
Hybrid Materials Based on Imidazo[4,5-b]porphyrins for Catalytic Oxidation of Sulfides
 , and
, and
Abstract
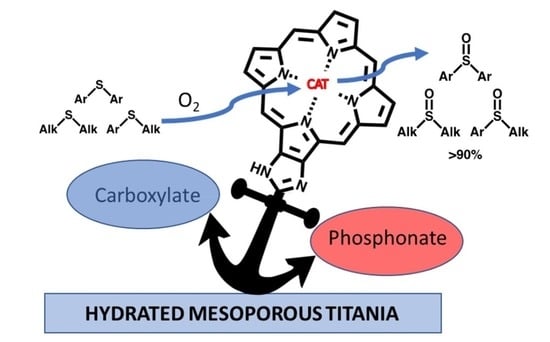
:
1. Introduction
2. Results and Discussion
2.1. Synthesis of (2-arylimidazo[4,5-b]porphyrinato)manganese(III) Chlorides and Their Characterization
2.2. Immobilization of Manganese(III) Complexes Mn(TMPIC) and Mn(TMPIP) on TiO2
2.3. Catalytic Reactions
3. Materials and Methods
3.1. General
3.2. Synthesis of Manganese(III) Complexes Mn(TMPIC), Mn(TMPIP) and Mn(TMPIP-OH)
3.3. Synthesis of Heterogenized Catalysts
3.4. Catalytic Reactions
3.5. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lu, W.; Zhou, L. Oxidation of C–H Bonds; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2017; p. 496. [Google Scholar]
- Clerici, M.G.; Kholdeeva, O.A. Liquid Phase Oxidation via Heterogeneous Catalysis: Organic Synthesis and Industrial Applications; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013. [Google Scholar]
- Teles, J.H.; Hermans, I.; Franz, G.; Sheldon, R.A. Oxidation. In Ullmann’s Encyclopedia of Industrial Chemistry; Verlag Chemie: Hoboken, NJ, USA, 2015; pp. 1–103. [Google Scholar]
- Mejia, E. (Ed.) Catalytic Aerobic Oxidations; The Royal Society of Chemistry: Cambridge, UK, 2020; p. 334. [Google Scholar]
- Punniyamurthy, T.; Velusamy, S.; Iqbal, J. Recent advances in transition metal catalyzed oxidation of organic Substrates with molecular oxygen. Chem. Rev. 2005, 105, 2329–2364. [Google Scholar] [CrossRef]
- Wang, D.; Weinstein, A.B.; White, P.B.; Stahl, S.S. Ligand-promoted palladium-catalyzed aerobic oxidation reactions. Chem. Rev. 2018, 118, 2636–2679. [Google Scholar] [CrossRef] [PubMed]
- Mansuy, D. A brief history of the contribution of metalloporphyrin models to cytochrome P450 chemistry and oxidation catalysis. Comptes Rendus Chim. 2007, 10, 392–413. [Google Scholar] [CrossRef]
- Pereira, M.M.; Dias, L.D.; Calvete, M.J.F. Metalloporphyrins: Bioinspired oxidation catalysts. ACS Catal. 2018, 8, 10784–10808. [Google Scholar] [CrossRef]
- Groves, J.T.; McClusky, G.A. Aliphatic hydroxylation via oxygen rebound. Oxygen transfer catalyzed by iron. J. Am. Chem. Soc. 1976, 98, 859–861. [Google Scholar] [CrossRef]
- Groves, J.T.; Nemo, T.E.; Myers, R.S. Hydroxylation and epoxidation catalyzed by iron-porphine complexes. Oxygen transfer from iodosylbenzene. J. Am. Chem. Soc. 1979, 101, 1032–1033. [Google Scholar] [CrossRef]
- Liu, W.; Groves, J.T. Manganese catalyzed C–H Halogenation. Acc. Chem. Res. 2015, 48, 1727–1735. [Google Scholar] [CrossRef]
- Haber, J.; Mlodnicka, T.; Poltowicz, J. Metal-dependent reactivity of some metalloporphyrins in oxidation with dioxygen. J. Mol. Catal. 1989, 54, 451–461. [Google Scholar] [CrossRef]
- Lu, W.Y.; Bartoli, J.F.; Battioni, P.; Mansuy, D. Selective oxygenation of hydrocarbons and sulfoxidation of thioethers by dioxygen with a Mn-porphyrin-based cytochrome P450 model system using zinc as electron donor. New J. Chem. 1992, 16, 621–628. [Google Scholar] [CrossRef]
- Guo, C.-C.; Yang, W.-J.; Mao, Y.-L. Selectively aerobic oxidation of CC and allylic CH bonds in α-pinene over simple metalloporphyrins. J. Mol. Catal. A Chem. 2005, 226, 279–284. [Google Scholar] [CrossRef]
- Liu, C.; Shen, D.-M.; Chen, Q.-Y. Fluorous biphasic catalytic oxidation of alkenes and aldehydes with air and 2-methylpropanal in the presence of (β-perfluoroalkylated tetraphenylporphyrin)cobalt complexes. Eur. J. Org. Chem. 2006, 2006, 2703–2706. [Google Scholar] [CrossRef]
- Ji, H.-B.; Yuan, Q.-L.; Zhou, X.-T.; Pei, L.-X.; Wang, L.-F. Highly efficient selective oxidation of alcohols to carbonyl compounds catalyzed by ruthenium (III) meso-tetraphenylporphyrin chloride in the presence of molecular oxygen. Bioorg. Med. Chem. Lett. 2007, 17, 6364–6368. [Google Scholar] [CrossRef] [PubMed]
- Meunier, B. Metalloporphyrins as versatile catalysts for oxidation reactions and oxidative DNA cleavage. Chem. Rev. 1992, 92, 1411–1456. [Google Scholar] [CrossRef]
