
Structure and Surface Behavior of Rh Complexes in Ionic Liquids Studied Using Angle-Resolved X-ray Photoelectron Spectroscopy

Abstract
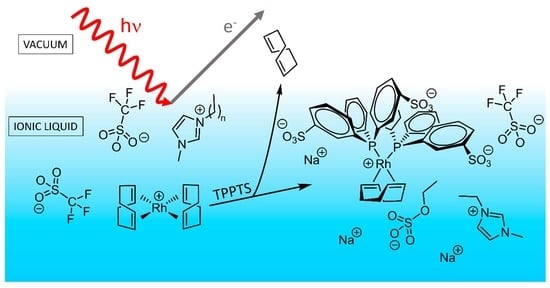
:
1. Introduction
2. Results and Discussion
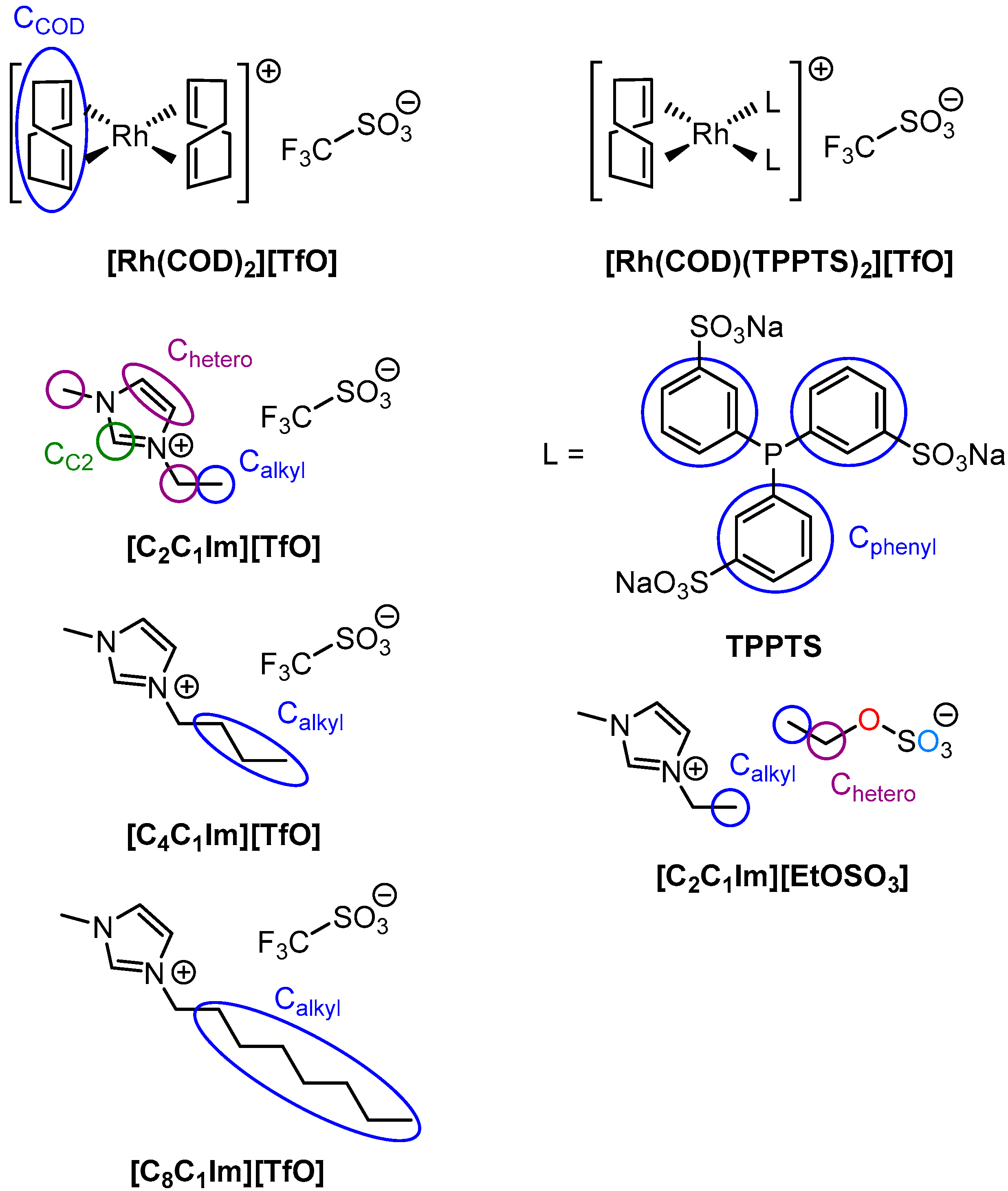
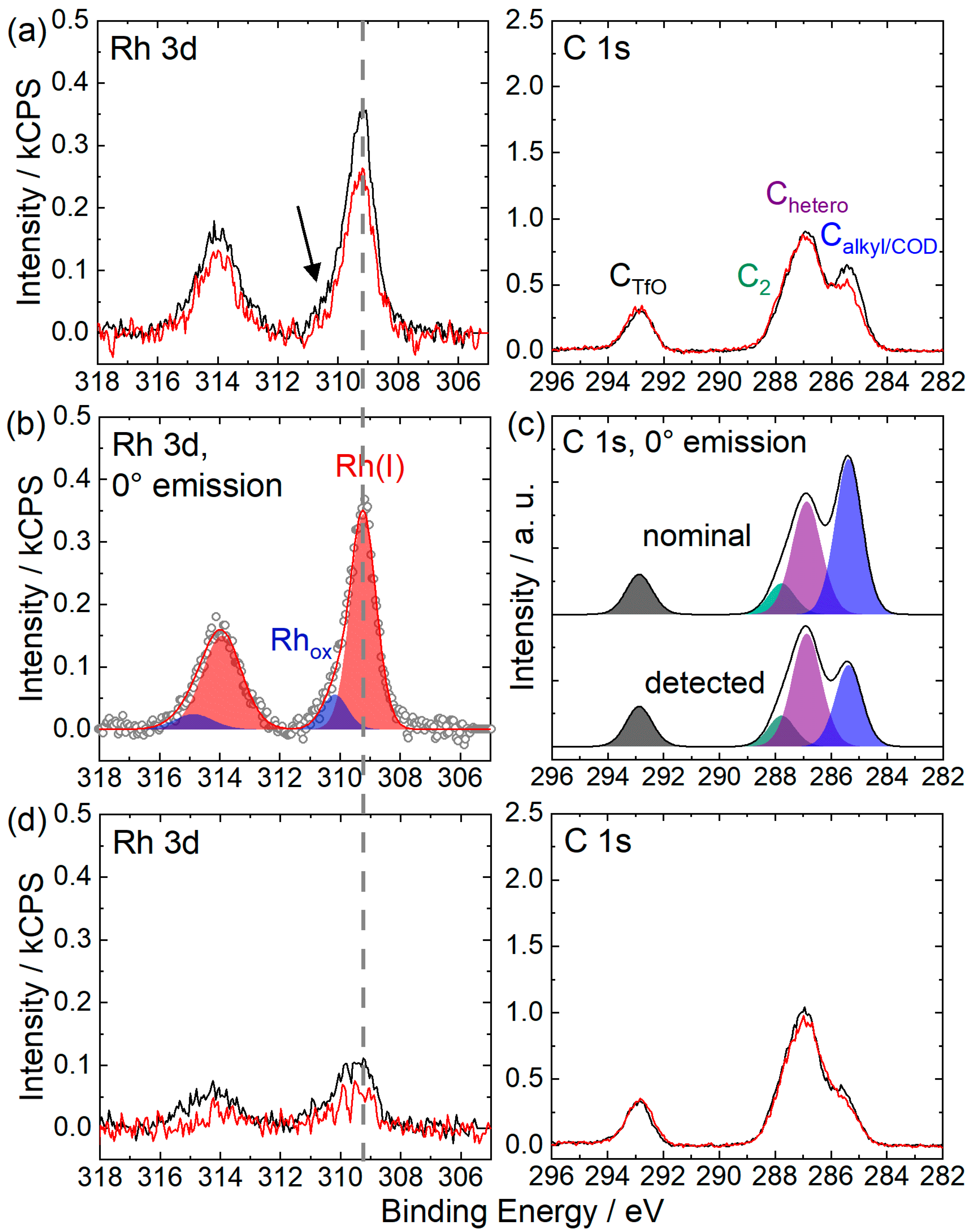
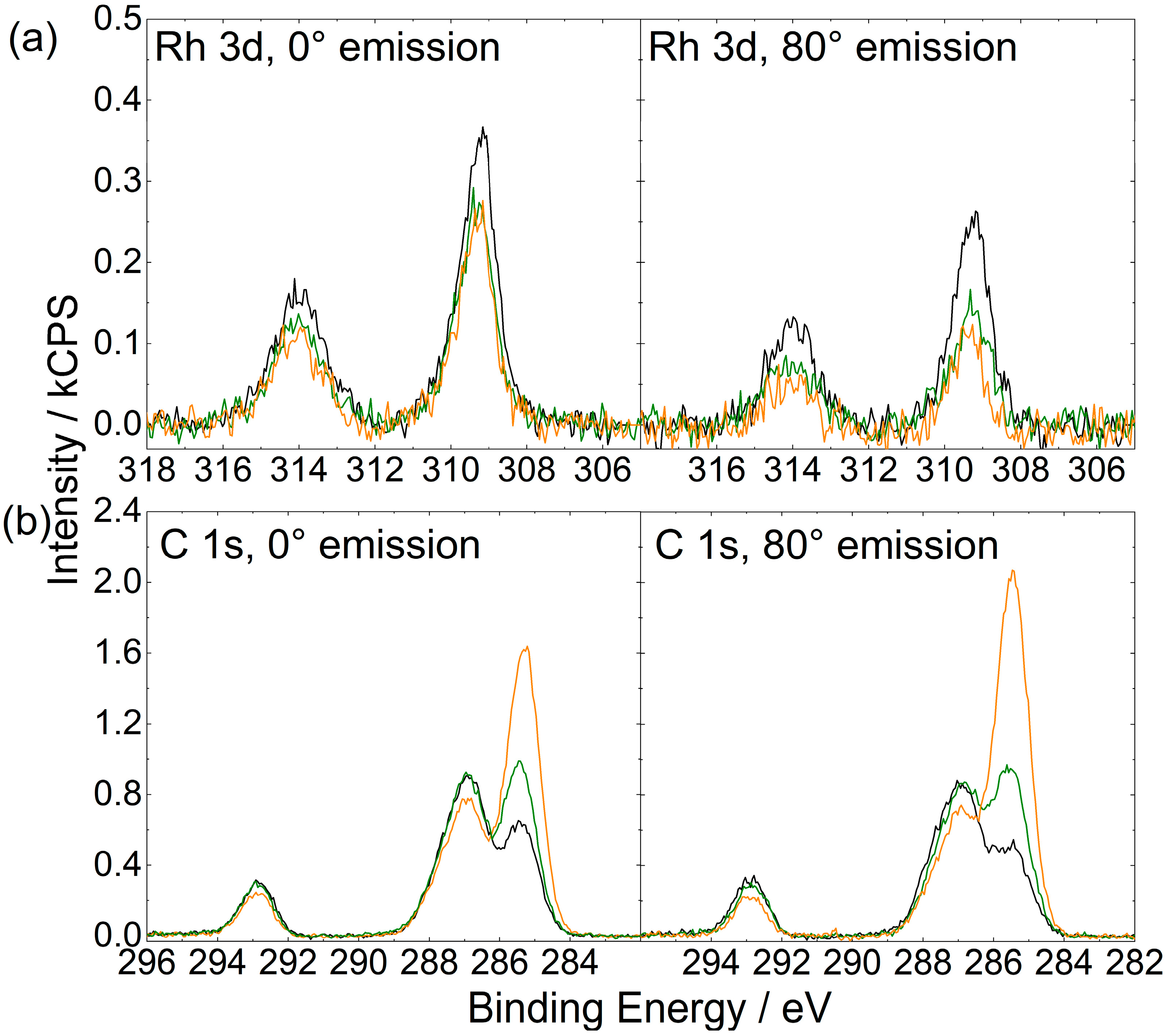
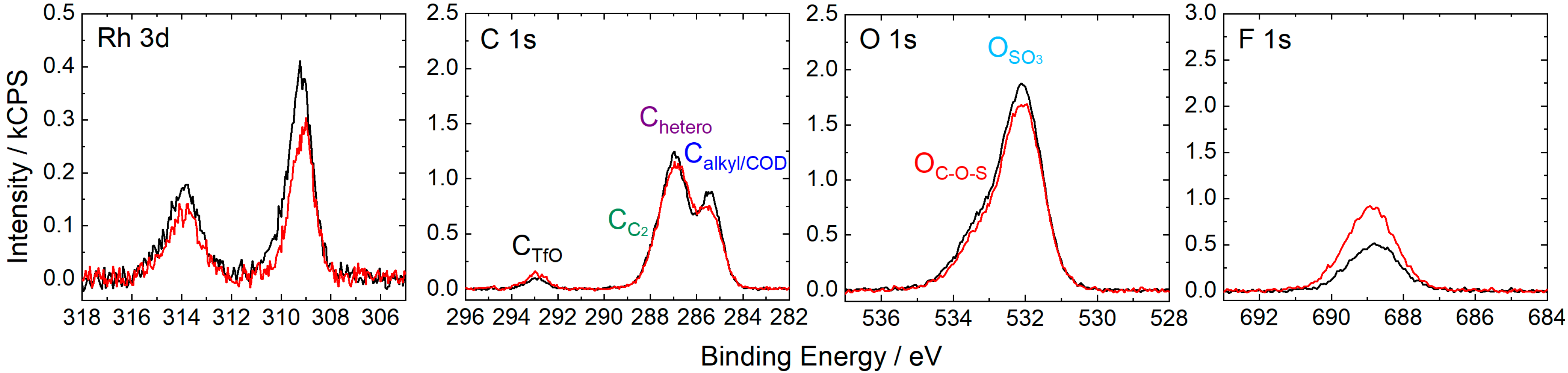
2.1. [Rh(COD)2][TfO] in [CnC1Im][TfO] (n = 2, 4, 8)
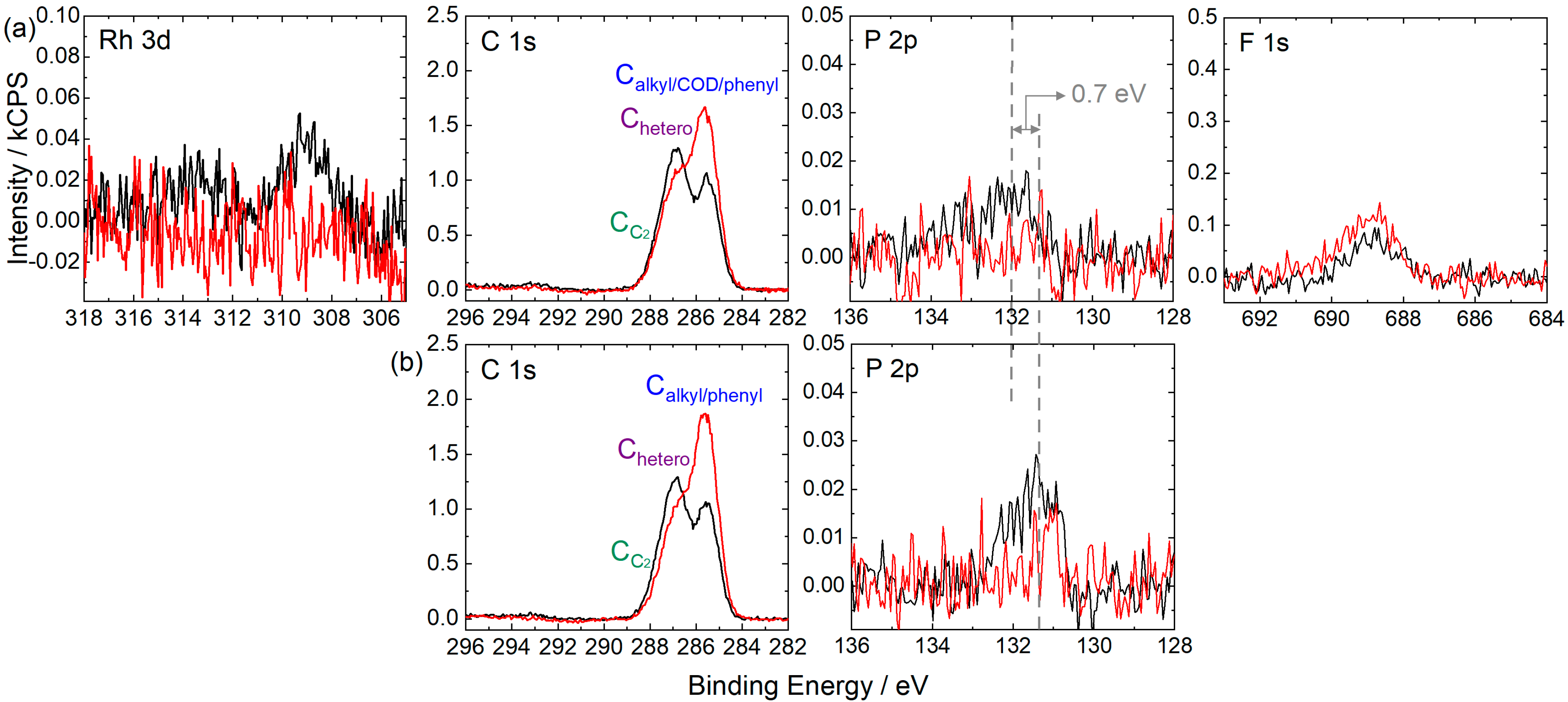
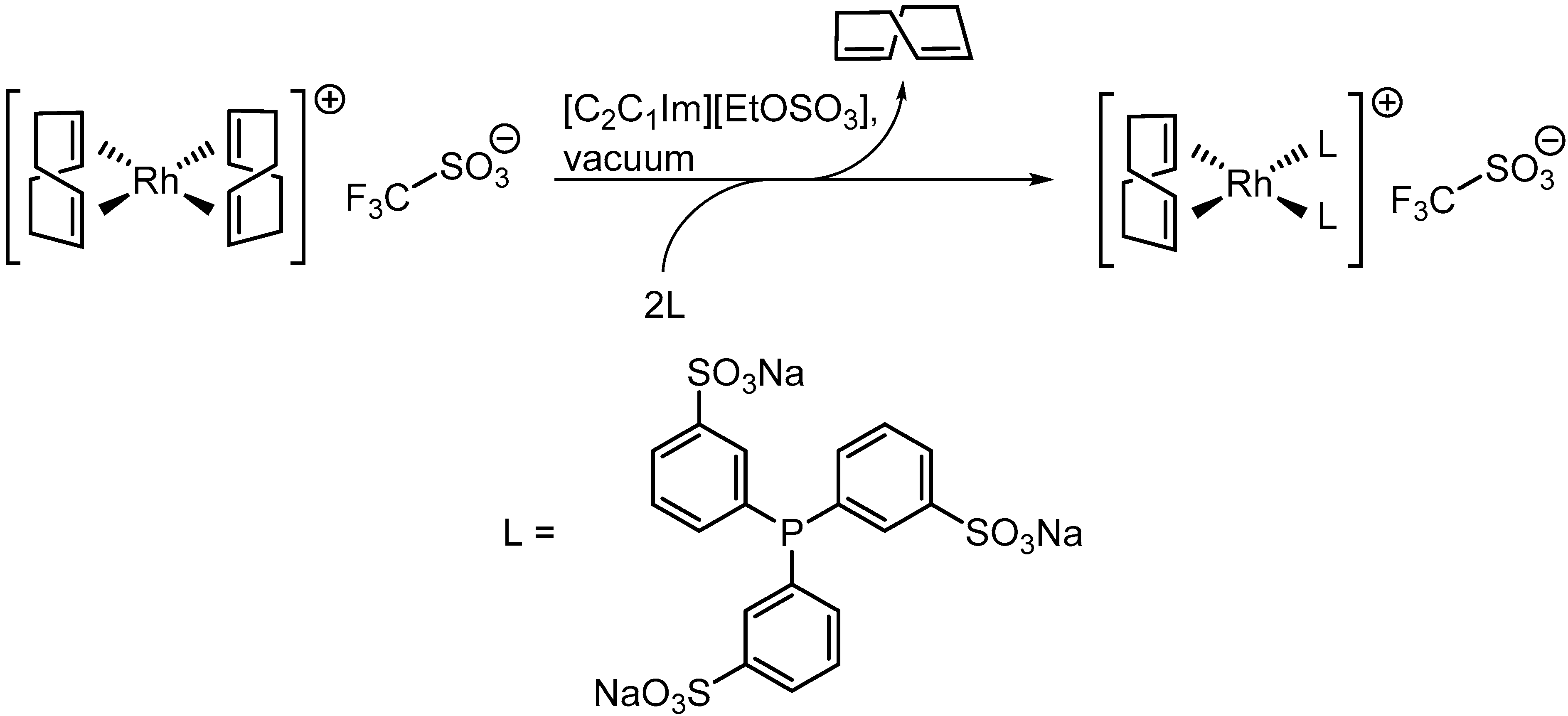
2.2. [Rh(COD)2][TfO] in [C2C1Im][EtOSO3]: Ligand Substitution Using TPPTS
3. Experimental Section
3.1. Materials and Synthesis
3.2. Sample Preparation
3.3. Solutions of [Rh(COD)2][TfO] in ILs
3.4. Saturated Solution of TPPTS in [C2C1Im][EtOSO3]
3.5. 5.9.%mol Solution of TPPTS in [C2C1Im][EtOSO3]
3.6. Solution for Ligand Substitution
3.7. ARXPS Measurements and Data Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Welton, T. Room-Temperature Ionic Liquids. Solvents for Synthesis and Catalysis. Chem. Rev. 1999, 99, 2071–2084. [Google Scholar] [CrossRef] [PubMed]
- Welton, T. Ionic liquids in catalysis. Coord. Chem. Rev. 2004, 248, 2459–2477. [Google Scholar] [CrossRef]
- Hallett, J.P.; Welton, T. Room-Temperature Ionic Liquids: Solvents for Synthesis and Catalysis. 2. Chem. Rev. 2011, 111, 3508–3576. [Google Scholar] [CrossRef] [PubMed]
- Hayes, R.; Warr, G.G.; Atkin, R. Structure and Nanostructure in Ionic Liquids. Chem. Rev. 2015, 115, 6357–6426. [Google Scholar] [CrossRef]
- Chiappe, C.; Pieraccini, D. Ionic liquids: Solvent properties and organic reactivity. J. Phys. Org. Chem. 2005, 18, 275–297. [Google Scholar] [CrossRef]
- Marullo, S.; D’Anna, F.; Rizzo, C.; Billeci, F. Ionic liquids: “Normal” solvents or nanostructured fluids? Org. Biomol. Chem. 2021, 19, 2076–2095. [Google Scholar] [CrossRef]
- Pešić, J.; Watson, M.; Papović, S.; Vraneš, M. Ionic Liquids: Review of their Current and Future Industrial Applications and their Potential Environmental Impact. Recent Pat. Nanotechnol. 2021, 15, 225–244. [Google Scholar] [CrossRef]
- Greer, A.J.; Jacquemin, J.; Hardacre, C. Industrial Applications of Ionic Liquids. Molecules 2020, 25, 5207. [Google Scholar] [CrossRef]
- Vekariya, R.L. A review of ionic liquids: Applications towards catalytic organic transformations. J. Mol. Liq. 2017, 227, 44–60. [Google Scholar] [CrossRef]
- Martins, M.A.P.; Frizzo, C.P.; Moreira, D.N.; Zanatta, N.; Bonacorso, H.G. Ionic Liquids in Heterocyclic Synthesis. Chem. Rev. 2008, 108, 2015–2050. [Google Scholar] [CrossRef]
- Dhameliya, T.M.; Nagar, P.R.; Bhakhar, K.A.; Jivani, H.R.; Shah, B.J.; Patel, K.M.; Patel, V.S.; Soni, A.H.; Joshi, L.P.; Gajjar, N.D. Recent advancements in applications of ionic liquids in synthetic construction of heterocyclic scaffolds: A spotlight. J. Mol. Liq. 2022, 348, 118329. [Google Scholar] [CrossRef]
- Dai, C.; Zhang, J.; Huang, C.; Lei, Z. Ionic Liquids in Selective Oxidation: Catalysts and Solvents. Chem. Rev. 2017, 117, 6929–6983. [Google Scholar] [CrossRef]
- Dyson, P. Review: Synthesis of organometallics and catalytic hydrogenations in ionic liquids. Appl. Organomet. Chem. Appl. Organometal. Chem. 2002, 16, 495–500. [Google Scholar] [CrossRef]
- Hemmeter, D.; Paap, U.; Taccardi, N.; Mehler, J.; Schulz, P.; Wasserscheid, P.; Maier, F.; Steinrück, H.-P. Formation and surface behavior of Pt and Pd complexes with ligand systems derived from nitrile-functionalized ionic liquids studied by XPS. ChemPhysChem 2022, 24, e202200391. [Google Scholar]
- Zhao, D.; Fei, Z.; Geldbach, T.J.; Scopelliti, R.; Dyson, P.J. Nitrile-Functionalized Pyridinium Ionic Liquids: Synthesis, Characterization, and Their Application in Carbon−Carbon Coupling Reactions. J. Am. Chem. Soc. 2004, 126, 15876–15882. [Google Scholar] [CrossRef]
