
In Situ Transition of a Nickel Metal–Organic Framework on TiO2 Photoanode towards Urea Photoelectrolysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Preparation of Ni(OH)2@TiO2 and T-Ni(OH)2@TiO2 Film
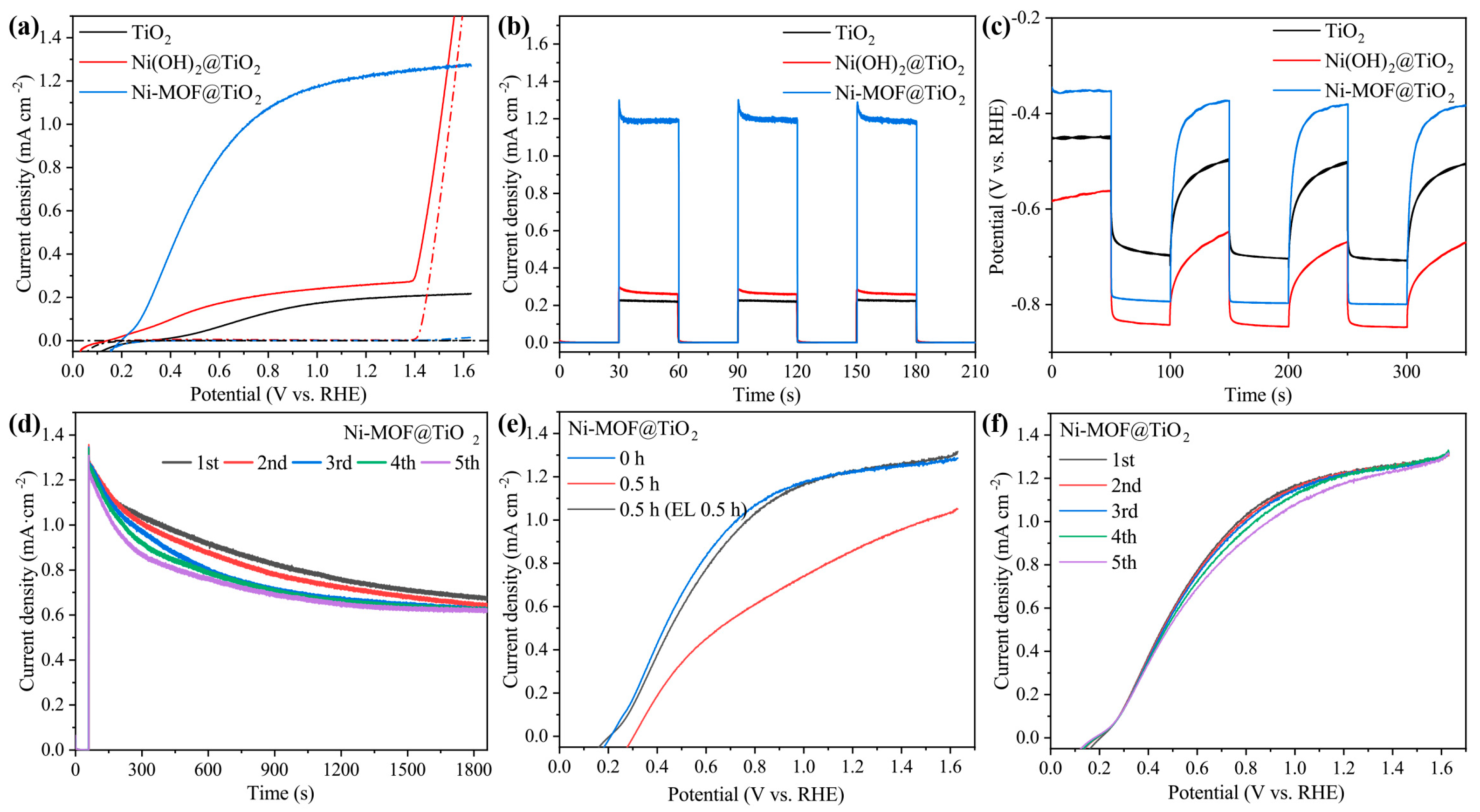
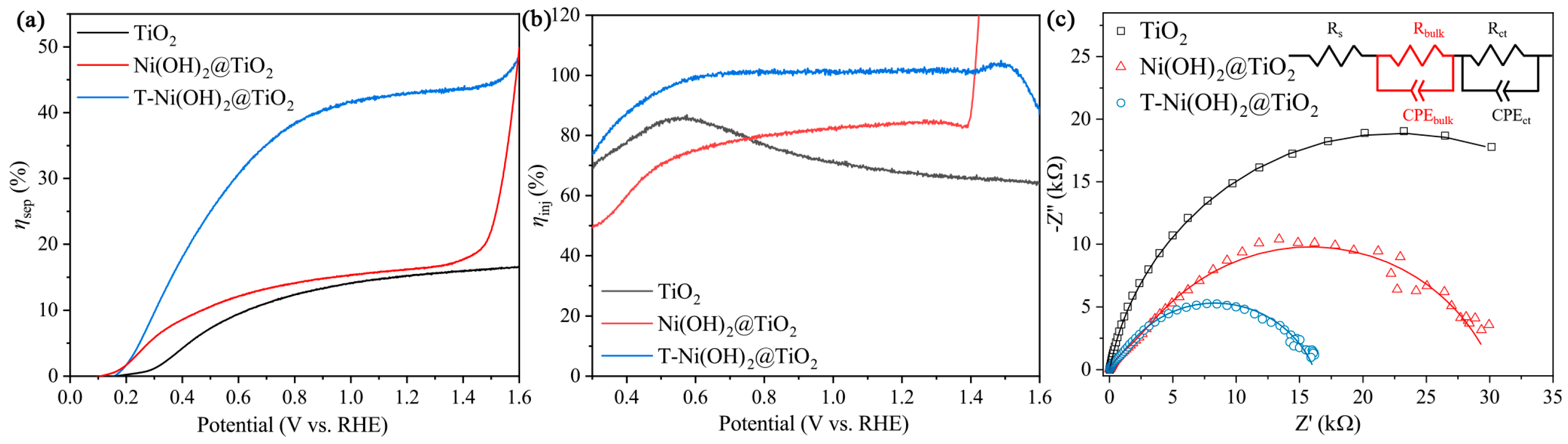
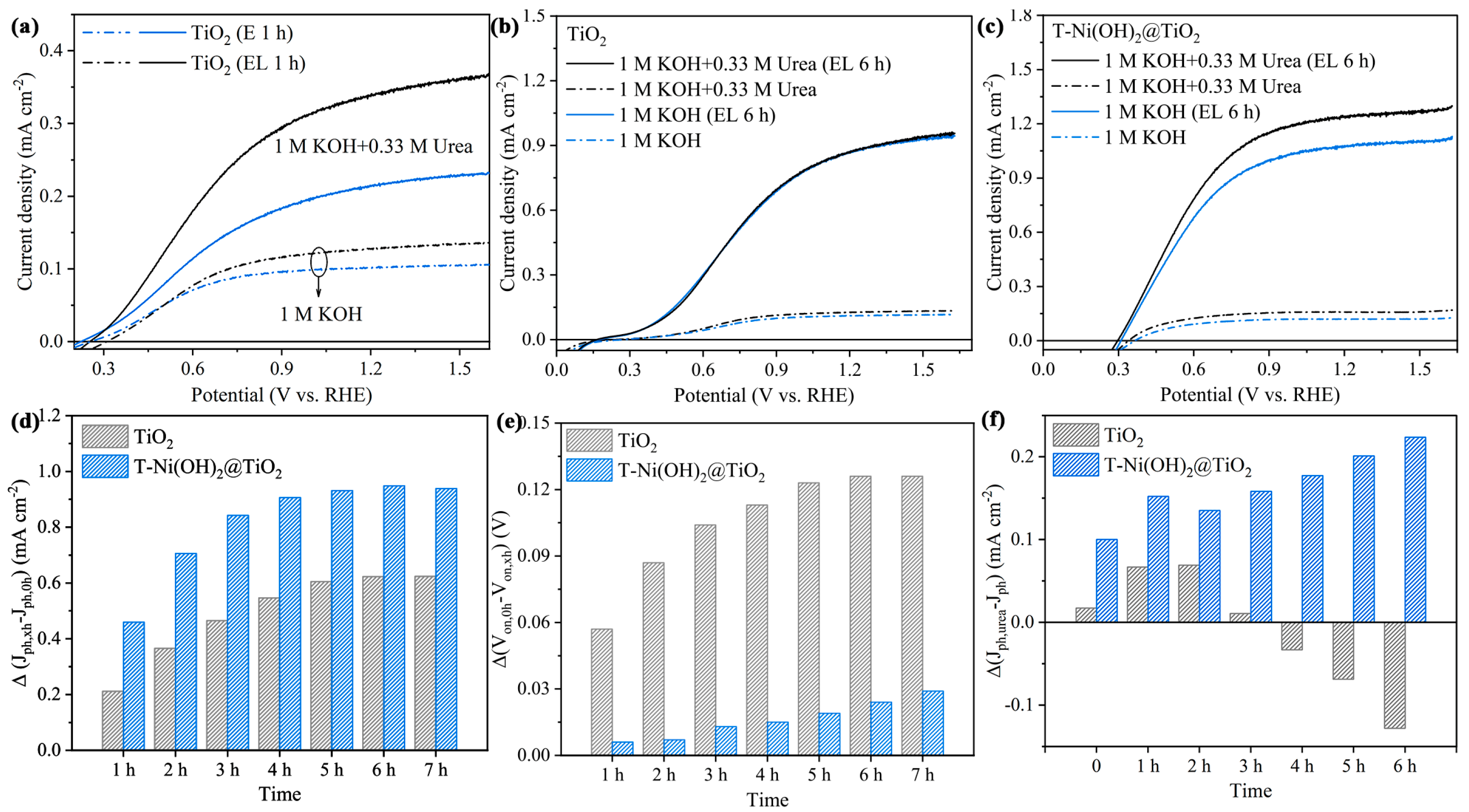
2.2. PEC UOR Performances of T-Ni(OH)2@TiO2
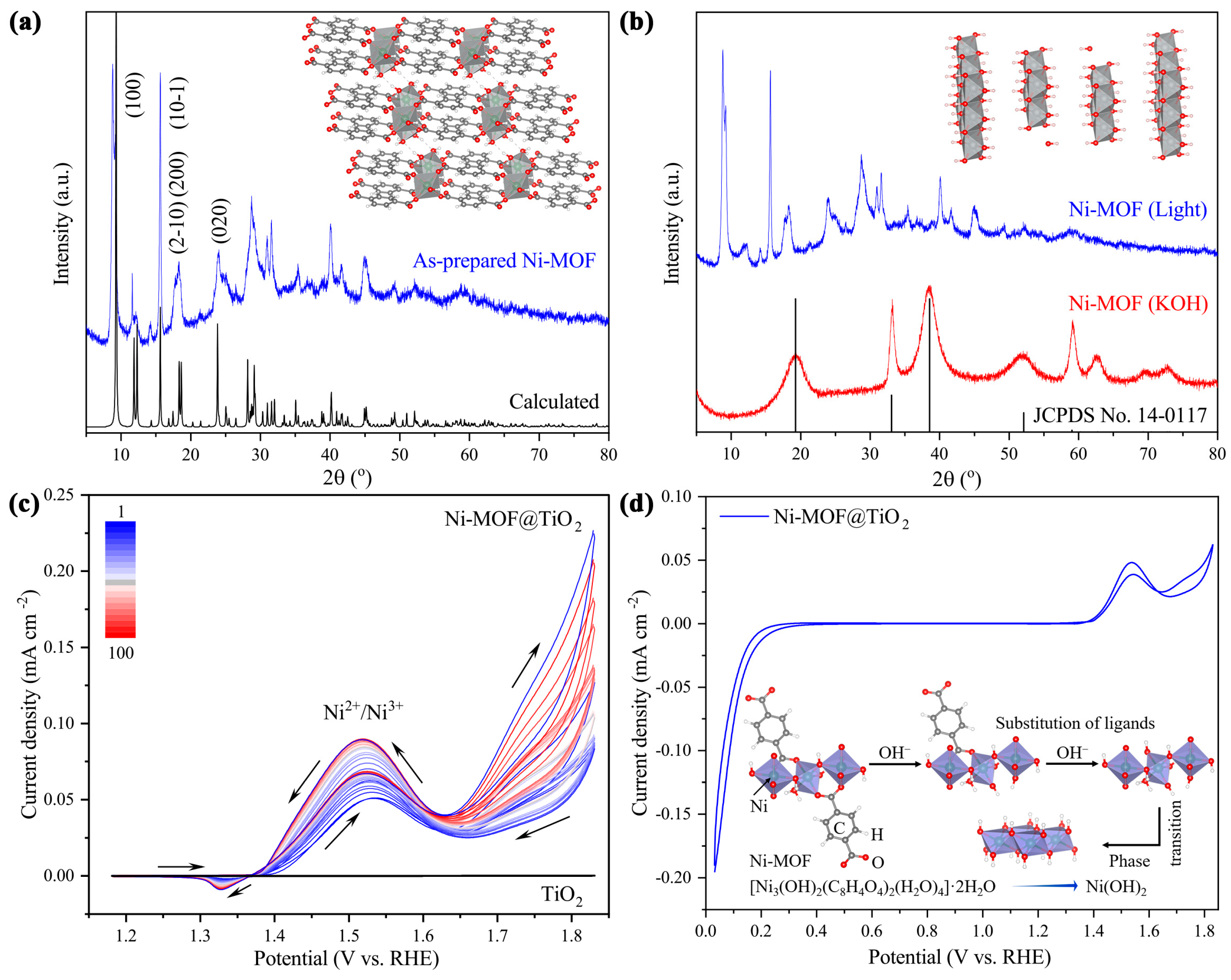
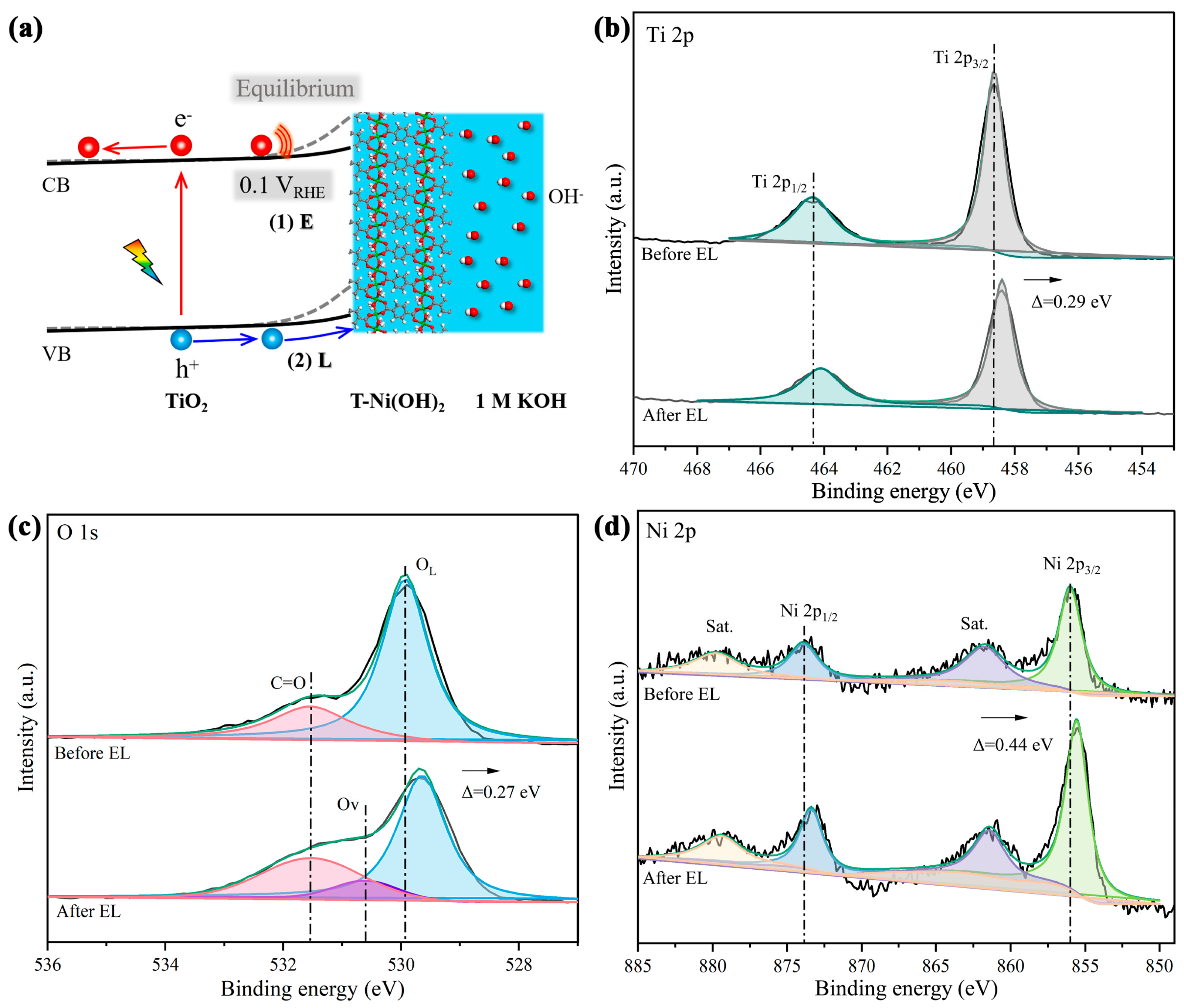
2.3. In Situ-Transition Mechanism of Ni-MOF
3. Materials and Methods
3.1. Materials
3.2. Preparation of TiO2 Photoanodes
3.3. Preparation of T-Ni(OH)2@TiO2 and Ni(OH)2@TiO2 Photoanodes
3.4. Material Characterizations
3.5. PEC Performance Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Pastor, E.; Sachs, M.; Selim, S.; Durrant, J.R.; Bakulin, A.A.; Walsh, A. Electronic defects in metal oxide photocatalysts. Nat. Rev. Mater. 2022, 7, 503–521. [Google Scholar] [CrossRef]
- Kundu, B.K.; Han, G.; Sun, Y. Derivatized Benzothiazoles as Two-Photon-Absorbing Organic Photosensitizers Active under Near Infrared Light Irradiation. J. Am. Chem. Soc. 2023, 145, 3535–3542. [Google Scholar] [CrossRef]
- Tang, R.; Zhou, S.; Zhang, Z.; Zheng, R.; Huang, J. Engineering Nanostructure–Interface of Photoanode Materials Toward Photoelectrochemical Water Oxidation. Adv. Mater. 2021, 33, 2005389. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Qi, J.; Yang, N.; Cui, W.; Wang, J.; Li, Q.; Zhang, Q.; Yu, X.; Gu, L.; Li, J.; et al. Dual-Defects Adjusted Crystal-Field Splitting of LaCo1−xNixO3−δ Hollow Multishelled Structures for Efficient Oxygen Evolution. Angew. Chem. Int. Ed. 2020, 59, 19691–19695. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Liang, Z.; Zou, R. Designing Advanced Catalysts for Energy Conversion Based on Urea Oxidation Reaction. Small 2020, 16, 1906133. [Google Scholar] [CrossRef]
- Zhu, D.; Zhang, H.; Miao, J.; Hu, F.; Wang, L.; Tang, Y.; Qiao, M.; Guo, C. Strategies for designing more efficient electrocatalysts towards the urea oxidation reaction. J. Mater. Chem. A 2022, 10, 3296–3313. [Google Scholar] [CrossRef]
- Radjenovic, J.; Sedlak, D.L. Challenges and Opportunities for Electrochemical Processes as Next-Generation Technologies for the Treatment of Contaminated Water. Environ. Sci. Technol. 2015, 49, 11292–11302. [Google Scholar] [CrossRef]
- Altalhi, T.A.; Ibrahim, M.M.; Mersal, G.A.M.; Mahmoud, M.H.H.; Kumeria, T.; El-Desouky, M.G.; El-Bindary, A.A.; El-Bindary, M.A. Adsorption of doxorubicin hydrochloride onto thermally treated green adsorbent: Equilibrium, kinetic and thermodynamic studies. J. Mol. Struct. 2022, 1263, 133160. [Google Scholar] [CrossRef]
- Kundu, B.K.; Pragti; Biswas, S.; Mondal, A.; Mazumdar, S.; Mobin, S.M.; Mukhopadhyay, S. Unveiling the urease like intrinsic catalytic activities of two dinuclear nickel complexes towards the in situ syntheses of aminocyanopyridines. Dalton Trans. 2021, 50, 4848–4858. [Google Scholar] [CrossRef]
