]
- Wang, X.; Huang, Z.; Jin, H.; Wang, S.; Zhang, Q.; Xiao, H.; Hu, X.; Li, J.; Sheng, S. Cistanches herba residues derived nitrogen and oxygen self-doped porous carbon for high performance supercapacitor. J. Alloys Compd. 2023, 956, 170264. [Google Scholar] [CrossRef]
- Zhang, Y.; Yue, P.; Zhang, C.; Wang, Y.; Wu, X. Facile synthesis of three-dimensional interconnected porous carbon for high performance supercapacitor. Diam. Relat. Mater. 2023, 136, 109941. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, X.; Shao, Z.; Jiang, S.P. Metal-organic frameworks derived porous carbon, metal oxides and metal sulfides-based compounds for supercapacitors application. Energy Storage Mater. 2020, 26, 1–22. [Google Scholar] [CrossRef]
- Yu, H.; Xu, Y.; Havener, K.; Zhang, L.; Wu, W.; Liao, X.; Huang, K. Efficient catalysis using honeycomb-like N-doped porous carbon supported Pt nanoparticles for the hydrogenation of cinnamaldehyde in water. Mol. Catal. 2022, 525, 112343. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, X.; Yao, S.; Hao, C.; Pan, C.; Xiang, X.; Tian, Z.Q.; Shen, P.K.; Shao, Z.; Jiang, S.P. Boosting Electrocatalytic Activity of Single Atom Catalysts Supported on Nitrogen-Doped Carbon through N Coordination Environment Engineering. Small 2022, 18, 2105329. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Huang, X.; Chen, Z.; Zhu, J.; Endrődi, B.; Janáky, C.; Deng, D. Membrane Electrode Assembly for Electrocatalytic CO2 Reduction: Principle and Application. Angew. Chem. Int. Ed. 2023, 62, e202302789. [Google Scholar] [CrossRef]
- Amer, M.S.; Arunachalam, P.; Ghanem, M.A.; Al-Mayouf, A.M.; Shar, M.A. Enriched active surface structure in nanosized tungsten-cobalt oxides electrocatalysts for efficient oxygen redox reactions. Appl. Surf. Sci. 2020, 513, 145831. [Google Scholar] [CrossRef]
- Chen, X.; Xie, Y.; Shao, Y.; Shen, K.; Li, Y. Facile Synthesis of Boron and Nitrogen Dual-Doped Hollow Mesoporous Carbons for Efficient Reduction of 4-Nitrophenol. ACS Appl. Mater. Interfaces 2021, 13, 42598–42604. [Google Scholar] [CrossRef]
- Amer, M.S.; Ghanem, M.A.; Al-Mayouf, A.M.; Arunachalam, P. Low-Symmetry Mesoporous Titanium Dioxide (lsm-TiO2) Electrocatalyst for Efficient and Durable Oxygen Evolution in Aqueous Alkali. J. Electrochem. Soc. 2018, 165, H300. [Google Scholar] [CrossRef]
- Wang, C.; Zhu, D.; Bi, H.; Zhang, Z.; Zhu, J. Synthesis of Nitrogen and Phosphorus/Sulfur Co-Doped Carbon Xerogels for the Efficient Electrocatalytic Reduction of p-Nitrophenol. Int. J. Mol. Sci. 2023, 24, 2432. [Google Scholar] [CrossRef]
- Shi, J.; Cui, H.; Xu, J.; Yan, N.; Liu, Y. Design and fabrication of hierarchically porous carbon frameworks with Fe2O3 cubes as hard template for CO2 adsorption. Chem. Eng. J. 2020, 389, 124459. [Google Scholar] [CrossRef]
- Wang, A.; Chen, W.; Liu, S.; Lin, W.; Tian, C. Layered porous carbon material derived from food residues and its application for elemental mercury adsorption in flue gas. Fuel 2023, 335, 126876. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhu, F.; Gong, R.; Chi, J.; Sun, J.; Xu, G.; Lu, P. Adsorption, regeneration and kinetic of gas phase elemental mercury capture on sulfur incorporated porous carbon synthesized by template method under simulated coal-fired flue gas. Fuel 2023, 342, 127925. [Google Scholar] [CrossRef]