- He, Z.; Alexandridis, P. Nanoparticles in ionic liquids: Interactions and organization. Phys. Chem. Chem. Phys. 2015, 17, 18238–18261. [Google Scholar] [CrossRef]
- Denicourt-Nowicki, A.; Léger, B.; Roucoux, A. N-Donor ligands based on bipyridine and ionic liquids: An efficient partnership to stabilize rhodium colloids. Focus on oxygen-containing compounds hydrogenation. Phys. Chem. Chem. Phys. 2011, 13, 13510–13517. [Google Scholar] [CrossRef]
- Antonietti, M.; Kuang, D.; Smarsly, B.; Zhou, Y. Ionic Liquids for the Convenient Synthesis of Functional Nanoparticles and Other Inorganic Nanostructures. Angew. Chem. Int. Ed. 2004, 43, 4988–4992. [Google Scholar] [CrossRef]
- Ejigu, A.; Walsh, D.A. Electrocatalysis in Room Temperature Ionic Liquids. In Electrochemistry in Ionic Liquids: Volume 2: Applications; Torriero, A.A.J., Ed.; Springer International Publishing: Heidelberg, Germany, 2015; pp. 483–506. [Google Scholar]
- Zhang, G.-R.; Etzold, B.J.M. Ionic liquids in electrocatalysis. J. Energy Chem. 2016, 25, 199–207. [Google Scholar] [CrossRef]
- Tan, X.; Sun, X.; Han, B. Ionic liquid-based electrolytes for CO2 electroreduction and CO2 electroorganic transformation. Natl. Sci. Rev. 2021, 9, nwab022. [Google Scholar] [CrossRef]
- Itoh, T. Ionic Liquids as Tool to Improve Enzymatic Organic Synthesis. Chem. Rev. 2017, 117, 10567–10607. [Google Scholar] [CrossRef] [PubMed]
- van Rantwijk, F.; Sheldon, R.A. Biocatalysis in Ionic Liquids. Chem. Rev. 2007, 107, 2757–2785. [Google Scholar] [CrossRef] [PubMed]
- Steinrück, H.-P.; Wasserscheid, P. Ionic Liquids in Catalysis. Catal. Lett. 2015, 145, 380–397. [Google Scholar] [CrossRef]
- Bartlewicz, O.; Dąbek, I.; Szymańska, A.; Maciejewski, H. Heterogeneous Catalysis with the Participation of Ionic Liquids. Catalysts 2020, 10, 1227. [Google Scholar] [CrossRef]
- Meyer, C.; Hager, V.; Schwieger, W.; Wasserscheid, P. Enhanced activity and selectivity in n-octane isomerization using a bifunctional SCILL catalyst. J. Catal. 2012, 292, 157–165. [Google Scholar] [CrossRef]
- Kernchen, U.; Etzold, B.; Korth, W.; Jess, A. Solid Catalyst with Ionic Liquid Layer (SCILL)—A New Concept to Improve Selectivity Illustrated by Hydrogenation of Cyclooctadiene. Chem. Eng. Technol. 2007, 30, 985–994. [Google Scholar] [CrossRef]
- Riisager, A.; Fehrmann, R.; Haumann, M.; Wasserscheid, P. Supported Ionic Liquid Phase (SILP) Catalysis: An Innovative Concept for Homogeneous Catalysis in Continuous Fixed-Bed Reactors. Eur. J. Inorg. Chem. 2006, 2006, 695–706. [Google Scholar] [CrossRef]
- Riisager, A.; Jørgensen, B.; Wasserscheid, P.; Fehrmann, R. First application of supported ionic liquid phase (SILP) catalysis for continuous methanol carbonylation. Chem. Commun. 2006, 9, 994–996. [Google Scholar] [CrossRef]
- Marinkovic, J.M.; Riisager, A.; Franke, R.; Wasserscheid, P.; Haumann, M. Fifteen Years of Supported Ionic Liquid Phase-Catalyzed Hydroformylation: Material and Process Developments. Ind. Eng. Chem. Res. 2019, 58, 2409–2420. [Google Scholar] [CrossRef]
- Baldelli, S. Interfacial Structure of Room-Temperature Ionic Liquids at the Solid–Liquid Interface as Probed by Sum Frequency Generation Spectroscopy. J. Phys. Chem. Lett. 2013, 4, 244–252. [Google Scholar] [CrossRef]
- Peñalber-Johnstone, C.; Adamová, G.; Plechkova, N.V.; Bahrami, M.; Ghaed-Sharaf, T.; Ghatee, M.H.; Seddon, K.R.; Baldelli, S. Sum frequency generation spectroscopy of tetraalkylphosphonium ionic liquids at the air–liquid interface. J. Chem. Phys. 2018, 148, 193841. [Google Scholar] [CrossRef]
- Huo, F.; Ding, J.; Tong, J.; He, H. Ionic liquid-air interface probed by sum frequency generation spectroscopy and molecular dynamics simulation: Influence of alkyl chain length and anion volume. Mol. Simul. 2022, 48, 844–854. [Google Scholar] [CrossRef]
- Iwahashi, T.; Ishiyama, T.; Sakai, Y.; Morita, A.; Kim, D.; Ouchi, Y. Bi-layering at ionic liquid surfaces: A sum-frequency generation vibrational spectroscopy- and molecular dynamics simulation-based study. Phys. Chem. Chem. Phys. 2020, 22, 12565–12576. [Google Scholar] [CrossRef]