- Vedharathinam, V.; Botte, G.G. Understanding the electro-catalytic oxidation mechanism of urea on nickel electrodes in alkaline medium. Electrochim. Acta 2012, 81, 292–300. [Google Scholar] [CrossRef]
- Vedharathinam, V.; Botte, G.G. Direct evidence of the mechanism for the electro-oxidation of urea on Ni(OH)2 catalyst in alkaline medium. Electrochim. Acta 2013, 108, 660–665. [Google Scholar] [CrossRef]
- El-Bindary, M.A.; El-Desouky, M.G.; El-Bindary, A.A. Metal–organic frameworks encapsulated with an anticancer compound as drug delivery system: Synthesis, characterization, antioxidant, anticancer, antibacterial, and molecular docking investigation. Appl. Organomet. Chem. 2022, 36, e6660. [Google Scholar] [CrossRef]
- Zhu, D.; Guo, C.; Liu, J.; Wang, L.; Du, Y.; Qiao, S.-Z. Two-dimensional metal–organic frameworks with high oxidation states for efficient electrocatalytic urea oxidation. Chem. Commun. 2017, 53, 10906–10909. [Google Scholar] [CrossRef] [PubMed]
- Maruthapandian, V.; Kumaraguru, S.; Mohan, S.; Saraswathy, V.; Muralidharan, S. An Insight on the Electrocatalytic Mechanistic Study of Pristine Ni MOF (BTC) in Alkaline Medium for Enhanced OER and UOR. ChemElectroChem 2018, 5, 2795–2807. [Google Scholar] [CrossRef]