- Hou, J.; Mao, X.; Wang, J.; Liang, C.; Liang, J. Preparation of rice husk-derived porous hard carbon: A self-template method for biomass anode material used for high-performance lithium-ion battery. Chem. Phys. 2021, 551, 111352. [Google Scholar] [CrossRef]
- Lesbayev, B.; Auyelkhankyzy, M.; Ustayeva, G.; Yeleuov, M.; Rakhymzhan, N.; Maltay, A.; Maral, Y. Recent advances: Biomass-derived porous carbon materials. S. Afr. J. Chem. Eng. 2023, 43, 327–336. [Google Scholar] [CrossRef]
- Zhang, B.; Jin, Y.; Huang, X.; Tang, S.; Chen, H.; Su, Y.; Yu, X.; Chen, S.; Chen, G. Biological self-assembled hyphae/starch porous carbon composites for removal of organic pollutants from water. Chem. Eng. J. 2022, 450, 138264. [Google Scholar] [CrossRef]
- Chen, W.; Zong, Y.; Zhou, Y.; Lu, W.; Zhang, Y.; Qian, J. The nature of MgO precursor decomposition and pore-forming in hard-templating of porous carbon derived from cotton. Colloids Surf. A Physicochem. Eng. Asp. 2019, 571, 160–167. [Google Scholar] [CrossRef]
- Zhang, W.; Cheng, R.; Bi, H.; Lu, Y.; Ma, L.; He, X. A review of porous carbons produced by template methods for supercapacitor applications. New Carbon Mater. 2021, 36, 69–81. [Google Scholar] [CrossRef]
- Pavlenko, V.; Khosravi, H.S.; Żółtowska, S.; Haruna, A.B.; Zahid, M.; Mansurov, Z.; Supiyeva, Z.; Galal, A.; Ozoemena, K.I.; Abbas, Q.; et al. A comprehensive review of template-assisted porous carbons: Modern preparation methods and advanced applications. Mater. Sci. Eng. R Rep. 2022, 149, 100682. [Google Scholar] [CrossRef]
- Subramani, K.; Shunmugasundaram, S.; Duraisamy, V.; Ilavarasi, R.; Murugesan Senthil Kumar, S.; Sathish, M. Dual heteroatoms doped SBA-15 templated porous carbon for symmetric supercapacitor in dual redox additive electrolyte. J. Colloid Interface Sci. 2022, 606, 286–297. [Google Scholar] [CrossRef]
- Xu, J.; Zhao, J.; Zhang, N.; Chen, X.; Ding, X. Improved electrochemical performance of SBA-15 based SiO2 anodes with N-doping porous carbon. J. Electroanal. Chem. 2023, 928, 117019. [Google Scholar] [CrossRef]
- Ruan, Y.; Zhao, Y.; Lu, Y.; Guo, D.; Zhao, Y.; Wang, S.; Ma, X. Mesoporous LaAl0.25Ni0.75O3 perovskite catalyst using SBA-15 as templating agent for methane dry reforming. Microporous Mesoporous Mater. 2020, 303, 110278. [Google Scholar] [CrossRef]
- Lei, Y.; Liang, X.; Yang, L.; Chen, J.; Qu, L.; Xu, K.; Feng, J. Li–Se batteries: Insights to the confined structure of selenium in hierarchical porous carbon and discharge mechanism in the carbonate electrolyte. Carbon 2022, 191, 122–131. [Google Scholar] [CrossRef]
- Wan, L.; Chen, D.; Liu, J.; Zhang, Y.; Chen, J.; Du, C.; Xie, M. Facile preparation of porous carbons derived from orange peel via basic copper carbonate activation for supercapacitors. J. Alloys Compd. 2020, 823, 153747. [Google Scholar] [CrossRef]