- Mansuy, D. Activation of alkanes: The biomimetic approach. Coord. Chem. Rev. 1993, 125, 129–141. [Google Scholar] [CrossRef]
- Dolphin, D.; Traylor, T.G.; Xie, L.Y. Polyhaloporphyrins: Unusual ligands for metals and metal-catalyzed oxidations. Acc. Chem. Res. 1997, 30, 251–259. [Google Scholar] [CrossRef]
- van der Made, A.W.; Smeets, J.W.H.; Nolte, R.J.M.; Drenth, W. Olefin epoxidation by a mono-oxygenase model. Effect of site isolation. J. Chem. Soc. Chem. Commun. 1983, 1204–1206. [Google Scholar] [CrossRef]
- Nakagaki, S.; Ferreira, G.K.B.; Ucoski, G.M.; Dias de Freitas Castro, K.A. Chemical reactions catalyzed by metalloporphyrin-based metal-organic frameworks. Molecules 2013, 18, 7279–7308. [Google Scholar] [CrossRef]
- Takagi, S.; Eguchi, M.; Tryk, D.A.; Inoue, H. Porphyrin photochemistry in inorganic/organic hybrid materials: Clays, layered semiconductors, nanotubes, and mesoporous materials. J. Photochem. Photobiol. C Photochem. Rev. 2006, 7, 104–126. [Google Scholar] [CrossRef]
- Santos, J.S.D.; Faria, A.L.; Amorin, P.M.D.S.; Luna, F.M.L.; Caiado, K.L.; Silva, D.O.C.E.; Sartoratto, P.P.C.; Assis, M.D. Iron(III) porphyrin covalently supported onto magnetic amino-functionalized nanospheres as catalyst for hydrocarbon and herbicide oxidations. J. Braz. Chem. Soc. 2012, 23, 1411–1420. [Google Scholar] [CrossRef]
- de Lima, O.J.; de Aguirre, D.P.; de Oliveira, D.C.; da Silva, M.A.; Mello, C.; Leite, C.A.P.; Sacco, H.C.; Ciuffi, K.J. Porphyrins entrapped in an alumina matrix. J. Mater. Chem. 2001, 11, 2476–2481. [Google Scholar] [CrossRef] [Green Version]
- Rayati, S.; Nejabat, F. Catalytic activity of Fe-porphyrins grafted on multiwalled carbon nanotubes in the heterogeneous oxidation of sulfides and degradation of phenols in water. Comptes Rendus Chim. 2017, 20, 967–974. [Google Scholar] [CrossRef]
- Chino, M.; Leone, L.; Zambrano, G.; Pirro, F.; D’Alonzo, D.; Firpo, V.; Aref, D.; Lista, L.; Maglio, O.; Nastri, F.; et al. Oxidation catalysis by iron and manganese porphyrins within enzyme-like cages. Biopolymers 2018, 109, e23107. [Google Scholar] [CrossRef]
- Neves, C.M.B.; Rebelo, S.L.H.; Faustino, M.A.F.; Neves, M.G.P.M.S.; Simões, M.M.Q. Second-generation manganese(III) porphyrins bearing 3,5-dichloropyridyl units: Innovative homogeneous and heterogeneous catalysts for the epoxidation of alkenes. Catalysts 2019, 9, 967. [Google Scholar] [CrossRef]
- Castro, K.A.D.F.; Westrup, K.C.M.; Silva, S.; Pereira, P.M.R.; Simões, M.M.Q.; Neves, M.D.G.P.; Cavaleiro, J.A.S.; Tomé, J.P.C.; Nakagaki, S. Iron(III) Complexation with galactodendritic porphyrin species and hydrocarbons’ oxidative transformations. Eur. J. Inorg. Chem. 2021, 2021, 2857–2869. [Google Scholar] [CrossRef]
- Qodrati-nasrabadi, F.; Sardivand-chegini, I.; Heydari-turkmani, A.; Zakavi, S. Acid-induced improvement of photosensitizing efficiency of porphyrins under heterogeneous aqueous conditions: High photosensitizing activity, dissociative stability and photooxidative durability. J. Environ. Chem. Eng. 2023, 11, 109022. [Google Scholar] [CrossRef]
- Bedioui, F. Zeolite-encapsulated and clay-intercalated metal porphyrin, phthalocyanine and Schiff-base complexes as models for biomimetic oxidation catalysts: An overview. Coord. Chem. Rev. 1995, 144, 39–68. [Google Scholar] [CrossRef]
- Nakagaki, S.; Xavier, C.R.; Wosniak, A.J.; Mangrich, A.S.; Wypych, F.; Cantão, M.P.; Denicoló, I.; Kubota, L.T. Synthesis and characterization of zeolite-encapsulated metalloporphyrins. Colloids Surf. A 2000, 168, 261–276. [Google Scholar] [CrossRef]
- Nakagaki, S.; Halma, M.; Bail, A.; Arízaga, G.G.; Wypych, F. First insight into catalytic activity of anionic iron porphyrins immobilized on exfoliated layered double hydroxides. J. Colloid Interface Sci. 2005, 281, 417–423. [Google Scholar] [CrossRef]
- Zhou, X.; Ji, H. Manganese porphyrin immobilized on montmorillonite: A highly efficient and reusable catalyst for the aerobic epoxidation of olefins under ambient conditions. J. Porphyr. Phthalocyanines 2012, 16, 1032–1039. [Google Scholar] [CrossRef]