- Xu, Y.; Chai, X.; Ren, T.; Yu, S.; Yu, H.; Wang, Z.; Li, X.; Wang, L.; Wang, H. Ir-Doped Ni-based metal–organic framework ultrathin nanosheets on Ni foam for enhanced urea electro-oxidation. Chem. Commun. 2020, 56, 2151–2154. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Fang, X.; Zhu, K.; Yuan, W.; Jiang, T.; Xue, H.; Tian, J. Fe-doping induced electronic structure reconstruction in Ni-based metal-organic framework for improved energy-saving hydrogen production via urea degradation. J. Power Sources 2022, 520, 230882. [Google Scholar] [CrossRef]
- Li, M.; Sun, H.; Yang, J.; Humayun, M.; Li, L.; Xu, X.; Xue, X.; Habibi-Yangjeh, A.; Temst, K.; Wang, C. Mono-coordinated metallocene ligands endow metal-organic frameworks with highly efficient oxygen evolution and urea electrolysis. Chem. Eng. J. 2022, 430, 132733. [Google Scholar] [CrossRef]
- Yuan, M.; Wang, R.; Sun, Z.; Lin, L.; Yang, H.; Li, H.; Nan, C.; Sun, G.; Ma, S. Morphology-Controlled Synthesis of Ni-MOFs with Highly Enhanced Electrocatalytic Performance for Urea Oxidation. Inorg. Chem. 2019, 58, 11449–11457. [Google Scholar] [CrossRef]
- Hu, S.; Wang, S.; Feng, C.; Wu, H.; Zhang, J.; Mei, H. Novel MOF-Derived Nickel Nitride as High-Performance Bifunctional Electrocatalysts for Hydrogen Evolution and Urea Oxidation. ACS Sustain. Chem. Eng. 2020, 8, 7414–7422. [Google Scholar] [CrossRef]