- Zhou, X.; Guo, L.; Wang, Q.; Wang, J.; Wang, X.; Yang, J.; Tang, J. Nitrogen-doped porous graphitized carbon from antibiotic bacteria residues induced by sodium carbonate and application in Li-ion battery. J. Electroanal. Chem. 2021, 889, 115179. [Google Scholar] [CrossRef]
- Yang, Y.; Yan, Y.; Ren, B.; Fan, C.; Liu, Y.; Deng, Q.; Zhong, L.; You, C.; Xu, Y.; Yang, R. Modified nano-CaCO3 hard template method for hierarchical porous carbon powder with enhanced electrochemical performance in lithium-sulfur battery. Adv. Powder Technol. 2021, 32, 3574–3584. [Google Scholar] [CrossRef]
- Yuan, Z.; Xu, Z.; Zhang, D.; Chen, W.; Huang, Y.; Zhang, T.; Tian, D.; Deng, H.; Zhou, Y.; Sun, Z. Mesoporous activated carbons synthesized by pyrolysis of waste polyester textiles mixed with Mg-containing compounds and their Cr(VI) adsorption. Colloids Surf. A Physicochem. Eng. Asp. 2018, 549, 86–93. [Google Scholar] [CrossRef]
- Xiao, P.; Wang, S.; Xu, X.; Zhu, J. In-situ template formation method to synthesize hierarchically porous carbon for electrocatalytic reduction of 4-nitrophenol. Carbon 2021, 184, 596–608. [Google Scholar] [CrossRef]
- Xu, G.; Shang, H.; Gong, W.; Zhang, X.; Shan, Y.; Ding, J.; Yu, W. One-pot fabrication of petroleum pitch derived hierarchical porous carbon via a recyclable MgO-templating strategy for p-nitrophenol removal. J. Environ. Chem. Eng. 2022, 10, 108458. [Google Scholar] [CrossRef]
- Lu, X.; Xu, H.; Yang, P.; Xiao, L.; Li, Y.; Ma, J.; Li, R.; Liu, L.; Liu, A.; Kondratiev, V.; et al. Zinc-assisted MgO template synthesis of porous carbon-supported Fe-Nx sites for efficient oxygen reduction reaction catalysis in Zn-air batteries. Appl. Catal. B 2022, 313, 121454. [Google Scholar] [CrossRef]
- Pan, C.; Yang, G.; Yang, H.; Wang, L.; Jiang, J.; Zhang, Y.; Wu, F. Facile fabrication of steam-exploded poplar loaded with non-metal-doped Ni-Fe nanoparticles: Catalytic activities in 4-nitrophenol reduction and electrocatalytic reaction. Arab. J. Chem. 2022, 15, 103944. [Google Scholar] [CrossRef]
- Donlon, B.A.; Razo-Flores, E.; Lettinga, G.; Field, J.A. Continuous detoxification, transformation, and degradation of nitrophenols in upflow anaerobic sludge blanket (UASB) reactors. Biotechnol. Bioeng. 1996, 51, 439–449. [Google Scholar] [CrossRef]
- Dinesh, B.; Saraswathi, R. Electrochemical synthesis of nanostructured copper-curcumin complex and its electrocatalytic application towards reduction of 4-nitrophenol. Sens. Actuators B 2017, 253, 502–512. [Google Scholar] [CrossRef]
- Chinnapaiyan, S.; Chen, T.W.; Chen, S.M.; Abdullah Alothman, Z.; Ajmal Ali, M.; Wabaidur, S.M.; Al-Hemaid, F.; Lee, S.Y.; Chang, W.H. Ultrasonic-assisted preparation and characterization of magnetic ZnFe2O4/g-C3N4 nanomaterial and their applications towards electrocatalytic reduction of 4-nitrophenol. Ultrason. Sonochem. 2020, 68, 105071. [Google Scholar] [CrossRef]
- Renu, R.; Komal, J.; Kumar, V.; Tikoo, K.B.; Rana, S.; Kaushik, A.; Singhal, S. Unfolding the electrocatalytic efficacy of highly conducting NiFe2O4-rGO nanocomposites on the road to rapid and sensitive detection of hazardous p-Nitrophenol. J. Electroanal. Chem. 2021, 887, 115161. [Google Scholar] [CrossRef]
