
Identification of AHL Synthase in Desulfovibrio vulgaris Hildenborough Using an In-Silico Methodology

 and
and 
Abstract
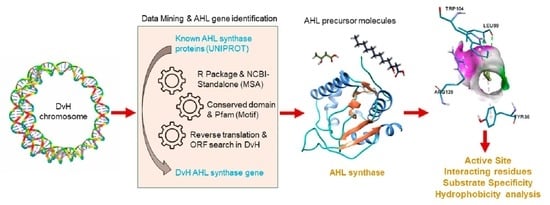
:
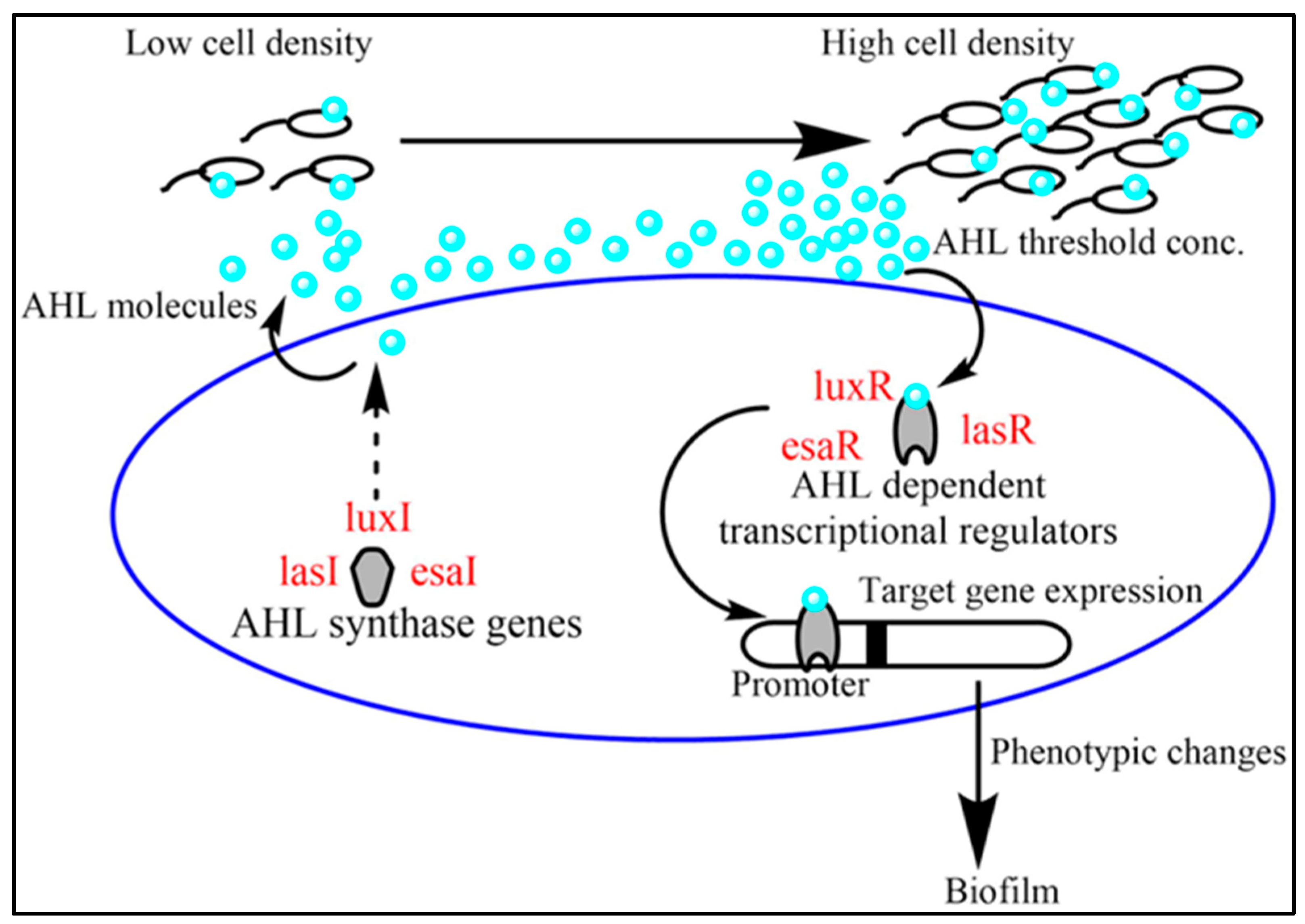
1. Introduction
2. Results and Discussion
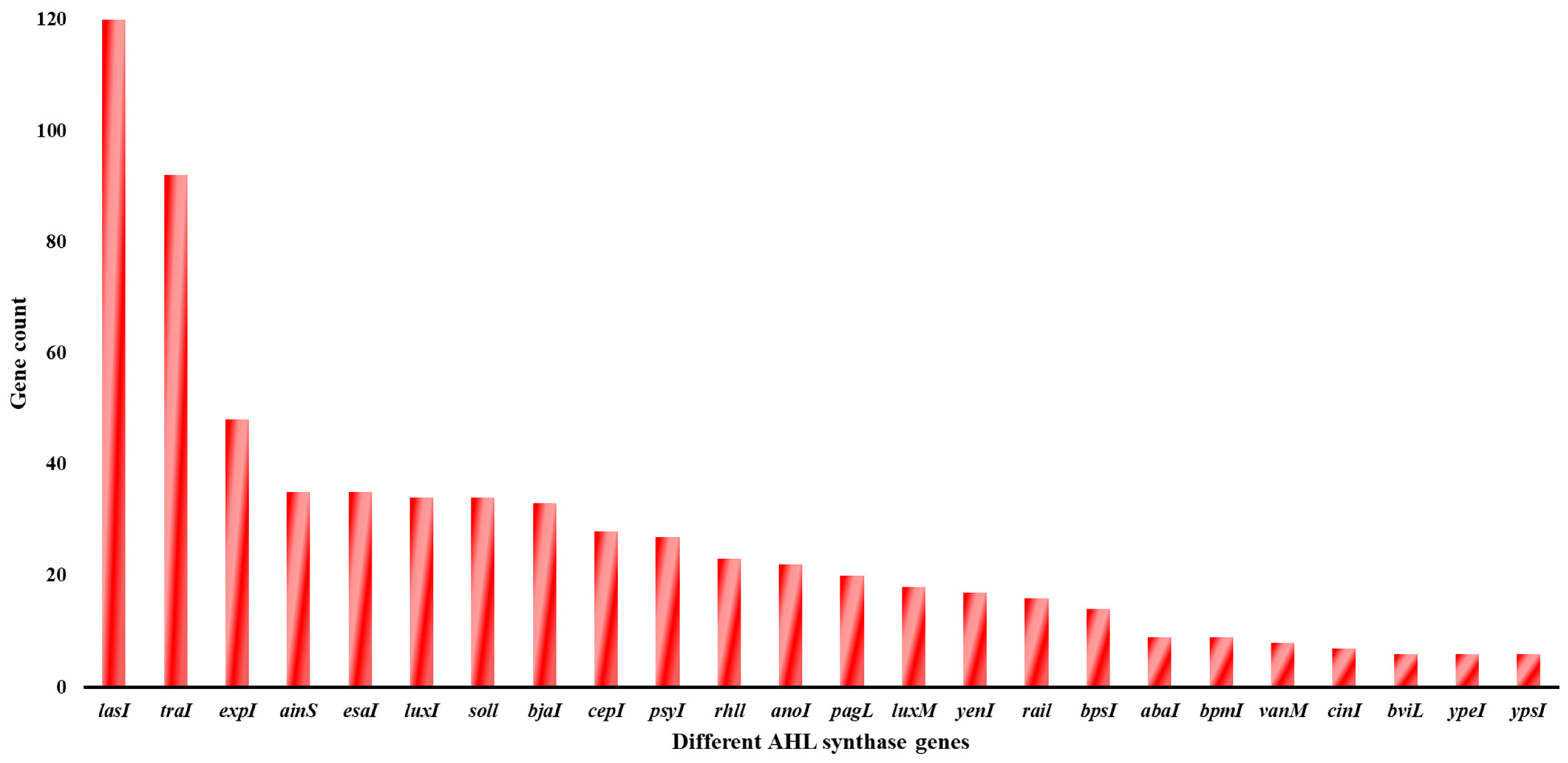