- Xu, H.; Ye, K.; Zhu, K.; Gao, Y.; Yin, J.; Yan, J.; Wang, G.; Cao, D. Transforming Carnation-Shaped MOF-Ni to Ni–Fe Prussian Blue Analogue Derived Efficient Bifunctional Electrocatalyst for Urea Electrolysis. ACS Sustain. Chem. Eng. 2020, 8, 16037–16045. [Google Scholar] [CrossRef]
- Li, M.; Ao, X.; Li, J.-G.; Sun, H.; Zheng, L.; Wang, C. NiCo-BDC nanosheets coated with amorphous Ni-S thin film for high-efficiency oxygen evolution reaction and urea oxidation reaction. FlatChem 2021, 25, 100222. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, C.; Shang, H.; Yuan, M.; Wu, Z.; Li, J.; Du, Y. Self-driven Ru-modified NiFe MOF nanosheet as multifunctional electrocatalyst for boosting water and urea electrolysis. J. Colloid Interface Sci. 2022, 605, 779–789. [Google Scholar] [CrossRef]
- Wang, X.; Xie, J.; Li, C.M. Architecting smart “umbrella” Bi2S3/rGO-modified TiO2 nanorod array structures at the nanoscale for efficient photoelectrocatalysis under visible light. J. Mater. Chem. A 2015, 3, 1235–1242. [Google Scholar] [CrossRef]
- Cho, I.S.; Chen, Z.; Forman, A.J.; Kim, D.R.; Rao, P.M.; Jaramillo, T.F.; Zheng, X. Branched TiO2 Nanorods for Photoelectrochemical Hydrogen Production. Nano Lett. 2011, 11, 4978–4984. [Google Scholar] [CrossRef] [PubMed]
- Challagulla, S.; Tarafder, K.; Ganesan, R.; Roy, S. Structure sensitive photocatalytic reduction of nitroarenes over TiO2. Sci. Rep. 2017, 7, 8783. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Zhao, M.; Zhou, J.; Li, W.; Wang, H.; Xu, Z.; Lu, L.; Pei, L.; Shi, Z.; Yan, S.; et al. Surface states as electron transfer pathway enhanced charge separation in TiO2 nanotube water splitting photoanodes. Appl. Catal. B Environ. 2018, 234, 100–108. [Google Scholar] [CrossRef]