- Sissaoui, J.; Budkina, D.S.; Vauthey, E. Probing Liquid Interfaces with Room-Temperature Ionic Liquids Using the Excited-State Dynamics of a Cationic Dye. J. Phys. Chem. B 2020, 124, 10546–10555. [Google Scholar] [CrossRef]
- Nishi, N.; Ishimatsu, R.; Yamamoto, M.; Kakiuchi, T. Orientation of 1-Dodecyl-4-phenylpyridinium Ions Constituting an Ionic Liquid at the Ionic Liquid|Water Interface Studied by Second Harmonic Generation. J. Phys. Chem. C 2007, 111, 12461–12466. [Google Scholar] [CrossRef]
- Costa, R.; Pereira, C.M.; Silva, A.F.; Brevet, P.-F.; Benichou, E. Ordering and Nonideality of Air–Ionic Liquid Interfaces in Surface Second Harmonic Generation. J. Phys. Chem. B 2020, 124, 3954–3961. [Google Scholar] [CrossRef]
- An, R.; Wei, Y.; Qiu, X.; Dai, Z.; Wu, M.; Gnecco, E.; Shah, F.U.; Zhang, W. Ionic liquids on uncharged and charged surfaces: In situ microstructures and nanofriction. Friction 2022, 10, 1893–1912. [Google Scholar] [CrossRef]
- Liu, S.; Peng, J.; Chen, L.; Sebastián, P.; Feliu, J.M.; Yan, J.; Mao, B. In-situ STM and AFM Studies on Electrochemical Interfaces in imidazolium-based ionic liquids. Electrochim. Acta 2019, 309, 11–17. [Google Scholar] [CrossRef]
- Endres, F.; Borisenko, N.; El Abedin, S.Z.; Hayes, R.; Atkin, R. The interface ionic liquid(s)/electrode(s): In situ STM and AFM measurements. Faraday Discuss. 2012, 154, 221–233; discussion 313–333, 465–471. [Google Scholar] [CrossRef]
- Rodenbücher, C.; Wippermann, K.; Korte, C. Atomic Force Spectroscopy on Ionic Liquids. Appl. Sci. 2019, 9, 2207. [Google Scholar] [CrossRef]
- Pajkossy, T.; Müller, C.; Jacob, T. The metal–ionic liquid interface as characterized by impedance spectroscopy and in situ scanning tunneling microscopy. Phys. Chem. Chem. Phys. 2018, 20, 21241–21250. [Google Scholar] [CrossRef] [PubMed]
- Sloutskin, E.; Ocko, B.M.; Tamam, L.; Kuzmenko, I.; Gog, T.; Deutsch, M. Surface Layering in Ionic Liquids: An X-ray Reflectivity Study. J. Am. Chem. Soc. 2005, 127, 7796–7804. [Google Scholar] [CrossRef] [PubMed]
- Mars, J.; Hou, B.; Weiss, H.; Li, H.; Konovalov, O.; Festersen, S.; Murphy, B.M.; Rütt, U.; Bier, M.; Mezger, M. Surface induced smectic order in ionic liquids—An X-ray reflectivity study of [C22C1im]+[NTf2]−. Phys. Chem. Chem. Phys. 2017, 19, 26651–26661, Erratum in Phys. Chem. Chem. Phys. 2018, 20, 24494–24495. [Google Scholar] [CrossRef] [PubMed]
- Villar-Garcia, I.J.; Fearn, S.; De Gregorio, G.F.; Ismail, N.L.; Gschwend, F.J.V.; McIntosh, A.J.S.; Lovelock, K.R.J. The ionic liquid–vacuum outer atomic surface: A low-energy ion scattering study. Chem. Sci. 2014, 5, 4404–4418. [Google Scholar] [CrossRef]
- Kauling, A.; Ebeling, G.; Morais, J.; Pádua, A.; Grehl, T.; Brongersma, H.H.; Dupont, J. Surface Composition/Organization of Ionic Liquids with Au Nanoparticles Revealed by High-Sensitivity Low-Energy Ion Scattering. Langmuir 2013, 29, 14301–14306. [Google Scholar] [CrossRef]
- Nakajima, K.; Miyashita, M.; Suzuki, M.; Kimura, K. Surface structures of binary mixtures of imidazolium-based ionic liquids using high-resolution Rutherford backscattering spectroscopy and time of flight secondary ion mass spectroscopy. J. Chem. Phys. 2013, 139, 224701. [Google Scholar] [CrossRef]
- Bomhardt, K.; Schneider, P.; Glaser, T.; Dürr, M. Surface Properties of Ionic Liquids: A Mass Spectrometric View Based on Soft Cluster-Induced Desorption. J. Am. Soc. Mass. Spectrom. 2022, 33, 974–980. [Google Scholar] [CrossRef]
- Krischok, S.; Eremtchenko, M.; Himmerlich, M.; Lorenz, P.; Uhlig, J.; Neumann, A.; Öttking, R.; Beenken, W.J.D.; Höfft, O.; Bahr, S.; et al. Temperature-Dependent Electronic and Vibrational Structure of the 1-Ethyl-3-methylimidazolium Bis(trifluoromethylsulfonyl)amide Room-Temperature Ionic Liquid Surface: A Study with XPS, UPS, MIES, and HREELS. J. Phys. Chem. B 2007, 111, 4801–4806. [Google Scholar] [CrossRef]