- Zhou, X.-T.; Ji, H.-B. Highly efficient selective oxidation of sulfides to sulfoxides by montmorillonite-immobilized metalloporphyrins in the presence of molecular oxygen. Catal. Commun. 2014, 53, 29–32. [Google Scholar] [CrossRef]
- Nakagaki, S.; Mantovani, K.M.; Sippel Machado, G.; Dias de Freitas Castro, K.A.; Wypych, F. Recent advances in solid catalysts obtained by metalloporphyrins immobilization on layered anionic exchangers: A short review and some new catalytic results. Molecules 2016, 21, 291. [Google Scholar] [CrossRef]
- Lindsay Smith, J.R.; Iamamoto, Y.; Vinhado, F.S. Oxidation of alkanes by iodosylbenzene (PhIO) catalysed by supported Mn(III) porphyrins: Activity and mechanism. J. Mol. Catal. A Chem. 2006, 252, 23–30. [Google Scholar] [CrossRef]
- Vinhado, F.S.; Prado-Manso, C.M.C.; Sacco, H.C.; Iamamoto, Y. Cationic manganese(III) porphyrins bound to a novel bis-functionalised silica as catalysts for hydrocarbons oxygenation by iodosylbenzene and hydrogen peroxide. J. Mol. Catal. A Chem. 2001, 174, 279–288. [Google Scholar] [CrossRef]
- Aparecida Vidoto, E.; Silvia Monsalves Moreira, M.; da Silva Vinhado, F.; Jorge Ciuffi, K.; Rangel Nascimento, O.; Iamamoto, Y. Immobilization of β halogenated ironporphyrin in the silica matrix by the sol–gel process. J. Non-Cryst. Solids 2002, 304, 151–159. [Google Scholar] [CrossRef]
- Ucoski, G.M.; Nunes, F.S.; DeFreitas-Silva, G.; Idemori, Y.M.; Nakagaki, S. Metalloporphyrins immobilized on silica-coated Fe3O4 nanoparticles: Magnetically recoverable catalysts for the oxidation of organic substrates. Appl. Catal. A 2013, 459, 121–130. [Google Scholar] [CrossRef]
- Gao, B.; Chen, Y.; Lei, Q. Hydroxylation of cyclohexane with molecular oxygen catalyzed by highly efficient heterogeneous Mn(III) porphyrin catalysts prepared by special synthesis and immobilization method. J. Incl. Phenom. Macrocycl. Chem. 2012, 74, 455–465. [Google Scholar] [CrossRef]
- Alkordi, M.H.; Liu, Y.; Larsen, R.W.; Eubank, J.F.; Eddaoudi, M. Zeolite-like metal−organic frameworks as platforms for applications: On metalloporphyrin-based catalysts. J. Am. Chem. Soc. 2008, 130, 12639–12641. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Qiu, W.; Long, W.; Deng, F.; Bai, G.; Zhang, G.; Zi, X.; He, H. Synthesis of porphyrin@MOFs type catalysts through “one-pot” self-assembly. J. Mol. Catal. A Chem. 2014, 393, 166–170. [Google Scholar] [CrossRef]
- Beyzavi, H.; Vermeulen, N.A.; Howarth, A.J.; Tussupbayev, S.; League, A.B.; Schweitzer, N.M.; Gallagher, J.R.; Platero-Prats, A.E.; Hafezi, N.; Sarjeant, A.A.; et al. A Hafnium-based metal–organic framework as a nature-inspired tandem reaction catalyst. J. Am. Chem. Soc. 2015, 137, 13624–13631. [Google Scholar] [CrossRef]
- Feng, L.; Wang, Y.; Yuan, S.; Wang, K.-Y.; Li, J.-L.; Day, G.S.; Qiu, D.; Cheng, L.; Chen, W.-M.; Madrahimov, S.T.; et al. Porphyrinic metal–organic frameworks installed with Brønsted acid sites for efficient tandem semisynthesis of artemisinin. ACS Catal. 2019, 9, 5111–5118. [Google Scholar] [CrossRef]
- Chen, L.; Yang, Y.; Jiang, D. CMPs as scaffolds for constructing porous catalytic frameworks: A built-in heterogeneous catalyst with high activity and selectivity based on nanoporous metalloporphyrin polymers. J. Am. Chem. Soc. 2010, 132, 9138–9143. [Google Scholar] [CrossRef] [PubMed]
- Amadelli, R.; Bregola, M.; Polo, E.; Carassiti, V.; Maldotti, A. Photooxidation of hydrocarbons on porphyrin-modified titanium dioxide powders. J. Chem. Soc. Chem. Commun. 1992, 1355–1357. [Google Scholar] [CrossRef]
- Mantovani, K.M.; Stival, J.F.; Wypych, F.; Bach, L.; Peralta Zamora, P.G.; Luiza Rocco, M.; Nakagaki, S. Unusual catalytic activity after simultaneous immobilization of two metalloporphyrins on hydrozincite/nanocrystalline anatase. J. Catal. 2017, 352, 442–451. [Google Scholar] [CrossRef]
- Machado, G.S.; Ucoski, G.M.; de Lima, O.J.; Ciuffi, K.J.; Wypych, F.; Nakagaki, S. Cationic and anionic metalloporphyrins simultaneously immobilized onto raw halloysite nanoscrolls catalyze oxidation reactions. Appl. Catal. A 2013, 460–461, 124–131. [Google Scholar] [CrossRef]