- Carton, A.; Mesbah, A.; Mazet, T.; Porcher, F.; François, M. Ab initio crystal structure of nickel(II) hydroxy-terephthalate by synchrotron powder diffraction and magnetic study. Solid State Sci. 2007, 9, 465–471. [Google Scholar] [CrossRef]
- Wang, G.; Ling, Y.; Lu, X.; Wang, H.; Qian, F.; Tong, Y.; Li, Y. Solar driven hydrogen releasing from urea and human urine. Energy Environ. Sci. 2012, 5, 8215–8219. [Google Scholar] [CrossRef]
- Yang, Y.; Ling, Y.; Wang, G.; Liu, T.; Wang, F.; Zhai, T.; Tong, Y.; Li, Y. Photohole Induced Corrosion of Titanium Dioxide: Mechanism and Solutions. Nano Lett. 2015, 15, 7051–7057. [Google Scholar] [CrossRef]
- Wang, L.; Zhu, S.; Marinkovic, N.; Kattel, S.; Shao, M.; Yang, B.; Chen, J.G. Insight into the synergistic effect between nickel and tungsten carbide for catalyzing urea electrooxidation in alkaline electrolyte. Appl. Catal. B Environ. 2018, 232, 365–370. [Google Scholar] [CrossRef] [Green Version]
- Safeer, N.K.M.; Alex, C.; Jana, R.; Datta, A.; John, N.S. Remarkable COx tolerance of Ni3+ active species in a Ni2O3 catalyst for sustained electrochemical urea oxidation. J. Mater. Chem. A 2022, 10, 4209–4221. [Google Scholar] [CrossRef]