- Krischok, S.; Ulbrich, A.; Ikari, T.; Kempter, V.; Marschewski, M.; Höfft, O. Surface structure of [XMIm]Tf2N ultrathin ionic liquid films probed by metastable He atoms and photoelectron spectroscopies (UPS and XPS). Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2014, 340, 51–57. [Google Scholar] [CrossRef]
- Biedron, A.B.; Garfunkel, E.L.; Castner, E.W., Jr.; Rangan, S. Ionic liquid ultrathin films at the surface of Cu(100) and Au(111). J. Chem. Phys. 2017, 146, 054704. [Google Scholar] [CrossRef]
- Smoll, E.J.; Tesa-Serrate, M.A.; Purcell, S.M.; D’Andrea, L.; Bruce, D.W.; Slattery, J.M.; Costen, M.L.; Minton, T.K.; McKendrick, K.G. Determining the composition of the vacuum–liquid interface in ionic-liquid mixtures. Faraday Discuss. 2018, 206, 497–522. [Google Scholar] [CrossRef]
- Tesa-Serrate, M.A.; Marshall, B.C.; Smoll, E.J., Jr.; Purcell, S.M.; Costen, M.L.; Slattery, J.M.; Minton, T.K.; McKendrick, K.G. Ionic Liquid–Vacuum Interfaces Probed by Reactive Atom Scattering: Influence of Alkyl Chain Length and Anion Volume. J. Phys. Chem. C 2015, 119, 5491–5505. [Google Scholar] [CrossRef]
- Purcell, S.M.; Lane, P.D.; D’Andrea, L.; Elstone, N.S.; Bruce, D.W.; Slattery, J.M.; Smoll, E.J., Jr.; Greaves, S.J.; Costen, M.L.; Minton, T.K.; et al. Surface Structure of Alkyl/Fluoroalkylimidazolium Ionic–Liquid Mixtures. J. Phys. Chem. B 2022, 126, 1962–1979. [Google Scholar] [CrossRef]
- Meusel, M.; Gezmis, A.; Jaekel, S.; Lexow, M.; Bayer, A.; Maier, F.; Steinrück, H.-P. Time- and Temperature-Dependent Growth Behavior of Ionic Liquids on Au(111) Studied by Atomic Force Microscopy in Ultrahigh Vacuum. J. Phys. Chem. C 2021, 125, 20439–20449. [Google Scholar] [CrossRef]
- Meusel, M.; Lexow, M.; Gezmis, A.; Bayer, A.; Maier, F.; Steinrück, H.-P. Growth of Multilayers of Ionic Liquids on Au(111) Investigated by Atomic Force Microscopy in Ultrahigh Vacuum. Langmuir 2020, 36, 13670–13681. [Google Scholar] [CrossRef]
- Meusel, M.; Lexow, M.; Gezmis, A.; Schötz, S.; Wagner, M.; Bayer, A.; Maier, F.; Steinrück, H.-P. Atomic Force and Scanning Tunneling Microscopy of Ordered Ionic Liquid Wetting Layers from 110 K up to Room Temperature. ACS Nano 2020, 14, 9000–9010. [Google Scholar] [CrossRef]
- Uhl, B.; Buchner, F.; Alwast, D.; Wgner, N.; Behm, R.J. Adsorption of the ionic liquid [BMP][TFSA] on Au(111) and Ag(111): Substrate effects on the structure formation investigated by STM. Beilstein J. Nanotechnol. 2013, 4, 903–918. [Google Scholar] [CrossRef]
- Waldmann, T.; Huang, H.-H.; Hoster, H.E.; Höfft, O.; Endres, F.; Behm, R.J. Imaging an Ionic Liquid Adlayer by Scanning Tunneling Microscopy at the Solid|Vacuum Interface. ChemPhysChem 2011, 12, 2565–2567. [Google Scholar] [CrossRef]
- Seidl, V.; Bosch, M.; Paap, U.; Livraghi, M.; Zhai, Z.; Wick, C.R.; Koller, T.M.; Wasserscheid, P.; Maier, F.; Smith, A.-S.; et al. Bis-polyethylene glycol-functionalized imidazolium ionic liquids: A multi-method approach towards bulk and surface properties. J. Ion. Liq. 2022, 2, 100041. [Google Scholar] [CrossRef]
- Kolbeck, C.; Niedermaier, I.; Deyko, A.; Lovelock, K.R.J.; Taccardi, N.; Wei, W.; Wasserscheid, P.; Maier, F.; Steinrück, H.-P. Influence of Substituents and Functional Groups on the Surface Composition of Ionic Liquids. Chem. Eur. J. 2014, 20, 3954–3965. [Google Scholar] [CrossRef] [PubMed]
- Kolbeck, C.; Killian, M.; Maier, F.; Paape, N.; Wasserscheid, P.; Steinrück, H.-P. Surface Characterization of Functionalized Imidazolium-Based Ionic Liquids. Langmuir 2008, 24, 9500–9507. [Google Scholar] [CrossRef] [PubMed]
- Dick, E.J.; Fouda, A.E.A.; Besley, N.A.; Licence, P. Probing the electronic structure of ether functionalised ionic liquids using X-ray photoelectron spectroscopy. Phys. Chem. Chem. Phys. 2020, 22, 1624–1631. [Google Scholar] [CrossRef] [PubMed]