- Machado, G.S.; Arízaga, G.G.C.; Wypych, F.; Nakagaki, S. Immobilization of anionic metalloporphyrins on zinc hydroxide nitrate and study of an unusual catalytic activity. J. Catal. 2010, 274, 130–141. [Google Scholar] [CrossRef]
- Perego, C.; Millini, R. Porous materials in catalysis: Challenges for mesoporous materials. Chem. Soc. Rev. 2013, 42, 3956–3976. [Google Scholar] [CrossRef] [PubMed]
- Suib, S.L.; Přech, J.; Čejka, J.; Kuwahara, Y.; Mori, K.; Yamashita, H. Some novel porous materials for selective catalytic oxidations. Mater. Today 2020, 32, 244–259. [Google Scholar] [CrossRef]
- El Mourabit, S.; Guillot, M.; Toquer, G.; Cambedouzou, J.; Goettmann, F.; Grandjean, A. Stability of mesoporous silica under acidic conditions. RSC Adv. 2012, 2, 10916–10924. [Google Scholar] [CrossRef]
- Yang, S.-A.; Choi, S.; Jeon, S.M.; Yu, J. Silica nanoparticle stability in biological media revisited. Sci. Rep. 2018, 8, 185. [Google Scholar] [CrossRef]
- Pereira, C.F.; Simões, M.M.Q.; Tomé, J.P.C.; Almeida Paz, F.A. Porphyrin-Based metal-organic frameworks as heterogeneous catalysts in oxidation reactions. Molecules 2016, 21, 1348. [Google Scholar] [CrossRef] [Green Version]
- Yakushev, A.A.; Abel, A.S.; Averin, A.D.; Beletskaya, I.P.; Cheprakov, A.V.; Ziankou, I.S.; Bonneviot, L.; Bessmertnykh-Lemeune, A. Visible light photocatalysis promoted by solid- and liquid-phase immobilized transition metal complexes in organic synthesis. Coord. Chem. Rev. 2022, 458, 214331. [Google Scholar] [CrossRef]
- Crossley, M.J.; McDonald, J.A. Fused porphyrin-imidazole systems: New building blocks for synthesis of porphyrin arrays. J. Chem. Soc. Perkin Trans. 1 1999, 17, 2429–2431. [Google Scholar] [CrossRef]
- Abdulaeva, I.A.; Birin, K.P.; Bessmertnykh-Lemeune, A.; Tsivadze, A.Y.; Gorbunova, Y.G. Heterocycle-appended porphyrins: Synthesis and challenges. Coord. Chem. Rev. 2020, 407, 213108. [Google Scholar] [CrossRef]
- Abdulaeva, I.A.; Birin, K.P.; Michalak, J.; Romieu, A.; Stern, C.; Bessmertnykh-Lemeune, A.; Guilard, R.; Gorbunova, Y.G.; Tsivadze, A.Y. On the synthesis of functionalized porphyrins and porphyrin conjugates via β-aminoporphyrins. New J. Chem. 2016, 40, 5758–5774. [Google Scholar] [CrossRef]
- Barona-Castaño, J.C.; Carmona-Vargas, C.C.; Brocksom, T.J.; De Oliveira, K.T. Porphyrins as catalysts in scalable organic reactions. Molecules 2016, 21, 310. [Google Scholar] [CrossRef]
- Adler, A.D.; Longo, F.R.; Kampas, F.; Kim, J. On the preparation of metalloporphyrins. J. Inorg. Nucl. Chem. 1970, 32, 2443–2445. [Google Scholar] [CrossRef]
- Boucher, L.J. Manganese porphyrin complexes. Coord. Chem. Rev. 1972, 7, 289–329. [Google Scholar] [CrossRef]
- Fagadar-Cosma, E.; Mirica, M.C.; Balcu, I.; Bucovicean, C.; Cretu, C.; Armeanu, I.; Fagadar-Cosma, G. Syntheses, spectroscopic and AFM characterization of some manganese porphyrins and their hybrid silica nanomaterials. Molecules 2009, 14, 1370–1388. [Google Scholar] [CrossRef]
- Queffélec, C.; Petit, M.; Janvier, P.; Knight, D.A.; Bujoli, B. Surface modification using phosphonic acids and esters. Chem. Rev. 2012, 112, 3777–3807. [Google Scholar] [CrossRef]
- Guerrero, G.; Alauzun, J.G.; Granier, M.; Laurencin, D.; Mutin, P.H. Phosphonate coupling molecules for the control of surface/interface properties and the synthesis of nanomaterials. Dalton Trans. 2013, 42, 12569–12585. [Google Scholar] [CrossRef]
- Zhu, Y.-P.; Ma, T.-Y.; Liu, Y.-L.; Ren, T.-Z.; Yuan, Z.-Y. Metal phosphonate hybrid materials: From densely layered to hierarchically nanoporous structures. Inorg. Chem. Front. 2014, 1, 360–383. [Google Scholar] [CrossRef]
- Cattani-Scholz, A. Functional organophosphonate interfaces for nanotechnology: A review. ACS Appl. Mater. Interfaces 2017, 9, 25643–25655. [Google Scholar] [CrossRef]