- Trotochaud, L.; Young, S.L.; Ranney, J.K.; Boettcher, S.W. Nickel–Iron Oxyhydroxide Oxygen-Evolution Electrocatalysts: The Role of Intentional and Incidental Iron Incorporation. J. Am. Chem. Soc. 2014, 136, 6744–6753. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Huang, K.; Wu, X.; Wang, M.; Feng, S. Surface polarization enables high charge separation in TiO2 nanorod photoanode. Nano Res. 2021, 14, 4056–4062. [Google Scholar] [CrossRef]
- Zheng, W.; Liu, M.; Lee, L.Y.S. Electrochemical Instability of Metal–Organic Frameworks: In Situ Spectroelectrochemical Investigation of the Real Active Sites. ACS Catal. 2020, 10, 81–92. [Google Scholar] [CrossRef]
- Yan, P.; Liu, G.; Ding, C.; Han, H.; Shi, J.; Gan, Y.; Li, C. Photoelectrochemical Water Splitting Promoted with a Disordered Surface Layer Created by Electrochemical Reduction. ACS Appl. Mater. Interfaces 2015, 7, 3791–3796. [Google Scholar] [CrossRef]
- Dai, W.; Zhou, J.; Bian, Y.; Hao, Z.; Cao, Y.; Xiao, J.; Gou, H.; Gao, F. A universal synthesis of MOF-Hydroxyl for highly active oxygen evolution. J. Colloid Interface Sci. 2022, 623, 318–326. [Google Scholar] [CrossRef]
- Tran, T.Q.N.; Das, G.; Yoon, H.H. Nickel-metal organic framework/MWCNT composite electrode for non-enzymatic urea detection. Sens. Actuators B Chem. 2017, 243, 78–83. [Google Scholar] [CrossRef]