- Kolbeck, C.; Cremer, T.; Lovelock, K.R.J.; Paape, N.; Schulz, P.S.; Wasserscheid, P.; Maier, F.; Steinrück, H.P. Influence of Different Anions on the Surface Composition of Ionic Liquids Studied Using ARXPS. J. Phys. Chem. B 2009, 113, 8682–8688. [Google Scholar] [CrossRef]
- Men, S.; Sun, Y.; Licence, P.; Qu, J. X-ray photoelectron spectroscopy of morpholinium ionic liquids: Impact of a long alkyl side substituent on the cation–anion interactions. Phys. Chem. Chem. Phys. 2022, 24, 24845–24851. [Google Scholar] [CrossRef]
- Lexow, M.; Maier, F.; Steinrück, H.-P. Ultrathin ionic liquid films on metal surfaces: Adsorption, growth, stability and exchange phenomena. Adv. Phys. X 2020, 5, 1761266. [Google Scholar] [CrossRef]
- Heller, B.S.J.; Paap, U.; Maier, F.; Steinrück, H.-P. Pronounced surface enrichment of fluorinated ionic liquids in binary mixtures with methoxy-functionalized ionic liquids. J. Mol. Liq. 2020, 305, 112783. [Google Scholar] [CrossRef]
- Heller, B.S.J.; Kolbeck, C.; Niedermaier, I.; Dommer, S.; Schatz, J.; Hunt, P.; Maier, F.; Steinrück, H.-P. Surface Enrichment in Equimolar Mixtures of Non-Functionalized and Functionalized Imidazolium-Based Ionic Liquids. ChemPhysChem 2018, 19, 1733–1745. [Google Scholar] [CrossRef]
- Heller, B.S.J.; Lexow, M.; Greco, F.; Shin, S.; Partl, G.; Maier, F.; Steinrück, H.-P. Temperature-Dependent Surface Enrichment Effects in Binary Mixtures of Fluorinated and Non-Fluorinated Ionic Liquids. Chem. Eur. J. 2020, 26, 1117–1126. [Google Scholar] [CrossRef]
- Paap, U.; Seidl, V.; Meyer, K.; Maier, F.; Steinrück, H.-P. Direct Correlation of Surface Tension and Surface Composition of Ionic Liquid Mixtures—A Combined Vacuum Pendant Drop and Angle-Resolved X-ray Photoelectron Spectroscopy Study. Molecules 2022, 27, 8561. [Google Scholar] [CrossRef]
- Oz, E.; Sahin, O.; Okur, H.I.; Suzer, S. Surface Propensity of Anions in a Binary Ionic-Liquid Mixture Assessed by Full-Range Angle-Resolved X-ray Photoelectron Spectroscopy and Surface-Tension Measurements. ChemPhysChem 2020, 21, 2397–2401. [Google Scholar] [CrossRef]
- Zhang, Y.; Khalifa, Y.; Maginn, E.J.; Newberg, J.T. Anion Enhancement at the Liquid–Vacuum Interface of an Ionic Liquid Mixture. J. Phys. Chem. C 2018, 122, 27392–27401. [Google Scholar] [CrossRef]
- Kolbeck, C.; Paape, N.; Cremer, T.; Schulz, P.S.; Maier, F.; Steinrück, H.-P.; Wasserscheid, P. Ligand Effects on the Surface Composition of Rh-Containing Ionic Liquid Solutions Used in Hydroformylation Catalysis. Chem. Eur. J. 2010, 16, 12083–12087. [Google Scholar] [CrossRef]
- Kolbeck, C.; Taccardi, N.; Paape, N.; Schulz, P.S.; Wasserscheid, P.; Steinrück, H.-P.; Maier, F. Redox chemistry, solubility, and surface distribution of Pt(II) and Pt(IV) complexes dissolved in ionic liquids. J. Mol. Liq. 2014, 192, 103–113. [Google Scholar] [CrossRef]
- Maier, F.; Gottfried, J.M.; Rossa, J.; Gerhard, D.; Schulz, P.S.; Schwieger, W.; Wasserscheid, P.; Steinrück, H.-P. Surface Enrichment and Depletion Effects of Ions Dissolved in an Ionic Liquid: An X-ray Photoelectron Spectroscopy Study. Angew. Chem. Int. Ed. 2006, 45, 7778–7780. [Google Scholar] [CrossRef]
- Hemmeter, D.; Kremitzl, D.; Schulz, P.S.; Wasserscheid, P.; Maier, F.; Steinrück, H.-P. The Buoy Effect: Surface Enrichment of a Pt Complex in IL Solution by Ligand Design. Chem. Eur. J. 2023, 29, e202203325. [Google Scholar] [CrossRef]
- Dimitrova, A.; Walle, M.; Himmerlich, M.; Krischok, S. Surface composition of [BMP][Tf2N] and [PMIm][Tf2N] in the presence of NbF5 and TaF5. A photoelectron spectroscopy study. J. Mol. Liq. 2017, 226, 78–84. [Google Scholar] [CrossRef]
- Kurisaki, T.; Tanaka, D.; Inoue, Y.; Wakita, H.; Minofar, B.; Fukuda, S.; Ishiguro, S.-i.; Umebayashi, Y. Surface Analysis of Ionic Liquids with and without Lithium Salt Using X-ray Photoelectron Spectroscopy. J. Phys. Chem. B 2012, 116, 10870–10875. [Google Scholar] [CrossRef] [PubMed]