- Maillet, C.; Janvier, P.; Pipelier, M.; Praveen, T.; Andres, Y.; Bujoli, B. Hybrid materials for catalysis? Design of new phosphonate-based supported catalysts for the hydrogenation of ketones under hydrogen pressure. Chem. Mater. 2001, 13, 2879–2884. [Google Scholar] [CrossRef]
- Maillet, C.; Janvier, P.; Bertrand, M.-J.; Praveen, T.; Bujoli, B. Phosphonate-based hybrid materials for catalysis? Supported rhodium/2,2′-bipyridine complexes as reduction catalysts under hydrogen pressure. Eur. J. Org. Chem. 2002, 2002, 1685–1689. [Google Scholar] [CrossRef]
- Schull, T.L.; Henley, L.; Deschamps, J.R.; Butcher, R.J.; Maher, D.P.; Klug, C.A.; Swider-Lyons, K.; Dressick, W.J.; Bujoli, B.; Greenwood, A.E.; et al. Organometallic supramolecular mixed-valence cobalt(I)/cobalt(II) aquo complexes stabilized with the water-soluble phosphine ligand p-TPPTP (p-triphenylphosphine triphosphonic acid). Organometallics 2007, 26, 2272–2276. [Google Scholar] [CrossRef]
- Guerrero, G.; Mutin, P.H.; Framery, E.; Vioux, A. Immobilization of platinum(II) and palladium(II) complexes on metal oxides by sol-gel processing and surface modification using bifunctional phosphine-phosphonate esters. New J. Chem. 2008, 32, 1519–1525. [Google Scholar] [CrossRef]
- Forato, F.; Belhboub, A.; Monot, J.; Petit, M.; Benoit, R.; Sarou-Kanian, V.; Fayon, F.; Jacquemin, D.; Queffelec, C.; Bujoli, B. Phosphonate-mediated immobilization of rhodium/bipyridine hydrogenation catalysts. Chem.-Eur. J. 2018, 24, 2457–2465. [Google Scholar] [CrossRef]
- Forato, F.; Talebzadeh, S.; Bujoli, B.; Queffélec, C.; Trammell, S.A.; Knight, D.A. Core-Shell Ag@TiO2 nanocomposites for low-power blue laser enhanced copper(I) catalyzed Ullmann coupling. ChemistrySelect 2017, 2, 769–773. [Google Scholar] [CrossRef]
- Zhu, Y.-P.; Ren, T.-Z.; Yuan, Z.-Y. Insights into mesoporous metal phosphonate hybrid materials for catalysis. Catal. Sci. Technol. 2015, 5, 4258–4279. [Google Scholar] [CrossRef]
- Bhanja, P.; Bhaumik, A. Organic–inorganic hybrid metal phosphonates as recyclable heterogeneous catalysts. ChemCatChem 2016, 8, 1607–1616. [Google Scholar] [CrossRef]
- Zhang, L.; Cole, J.M. Anchoring groups for dye-sensitized solar cells. ACS Appl. Mater. Interfaces 2015, 7, 3427–3455. [Google Scholar] [CrossRef]
- Mitrofanov, A.; Brandes, S.; Herbst, F.; Rigolet, S.; Bessmertnykh-Lemeune, A.; Beletskaya, I. Immobilization of copper complexes with (1,10-phenanthrolinyl)phosphonates on titania supports for sustainable catalysis. J. Mater. Chem. A 2017, 5, 12216–12235. [Google Scholar] [CrossRef]
- DiGiacomo, P.M.; Dines, M.B. Nonaqueous Preparation of Layered or Amorphous Organometallic Inorganic Polymers. U.S. Patent 4299943, 10 November 1981. [Google Scholar]
- Guerrero, G.; Mutin, P.H.; Vioux, A. Anchoring of phosphonate and phosphinate coupling molecules on titania particles. Chem. Mater. 2001, 13, 4367–4373. [Google Scholar] [CrossRef]
- Kamm, J.M.; Iverson, C.P.; Lau, W.-Y.; Hopkins, M.D. Axial ligand effects on the structures of self-assembled gallium–porphyrin monolayers on highly oriented pyrolytic graphite. Langmuir 2016, 32, 487–495. [Google Scholar] [CrossRef]
- Yamada, T.; Takai, T.; Rhode, O.; Mukaiyama, T. Highly efficient method for epoxidation of olefms with molecular oxygen and aldehydes catalyzed by nickel(II) complexes. Chem. Lett. 1991, 20, 1–4. [Google Scholar] [CrossRef]
- Murahashi, S.-I.; Naota, T.; Komiya, N. Metalloporphyrin-catalyzed oxidation of alkanes with molecular oxygen in the presence of acetaldehyde. Tetrahedron Lett. 1995, 36, 8059–8062. [Google Scholar] [CrossRef]
- Mastrorilli, P.; Nobile, C.F. Catalytic activity of a polymerizable tris(β-ketoesterate)iron(III) complex towards the oxidation of organic substrates. Tetrahedron Lett. 1994, 35, 4193–4196. [Google Scholar] [CrossRef]
- Brown, J.W.; Nguyen, Q.T.; Otto, T.; Jarenwattananon, N.N.; Glöggler, S.; Bouchard, L.-S. Epoxidation of alkenes with molecular oxygen catalyzed by a manganese porphyrin-based metal–organic framework. Catal. Commun. 2015, 59, 50–54. [Google Scholar] [CrossRef]