- Cao, Q.; Yuan, Y.; Wang, K.; Huang, W.; Zhao, Y.; Sun, X.; Ding, R.; Lin, W.; Liu, E.; Gao, P. Phase and crystallinity regulations of Ni(OH)2 by vanadium doping boost electrocatalytic urea oxidation reaction. J. Colloid Interface Sci. 2022, 618, 411–418. [Google Scholar] [CrossRef]
- Gao, R.-T.; Wang, L. Stable Cocatalyst-Free BiVO4 Photoanodes with Passivated Surface States for Photocorrosion Inhibition. Angew. Chem. Int. Ed. 2020, 59, 23094–23099. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, L.; Lin, H.; Liu, Y.; Ye, J.; Wen, Y.; Chen, A.; Wang, L.; Ni, F.; Zhou, Z.; et al. A Lattice-Oxygen-Involved Reaction Pathway to Boost Urea Oxidation. Angew. Chem. Int. Ed. 2019, 58, 16820–16825. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, J.; Yang, P.; Wang, L.; Chen, H.; Lu, X.; Yang, Q.; Zou, L.; Huang, C.; Xie, J. In Situ Transition of a Nickel Metal–Organic Framework on TiO2 Photoanode towards Urea Photoelectrolysis. Catalysts 2023, 13, 727. https://doi.org/10.3390/catal13040727
Ren J, Yang P, Wang L, Chen H, Lu X, Yang Q, Zou L, Huang C, Xie J. In Situ Transition of a Nickel Metal–Organic Framework on TiO2 Photoanode towards Urea Photoelectrolysis. Catalysts. 2023; 13(4):727. https://doi.org/10.3390/catal13040727
Chicago/Turabian StyleRen, Jie, Pingping Yang, Liuliu Wang, Hongyu Chen, Xingyu Lu, Qing Yang, Li Zou, Cheng Huang, and Jiale Xie. 2023. "In Situ Transition of a Nickel Metal–Organic Framework on TiO2 Photoanode towards Urea Photoelectrolysis" Catalysts 13, no. 4: 727. https://doi.org/10.3390/catal13040727