- Smoll, E.J.; Chen, X.; Hall, L.M.; D’Andrea, L.; Slattery, J.M.; Minton, T.K. Probing a Ruthenium Coordination Complex at the Ionic Liquid–Vacuum Interface with Reactive-Atom Scattering, X-ray Photoelectron Spectroscopy, and Time-of-Flight Secondary Ion Mass Spectrometry. J. Phys. Chem. C 2020, 124, 382–397. [Google Scholar] [CrossRef]
- Men, S.; Jiang, J. X-ray photoelectron spectroscopy as a probe of the interaction between rhodium acetate and ionic liquids. Chem. Phys. Lett. 2016, 646, 125–129. [Google Scholar] [CrossRef]
- Men, S.; Jin, Y. Rhodium Catalyst-Ionic Liquids Interaction by X-ray Photoelectron Spectroscopy Data. Russ. J. Phys. Chem. A 2018, 92, 2472. [Google Scholar]
- Men, S.; Lovelock, K.R.J.; Licence, P. Directly probing the effect of the solvent on a catalyst electronic environment using X-ray photoelectron spectroscopy. RSC Adv. 2015, 5, 35958–35965. [Google Scholar] [CrossRef]
- Men, S.; Jiang, J. Probing the Formation of the NHC-Palladium Species in Ionic Liquids by X-ray Photoelectron Spectroscopy. Russ. J. Phys. Chem. A 2018, 92, 1627–1630. [Google Scholar]
- Niedermaier, I.; Kolbeck, C.; Steinrück, H.-P.; Maier, F. Dual analyzer system for surface analysis dedicated for angle-resolved photoelectron spectroscopy at liquid surfaces and interfaces. Rev. Sci. Instrum. 2016, 87, 045105. [Google Scholar] [CrossRef]
- Steinrück, H.-P. Recent developments in the study of ionic liquid interfaces using X-ray photoelectron spectroscopy and potential future directions. Phys. Chem. Chem. Phys. 2012, 14, 5010–5029. [Google Scholar] [CrossRef]
- Steinrück, H.-P.; Libuda, J.; Wasserscheid, P.; Cremer, T.; Kolbeck, C.; Laurin, M.; Maier, F.; Sobota, M.; Schulz, P.S.; Stark, M. Surface Science and Model Catalysis with Ionic Liquid-Modified Materials. Adv. Mater. 2011, 23, 2571–2587. [Google Scholar] [CrossRef]
- Blundell, R.K.; Delorme, A.E.; Smith, E.F.; Licence, P. An ARXPS and ERXPS study of quaternary ammonium and phosphonium ionic liquids: Utilising a high energy Ag Lα′ X-ray source. Phys. Chem. Chem. Phys. 2016, 18, 6122–6131. [Google Scholar] [CrossRef]
- Men, S.; Hurisso, B.B.; Lovelock, K.R.J.; Licence, P. Does the influence of substituents impact upon the surface composition of pyrrolidinium-based ionic liquids? An angle resolved XPS study. Phys. Chem. Chem. Phys. 2012, 14, 5229–5238. [Google Scholar] [CrossRef]
- Lockett, V.; Sedev, R.; Harmer, S.; Ralston, J.; Horne, M.; Rodopoulos, T. Orientation and mutual location of ions at the surface of ionic liquids. Phys. Chem. Chem. Phys. 2010, 12, 13816–13827. [Google Scholar] [CrossRef]
- Suarez, P.A.Z.; Dullius, J.E.L.; Einloft, S.; De Souza, R.F.; Dupont, J. The use of new ionic liquids in two-phase catalytic hydrogenation reaction by rhodium complexes. Polyhedron 1996, 15, 1217–1219. [Google Scholar] [CrossRef]
- Nindakova, L.O.; Shainyan, B.A.; Albanov, A.I.; Shmidt, F.K. Double Stereoselection in the Hydrogenation over Cationic Rh(I) Complexes with Two Different Chiral Ligands. Russ. J. Org. Chem. 2003, 39, 926–932. [Google Scholar] [CrossRef]
- Carvalho, M.; Wieserman, L.F.; Hercules, D.M. Spectroscopic Characterization of Wilkinson’s Catalyst Using X-ray Photoelectron Spectroscopy (ESCA). Appl. Spectrosc. 1982, 36, 290–296. [Google Scholar] [CrossRef]
- Díaz, J.; Paolicelli, G.; Ferrer, S.; Comin, F. Separation of the sp3 and sp2 components in the C1s photoemission spectra of amorphous carbon films. Phys. Rev. B Condens. Matter 1996, 54, 8064–8069. [Google Scholar] [CrossRef]
- Fujimoto, A.; Yamada, Y.; Koinuma, M.; Sato, S. Origins of sp3C peaks in C1s X-ray Photoelectron Spectra of Carbon Materials. Anal. Chem. 2016, 88, 6110–6114. [Google Scholar] [CrossRef]
- Dewar, M.J.S. A Review of the-Complex Theory. Bull. Soc. Chim. Fr. 1951, 18, C79. [Google Scholar]