- Malko, D.; Guo, Y.; Jones, P.; Britovsek, G.; Kucernak, A. Heterogeneous iron containing carbon catalyst (Fe-N/C) for epoxidation with molecular oxygen. J. Catal. 2019, 370, 357–363. [Google Scholar] [CrossRef]
- Gong, M.; Guo, Y.; Malko, D.; Rubio-Garcia, J.; Dawson, J.M.S.; Britovsek, G.J.P.; Kucernak, A. Using molecular oxygen and Fe–N/C heterogeneous catalysts to achieve Mukaiyama epoxidations via in situ produced organic peroxy acids and acylperoxy radicals. Catal. Sci. Technol. 2022, 12, 2978–2989. [Google Scholar] [CrossRef]
- Rahimi, R.; Gholamrezapor, E.; Naimi-jamal, M.R. Oxidation of benzyl alcohols to the corresponding carbonyl compounds catalyzed by copper (II) meso-tetraphenylporphyrin as cytochrome P-450 model reaction. Inorg. Chem. Commun. 2011, 14, 1561–1568. [Google Scholar] [CrossRef]
- Chen, L.; Yang, Y.; Guo, Z.; Jiang, D. Highly efficient activation of molecular oxygen with nanoporous metalloporphyrin frameworks in heterogeneous systems. Adv. Mater. 2011, 23, 3149–3154. [Google Scholar] [CrossRef]
- Miller, S. ES&T Views: In a faraway state. Environ. Sci. Technol. 1990, 24, 1286–1289. [Google Scholar]
- Stingl, K.A.; Tsogoeva, S.B. Recent advances in sulfoxidation reactions: A metal-free approach. Tetrahedron Asymmetry 2010, 21, 1055–1074. [Google Scholar] [CrossRef]
- Liu, Y.; Howarth, A.J.; Hupp, J.T.; Farha, O.K. Selective photooxidation of a mustard-gas simulant catalyzed by a porphyrinic metal–organic framework. Angew. Chem. Int. Ed. 2015, 54, 9001–9005. [Google Scholar] [CrossRef] [PubMed]
- Lang, X.; Hao, W.; Leow, W.R.; Li, S.; Zhao, J.; Chen, X. Tertiary amine mediated aerobic oxidation of sulfides into sulfoxides by visible-light photoredox catalysis on TiO2. Chem. Sci. 2015, 6, 5000–5005. [Google Scholar] [CrossRef]
- Sipos, G.; Drinkel, E.E.; Dorta, R. The emergence of sulfoxides as efficient ligands in transition metal catalysis. Chem. Soc. Rev. 2015, 44, 3834–3860. [Google Scholar] [CrossRef]
- Laudadio, G.; Straathof, N.J.W.; Lanting, M.D.; Knoops, B.; Hessel, V.; Noël, T. An environmentally benign and selective electrochemical oxidation of sulfides and thiols in a continuous-flow microreactor. Green Chem. 2017, 19, 4061–4066. [Google Scholar] [CrossRef]
- Jiang, X. (Ed.) Sulfur Chemistry; Springer: Berlin, Germany, 2019. [Google Scholar]
- Matavos-Aramyan, S.; Soukhakian, S.; Jazebizadeh, M.H. Selected methods for the synthesis of sulfoxides and sulfones with emphasis on oxidative protocols. Phosphorus Sulfur Silicon Relat. Elem. 2020, 195, 181–193. [Google Scholar] [CrossRef]
- Baciocchi, E.; Rol, C.; Scamosci, E.; Sebastiani, G.V. Medium and structure effects on the anodic oxidation of aryl arylmethyl sulfides. J. Org. Chem. 1991, 56, 5498–5502. [Google Scholar] [CrossRef]
- Baciocchi, E.; Crescenzi, C.; Lanzalunga, O. Photoinduced electron transfer reactions of benzyl phenyl sulfides promoted by 9,10-dicyanoanthracene. Tetrahedron 1997, 53, 4469–4478. [Google Scholar] [CrossRef]
- Baciocchi, E.; Del Giacco, T.; Gerini, M.F.; Lanzalunga, O. Rates of C−S bond cleavage in tert-alkyl phenyl sulfide radical cations. Org. Lett. 2006, 8, 641–644. [Google Scholar] [CrossRef]
- Mata, E.G. Recent advances in the synthesis of sulfoxides from sulfides. Phosphorus Sulfur Silicon Relat. Elem. 1996, 117, 231–286. [Google Scholar] [CrossRef]
- Legros, J.; Dehli, J.R.; Bolm, C. Applications of catalytic asymmetric sulfide oxidations to the syntheses of biologically active sulfoxides. Adv. Synth. Catal. 2005, 347, 19–31. [Google Scholar] [CrossRef]
- Srour, H.; Le Maux, P.; Chevance, S.; Simonneaux, G. Metal-catalyzed asymmetric sulfoxidation, epoxidation and hydroxylation by hydrogen peroxide. Coord. Chem. Rev. 2013, 257, 3030–3050. [Google Scholar] [CrossRef]
- Khanna, V.; Maikap, G.C.; Iqbal, J. An efficient oxidation of sulfides to sulfones using 2-methylpropanal and dioxygen. Tetrahedron Lett. 1996, 37, 3367–3370. [Google Scholar] [CrossRef]
- Tumula, V.R.; Bondwal, S.; Bisht, P.; Pendem, C.; Kumar, J. Oxidation of sulfides to sulfones with hydrogen peroxide in the presence of acetic acid and Amberlyst 15. React. Kinet. Mech. Catal. 2012, 107, 449–466. [Google Scholar] [CrossRef]