- Shi, Q.; Chen, M.; Diao, G. Electrocatalytical reduction of m-nitrophenol on reduced graphene oxide modified glassy carbon electrode. Electrochim. Acta 2013, 114, 693–699. [Google Scholar] [CrossRef]
- Zhou, A.; Dou, Y.; Zhao, C.; Zhou, J.; Wu, X.; Li, J. A leaf-branch TiO2/carbon@MOF composite for selective CO2 photoreduction. Appl. Catal. B 2020, 264, 118519. [Google Scholar] [CrossRef]
- Lazzarini, A.; Piovano, A.; Pellegrini, R.; Agostini, G.; Rudić, S.; Lamberti, C.; Groppo, E. Graphitization of Activated Carbons: A Molecular-level Investigation by INS, DRIFT, XRD and Raman Techniques. Phys. Procedia 2016, 85, 20–26. [Google Scholar] [CrossRef]
- Enterría, M.; Figueiredo, J.L. Nanostructured mesoporous carbons: Tuning texture and surface chemistry. Carbon 2016, 108, 79–102. [Google Scholar] [CrossRef]
- Figueiredo, J.L.; Pereira, M.F.R. The role of surface chemistry in catalysis with carbons. Catal. Today 2010, 150, 2–7. [Google Scholar] [CrossRef]
- Moreno-Castilla, C.; Ferro-Garcia, M.A.; Joly, J.P.; Bautista-Toledo, I.; Carrasco-Marin, F.; Rivera-Utrilla, J. Activated Carbon Surface Modifications by Nitric Acid, Hydrogen Peroxide, and Ammonium Peroxydisulfate Treatments. Langmuir 1995, 11, 4386–4392. [Google Scholar] [CrossRef]
- Chiang, Y.; Lee, C.; Lee, H. Surface chemistry of polyacrylonitrile- and rayon-based activated carbon fibers after post-heat treatment. Mater. Chem. Phys. 2007, 101, 199–210. [Google Scholar] [CrossRef]
- Yoon, C.; Long, D.; Jang, S.; Qiao, W.; Ling, L.; Miyawaki, J.; Rhee, C.; Mochida, I.; Yoon, S. Electrochemical surface oxidation of carbon nanofibers. Carbon 2011, 49, 96–105. [Google Scholar] [CrossRef]
- Ho, V.M.H.; Vo, T.K.; Nguyen, T.H.A.; Dinh, Q.K.; Nguyen, V.C. Dual role of flower-like Fe3O4/Ag microstructure in electrocatalytic detection and catalytic reduction of 4-nitrophenol. Mater. Sci. Semicond. Process. 2023, 160, 107441. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Potential/V | Current/μA | Slope/(μA V−1) | Current Density /(μA cm−2) |
|---|---|---|---|---|
| Glassy carbon | -- | -- | -- | -- |
| PC-oxi | −0.621 | −45.1 | 650 | −229.7 |
| PC-oxa | −0.646 | −65.5 | 825 | −333.6 |
| PC-bic | −0.679 | −135.5 | 1500 | −690.1 |
| 2nd | −0.685 | −155.1 | 1600 | −789.9 |
| 3rd | −0.683 | −169.7 | 1800 | −864.3 |
| 4th | −0.681 | −189.0 | 1900 | −962.6 |
| 5th | −0.683 | −167.5 | 1700 | −853.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gan, W.; Xiao, P.; Zhu, J. Templating Synthesis of Hierarchically Porous Carbon with Magnesium Salts for Electrocatalytic Reduction of 4-Nitrophenol. Catalysts 2023, 13, 1132. https://doi.org/10.3390/catal13071132
Gan W, Xiao P, Zhu J. Templating Synthesis of Hierarchically Porous Carbon with Magnesium Salts for Electrocatalytic Reduction of 4-Nitrophenol. Catalysts. 2023; 13(7):1132. https://doi.org/10.3390/catal13071132
Chicago/Turabian StyleGan, Wanyi, Ping Xiao, and Junjiang Zhu. 2023. "Templating Synthesis of Hierarchically Porous Carbon with Magnesium Salts for Electrocatalytic Reduction of 4-Nitrophenol" Catalysts 13, no. 7: 1132. https://doi.org/10.3390/catal13071132