- Chatt, J.; Duncanson, L.A. 586. Olefin co-ordination compounds. Part III. Infra-red spectra and structure: Attempted preparation of acetylene complexes. J. Chem. Soc. (Resumed) 1953, 2939–2947. [Google Scholar] [CrossRef]
- Werner, H.; Bosch, M.; Schneider, M.E.; Hahn, C.; Kukla, F.; Manger, M.; Windmüller, B.; Weberndörfer, B.; Laubender, M. Preparation, molecular structure and reactivity of mono- and di-nuclear sulfonato rhodium(I) complexes. J. Chem. Soc. Dalton Trans. 1998, 21, 3549–3558. [Google Scholar] [CrossRef]
- Lovelock, K.R.J.; Kolbeck, C.; Cremer, T.; Paape, N.; Schulz, P.S.; Wasserscheid, P.; Maier, F.; Steinrück, H.P. Influence of Different Substituents on the Surface Composition of Ionic Liquids Studied Using ARXPS. J. Phys. Chem. B 2009, 113, 2854–2864. [Google Scholar] [CrossRef]
- Nakajima, K.; Ohno, A.; Suzuki, M.; Kimura, K. Observation of Molecular Ordering at the Surface of Trimethylpropylammonium Bis(trifluoromethanesulfonyl)imide Using High-Resolution Rutherford Backscattering Spectroscopy. Langmuir 2008, 24, 4482–4484. [Google Scholar] [CrossRef]
- Nakajima, K.; Lísal, M.; Kimura, K. Surfaces of Ionic Liquids. In Surface and Interface Science; Wandelt, K., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2020; pp. 351–389. [Google Scholar]
- Maier, F.; Cremer, T.; Kolbeck, C.; Lovelock, K.R.J.; Paape, N.; Schulz, P.S.; Wasserscheid, P.; Steinrück, H.P. Insights into the surface composition and enrichment effects of ionic liquids and ionic liquid mixtures. Phys. Chem. Chem. Phys. 2010, 12, 1905–1915. [Google Scholar] [CrossRef]
- Sanmartín Pensado, A.; Malfreyt, P.; Pádua, A.A.H. Molecular Dynamics Simulations of the Liquid Surface of the Ionic Liquid 1-Hexyl-3-methylimidazolium Bis(trifluoromethanesulfonyl)amide: Structure and Surface Tension. J. Phys. Chem. B 2009, 113, 14708–14718. [Google Scholar] [CrossRef] [PubMed]
- Iwahashi, T.; Miyamae, T.; Kanai, K.; Seki, K.; Kim, D.; Ouchi, Y. Anion Configuration at the Air/Liquid Interface of Ionic Liquid [bmim]OTf Studied by Sum-Frequency Generation Spectroscopy. J. Phys. Chem. B 2008, 112, 11936–11941. [Google Scholar] [CrossRef] [PubMed]
- Kolbeck, C.; Lehmann, J.; Lovelock, K.R.J.; Cremer, T.; Paape, N.; Wasserscheid, P.; Fröba, A.P.; Maier, F.; Steinrück, H.P. Density and Surface Tension of Ionic Liquids. J. Phys. Chem. B 2010, 114, 17025–17036. [Google Scholar] [CrossRef]
- Wagner, C.D.; Davis, L.E.; Zeller, M.V.; Taylor, J.A.; Raymond, R.H.; Gale, L.H. Empirical Atomic Sensitivity Factors for Quantitative Analysis by Electron Spectroscopy for Chemical Analysis. Surf. Interface Anal. 1981, 3, 211–225. [Google Scholar] [CrossRef]
- Gottfried, J.M.; Maier, F.; Rossa, J.; Gerhard, D.; Schulz, P.S.; Wasserscheid, P.; Steinrück, H.-P. Surface Studies on the Ionic Liquid 1-Ethyl-3-Methylimidazolium Ethylsulfate Using X-Ray Photoelectron Spectroscopy (XPS). Z. Phys. Chem. 2006, 220, 1439–1453. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (a) 9%mol [Rh(COD)2][TfO] in [C2C1Im][TfO] | Rh 3d5/2 | Rh 3d5/2 ox | C 1s TfO | C 1s C2 | C 1s hetero | C 1s alkyl/COD | N 1s | F 1s | O 1s | S 2p |
| Binding Energy/eV | 309.4 | 310.7 | 292.9 | 287.8 | 286.9 | 285.5 | 402.2 | 688.8 | 532.3 | 169.4 |
| Nominal | 0.099 | 0.00 | 1.10 | 1.00 | 4.00 | 2.58 1.00/1.58 | 2.00 | 3.30 | 3.30 | 1.10 |
| Experimental, 0° | 0.053 | 0.012 | 1.22 | 1.04 | 4.15 | 1.43 | 2.09 | 3.57 | 3.70 | 1.20 |
| Experimental, 80° | 0.033 | 1.33 | 1.02 | 4.09 | 1.23 | 1.99 | 4.17 | 3.40 | 1.21 | |
| (b) 20%mol [Rh(COD)2][TfO] in [C2C1Im][TfO] | Rh 3d5/2 | Rh 3d5/2 ox | C 1s TfO | C 1s C2 | C 1s hetero | C 1s alkyl/COD | N 1s | F 1s | O 1s | S 2p |
| Binding Energy/eV | 309.2 | 310.2 | 292.9 | 287.8 | 286.9 | 285.4 | 402.2 | 688.8 | 532.4 | 169.4 |
| Nominal | 0.250 | 0.00 | 1.25 | 1.00 | 4.00 | 5.00 1.00/4.00 | 2.00 | 3.75 | 3.75 | 1.25 |