- Morozkov, G.V.; Abel, A.S.; Filatov, M.A.; Nefedov, S.E.; Roznyatovsky, V.A.; Cheprakov, A.V.; Mitrofanov, A.Y.; Ziankou, I.S.; Averin, A.D.; Beletskaya, I.P.; et al. Ruthenium(II) complexes with phosphonate-substituted phenanthroline ligands: Synthesis, characterization and use in organic photocatalysis. Dalton Trans. 2022, 51, 13612–13630. [Google Scholar] [CrossRef] [PubMed]
- Tabushi, I. Reductive dioxygen activation by use of artificial P-450 systems. Coord. Chem. Rev. 1988, 86, 1–42. [Google Scholar] [CrossRef]
- Laszlo, P.; Levart, M. “Clayniac”-catalyzed epoxidation: The role of the aldehyde as co-reducer of molecular oxygen. Tetrahedron Lett. 1993, 34, 1127–1130. [Google Scholar] [CrossRef]
- Wentzel, B.B.; Gosling, P.A.; Feiters, M.C.; Nolte, R.J.M. Mechanistic studies on the epoxidation of alkenes with molecular oxygen and aldehydes catalysed by transition metal–β-diketonate complexes. J. Chem. Soc. Dalton Trans. 1998, 13, 2241–2246. [Google Scholar] [CrossRef]
- Boring, E.; Geletii, Y.V.; Hill, C.L. Catalysts for selective aerobic oxidation under ambient conditions. In Advances in Catalytic Activation of Dioxygen by Metal Complexes; Simándi, L.I., Ed.; Springer: Boston, MA, USA, 2002; pp. 227–264. [Google Scholar]
- Livingston, S.R.; Landry, C.C. Oxidation of a mustard gas analogue using an aldehyde/O2 system catalyzed by V-doped mesoporous silica. J. Am. Chem. Soc. 2008, 130, 13214–13215. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Ji, H. Biomimetic kinetics and mechanism of cyclohexene epoxidation catalyzed by metalloporphyrins. Chem. Eng. J. 2010, 156, 411–417. [Google Scholar] [CrossRef]
- Lanucara, F.; Crestoni, M.E. Biomimetic oxidation reactions of a naked manganese(V)–oxo porphyrin complex. Chem.-Eur. J. 2011, 17, 12092–12100. [Google Scholar] [CrossRef] [PubMed]
- Klaine, S.; Bratcher, F.; Winchester, C.M.; Zhang, R. Formation and kinetic studies of manganese(IV)-oxo porphyrins: Oxygen atom transfer mechanism of sulfide oxidations. J. Inorg. Biochem. 2020, 204, 110986. [Google Scholar] [CrossRef]
- Xiang, G.; Wu, D.; He, J.; Wang, X. Acquired pH-responsive and reversible enrichment of organic dyes by peroxide modified ultrathin TiO2 nanosheets. Chem. Commun. 2011, 47, 11456–11458. [Google Scholar] [CrossRef]
- Moghadam, M.; Tangestaninejad, S.; Mirkhani, V.; Mohammadpoor-Baltork, I.; Abbasi-Larki, A.A. Biomimetic oxidation of sulfides with sodium periodate catalyzed by polystyrene-bound manganese(III) tetrapyridylporphyrin. Appl. Catal. A 2008, 349, 177–181. [Google Scholar] [CrossRef]
- Mirkhani, V.; Moghadam, M.; Tangestaninejad, S.; Mohammdpoor-Baltork, I.; Kargar, H.; Araghi, M. Highly efficient oxidation of sulfides with sodium periodate catalyzed by reusable silica supported Mn(Br8TPP)Cl and Mn(TPP)Cl catalysts under various reaction conditions. Appl. Catal. A 2009, 353, 61–67. [Google Scholar] [CrossRef]
- Rayati, S.; Rezaie, S.; Nejabat, F. Catalytic activity of Mn(III) porphyrins supported onto graphene oxide nano-sheets for green oxidation of sulfides. J. Coord. Chem. 2019, 72, 1466–1479. [Google Scholar] [CrossRef]
- Rezaeifard, A.; Jafarpour, M. The catalytic efficiency of Fe-porphyrins supported on multi-walled carbon nanotubes in the heterogeneous oxidation of hydrocarbons and sulfides in water. Catal. Sci. Technol. 2014, 4, 1960–1969. [Google Scholar] [CrossRef]
- Deniaud, D.; Spyroulias, G.A.; Bartoli, J.-F.; Battioni, P.; Mansuy, D.; Pinel, C.; Odobel, F.; Bujoli, B. Shape selectivity for alkane hydroxylation with a new class of phosphonate-based heterogenised manganese porphyrins. New J. Chem. 1998, 22, 901–905. [Google Scholar] [CrossRef]
- Benítez, I.O.; Bujoli, B.; Camus, L.J.; Lee, C.M.; Odobel, F.; Talham, D.R. Monolayers as models for supported catalysts: Zirconium phosphonate films containing manganese(III) porphyrins. J. Am. Chem. Soc. 2002, 124, 4363–4370. [Google Scholar] [CrossRef] [PubMed]