| Experimental, 0° | 0.158 | 0.026 | 1.40 | 1.11 | 4.44 | 2.91 | 2.22 | 4.20 | 4.37 | 1.40 |
| Experimental, 80° | 0.125 | 1.54 | 1.13 | 4.50 | 2.35 | 1.97 | 5.03 | 4.05 | 1.40 | |
| (c) 20%mol [Rh(COD)2][TfO] in [C4C1Im][TfO] | Rh 3d5/2 | Rh 3d5/2 ox | C 1s TfO | C 1s C2 | C 1s hetero | C 1s alkyl/COD | N 1s | F 1s | O 1s | S 2p |
| Binding Energy / eV | 309.3 | 310.2 | 292.9 | 287.8 | 286.9 | 285.4 | 402.3 | 688.8 | 532.4 | 169.4 |
| Nominal | 0.256 | 0.00 | 1.26 | 1.00 | 4.00 | 7.10 3.00/4.10 | 2.00 | 3.77 | 3.77 | 1.26 |
| Experimental, 0° | 0.140 | 0.017 | 1.46 | 1.18 | 4.72 | 4.83 | 2.28 | 4.06 | 4.34 | 1.39 |
| Experimental, 80° | 0.083 | 1.48 | 1.16 | 4.62 | 4.83 | 2.22 | 4.68 | 3.93 | 1.41 | |
| (d) 20%mol [Rh(COD)2][TfO] in [C8C1Im][TfO] | Rh 3d5/2 | Rh 3d5/2 ox | C 1s TfO | C 1s C2 | C 1s hetero | C 1s alkyl/COD | N 1s | F 1s | O 1s | S 2p |
| Binding Energy/eV | 309.3 | 310.4 | 292.9 | 287.8 | 286.9 | 285.3 | 402.3 | 688.8 | 532.4 | 169.4 |
| Nominal | 0.250 | 0.00 | 1.25 | 1.00 | 4.00 | 11.0 7.00/4.00 | 2.00 | 3.75 | 3.75 | 1.25 |
| Experimental, 0° | 0.144 | 0.017 | 1.38 | 1.16 | 4.65 | 9.33 | 2.14 | 3.87 | 4.16 | 1.39 |
| Experimental, 80° | 0.066 | 1.19 | 1.06 | 4.23 | 11.7 | 1.97 | 3.56 | 3.18 | 1.29 |
| (a) 20%mol [Rh(COD)2][TfO] in [C2C1Im][EtOSO3] | Rh 3d5/2 | Rh 3d5/2 ox | C 1s TfO | C 1s C2 | C 1s hetero | C 1s alkyl/ COD | N 1s | F 1s | O 1s C-O-S | O 1s SO3 | S 2p | P 2p | Na 1s |
| Binding Energy/eV | 309.2 | 310.2 | 292.9 | 287.8 | 286.9 | 285.4 | 402.2 | 688.8 | 533.4 | 532.1 | 169.5 | ||
| Nominal | 0.250 | 0.00 | 0.250 | 1.00 | 5.00 | 6.00 2.00/4.00 | 2.00 | 0.750 | 1.00 | 3.75 | 1.25 | ||
| Experimental, 0° | 0.171 | 0.025 | 0.400 | 1.14 | 5.70 | 3.80 | 2.22 | 0.983 | 1.19 | 4.24 | 1.39 | ||
| Experimental, 80° | 0.133 | 0.634 | 1.15 | 5.76 | 3.29 | 2.13 | 1.75 | 1.02 | 3.92 | 1.47 | |||
| (b) 5.9%mol solution of TPPTS in [C2C1Im][EtOSO3] (2:31.6 TPPTS:IL ratio) | Rh 3d5/2 | Rh 3d5/2 ox | C 1s TfO | C 1s C2 | C 1s hetero | C 1s alkyl/ phenyl | N 1s | F 1s | O 1s C-O-S | O 1s SO3 | S 2p | P 2p | Na 1s |
| Binding Energy/eV | 287.8 | 286.9 | 285.5 | 402.2 | 533.4 | 531.9 | 169.5 | 131.7 | 1071.9 | ||||
| Nominal | 1.00 | 5.00 | 3.14 | 2.00 | 1.00 | 3.57 | 1.19 | 0.063 | 0.190 | ||||
| Experimental, 0° | 0.959 | 4.80 | 3.58 | 1.74 | 1.12 | 3.58 | 1.20 | 0.060 | 0.122 | ||||
| Experimental, 80° | 0.774 | 3.87 | 6.80 | 1.12 | 1.01 | 2.56 | 0.96 | 0.063 | |||||
| (c) [Rh(COD)2][TfO] and TPPTS in [C2C1Im][EtOSO3] (1:2:31.6 Rh:TPPTS:IL ratio) | Rh 3d5/2 | Rh 3d5/2 ox | C 1s TfO | C 1s C2 | C 1s hetero | C 1s alkyl/ COD/ phenyl | N 1s | F 1s | O 1s C-O-S | O 1s SO3 | S 2p | P 2p | Na 1s |
| Binding Energy/eV | 309.0 | 287.7 | 286.8 | 285.5 | 402.2 | 688.8 | 533.3 | 531.9 | 169.5 | 132.5 | 1071.9 | ||
| Nominal | 0.032 | 0.00 | 0.032 | 1.00 | 5.00 | 3.39 | 2.00 | 0.095 | 1.00 | 3.66 | 1.22 | 0.063 | 0.190 |
| Experimental, 0° | 0.031 | 1.00 | 4.99 | 3.55 | 1.85 | 0.122 | 1.10 | 3.61 | 1.25 | 0.054 | 0.122 | ||
| Experimental, 80° | 0.84 | 4.19 | 6.29 | 1.36 | 0.200 | 0.962 | 2.69 | 1.09 | 0.062 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hemmeter, D.; Paap, U.; Maier, F.; Steinrück, H.-P. Structure and Surface Behavior of Rh Complexes in Ionic Liquids Studied Using Angle-Resolved X-ray Photoelectron Spectroscopy. Catalysts 2023, 13, 871. https://doi.org/10.3390/catal13050871
Hemmeter D, Paap U, Maier F, Steinrück H-P. Structure and Surface Behavior of Rh Complexes in Ionic Liquids Studied Using Angle-Resolved X-ray Photoelectron Spectroscopy. Catalysts. 2023; 13(5):871. https://doi.org/10.3390/catal13050871
Chicago/Turabian StyleHemmeter, Daniel, Ulrike Paap, Florian Maier, and Hans-Peter Steinrück. 2023. "Structure and Surface Behavior of Rh Complexes in Ionic Liquids Studied Using Angle-Resolved X-ray Photoelectron Spectroscopy" Catalysts 13, no. 5: 871. https://doi.org/10.3390/catal13050871