- Eu, S.; Hayashi, S.; Umeyama, T.; Matano, Y.; Araki, Y.; Imahori, H. Quinoxaline-fused porphyrins for dye-sensitized solar cells. J. Phys. Chem. C 2008, 112, 4396–4405. [Google Scholar] [CrossRef]
- Kálmán, F.K.; Woods, M.; Caravan, P.; Jurek, P.; Spiller, M.; Tircsó, G.; Király, R.; Brücher, E.; Sherry, A.D. Potentiometric and relaxometric properties of a gadolinium-based MRI contrast agent for sensing tissue pH. Inorg. Chem. 2007, 46, 5260–5270. [Google Scholar] [CrossRef] [PubMed]
- Neveselý, T.; Svobodová, E.; Chudoba, J.; Sikorski, M.; Cibulka, R. Efficient Metal-free aerobic photooxidation of sulfides to sulfoxides mediated by a vitamin B2 derivative and visible light. Adv. Synth. Catal. 2016, 358, 1654–1663. [Google Scholar] [CrossRef]
- Kamata, K.; Hirano, T.; Mizuno, N. Highly efficient oxidation of sulfides with hydrogen peroxide catalyzed by [SeO4{WO(O2)2}2]2−. Chem. Commun. 2009, 3958–3960. [Google Scholar] [CrossRef]
- Hosseini-Eshbala, F.; Sedrpoushan, A.; Dehdashti, M.N.; Breit, B.; Mohanazadeh, F.; Veisi, H. Needle ball-like nanostructured mixed Cu-Ni-Co oxides: Synthesis, characterization and application to the selective oxidation of sulfides to sulfoxides. Mater. Sci. Eng. C 2019, 103, 109814. [Google Scholar] [CrossRef]
- Gao, Y.; Lam, Y. Polymer-supported N-phenylsulfonyloxaziridine (Davis reagent): A versatile oxidant. Adv. Synth. Catal. 2008, 350, 2937–2946. [Google Scholar] [CrossRef]
- Numata, T.; Itoh, O.; Yoshimura, T.; Oae, S. Intramolecular stereospecific Pummerer reactions of aryl (substitutedmethyl) sulfoxides bearing electron-withdrawing groups with acetic anhydride. Bull. Chem. Soc. Jpn. 1983, 56, 257–265. [Google Scholar] [CrossRef]
- Rostami, A.; Moradi, S.; Shokri, Z. Fe3O4 as a magnetically reusable Fenton nanocatalyst for the oxidation of thiols to sulfonic acid and sulfides to sulfoxides and sulfones. Comptes Rendus Chim. 2018, 21, 80–87. [Google Scholar] [CrossRef]
- Lindén, A.A.; Krüger, L.; Bäckvall, J.-E. Highly selective sulfoxidation of allylic and vinylic sulfides by hydrogen peroxide using a flavin as catalyst. J. Org. Chem. 2003, 68, 5890–5896. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Entry | IBA (Equiv.) | Temperature (°C) | Time (h) | Conversion 2 (%) | Selectivity 2 (%) | |
| Sulfoxide | Sulfone | |||||
| 1 | 6 | 80 | 0.5 | 100 | 58 | 42 |
| 2 | 6 | 40 | 2 | 100 | 63 | 37 |
| 3 | 6 | 30 | 2.5 | 92 | 99 | 1 |
| 3 | 100 | 53 | 47 | |||
| 4 | 6 | 20 | 2 | 85 | 100 | 0 |
| 2.5 | 100 | 82 | 12 | |||
| 5 | 3 | 80 | 1.5 | 76 | 96 | 4 |
| 3.5 | 86 | 86 | 14 | |||
| 6 | 3 | 50 | 1.5 | 52 | 100 | 0 |
| 8 | 94 | 95 | 5 | |||
| 7 | 5 | 20 | 3 | 100 | 98 | 2 |
| 8 3 | 5 | 20 | 3 | 0 | ||
| 9 | 0 | 20 | 3 | 0 | ||
| 10 4 | 5 | 20 | 3 | 0 | ||
| 11 5 | 5 | 20 | 3 | 0 | ||
| 12 6 | 5 | 20 | 3 | 0 | ||
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Sulfide | IBA (Equiv.) | Time (h) | Conversion 2 (%) | Selectivity 2 (%) | |
| Sulfoxide | Sulfone | |||||
| 1 |  | 5 | 2.5 | 100 | 98 | 2 |
| 2 |  | 5 | 2 | 100 | 98 | 2 |
| 3 |  | 5 | 7 | 100 | 97 | 3 |
| 4 |  | 3.3 | 6 | 100 | 92 | 8 |
| 5 |  | 6 | 4 | 94 | 98 | 2 |
| 6 |  | 9 | 2.5 | 92 | 92 | 8 |
| 7 |  | 3 | 3 | 99 | 94 | 6 |
| 8 |  | 10 | 4 | 98 | 97 | 3 |
| 9 |  | 7 | 4 | 98 | 89 | 11 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdulaeva, I.A.; Birin, K.P.; Chassagnon, R.; Bessmertnykh-Lemeune, A. Hybrid Materials Based on Imidazo[4,5-b]porphyrins for Catalytic Oxidation of Sulfides. Catalysts 2023, 13, 402. https://doi.org/10.3390/catal13020402
Abdulaeva IA, Birin KP, Chassagnon R, Bessmertnykh-Lemeune A. Hybrid Materials Based on Imidazo[4,5-b]porphyrins for Catalytic Oxidation of Sulfides. Catalysts. 2023; 13(2):402. https://doi.org/10.3390/catal13020402
Chicago/Turabian StyleAbdulaeva, Inna A., Kirill P. Birin, Remi Chassagnon, and Alla Bessmertnykh-Lemeune. 2023. "Hybrid Materials Based on Imidazo[4,5-b]porphyrins for Catalytic Oxidation of Sulfides" Catalysts 13, no. 2: 402. https://doi.org/10.3390/catal13020402