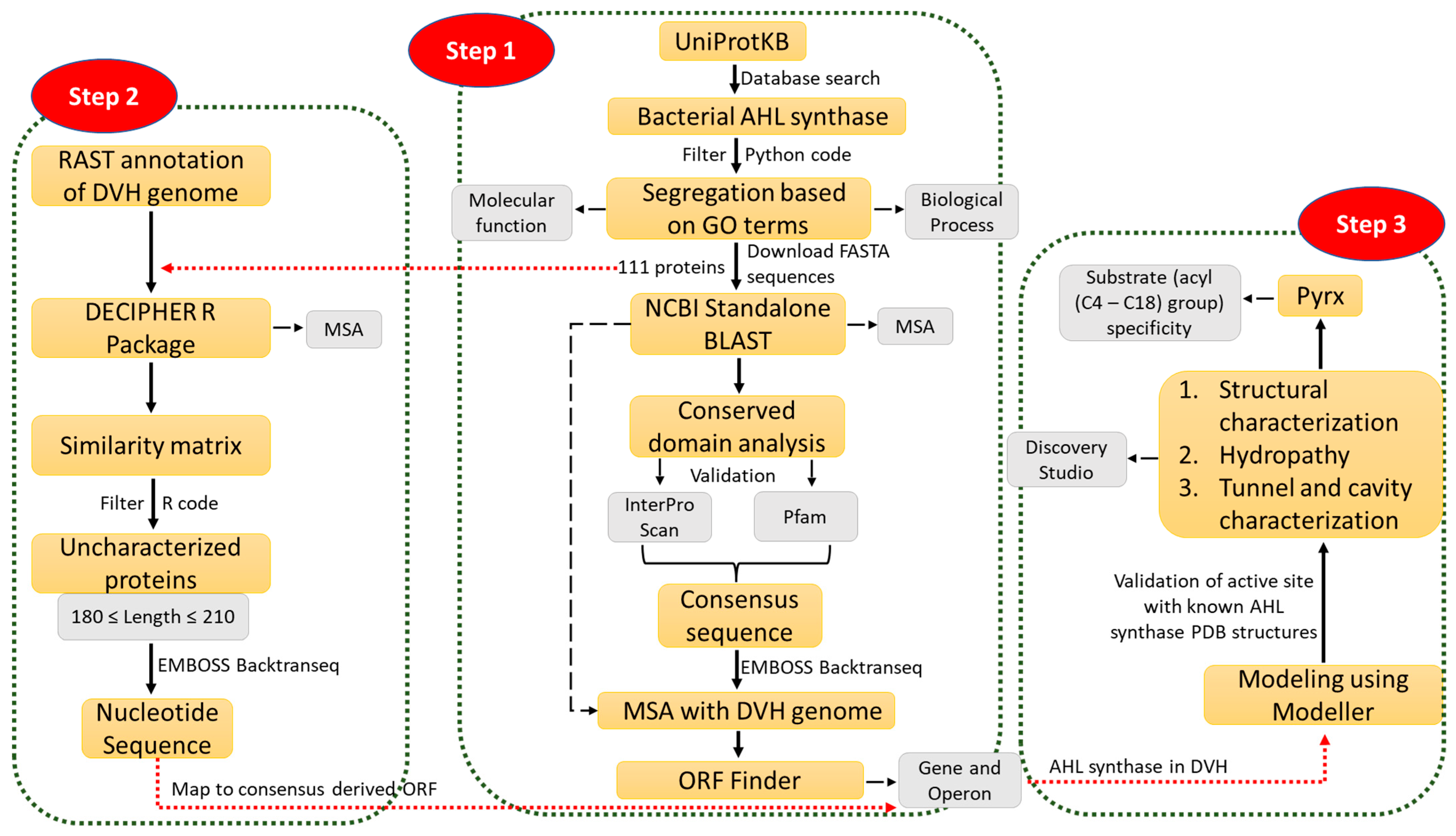
2.1. Data Mining and Prediction of AHL Synthase Using Multiple Sequence Alignment
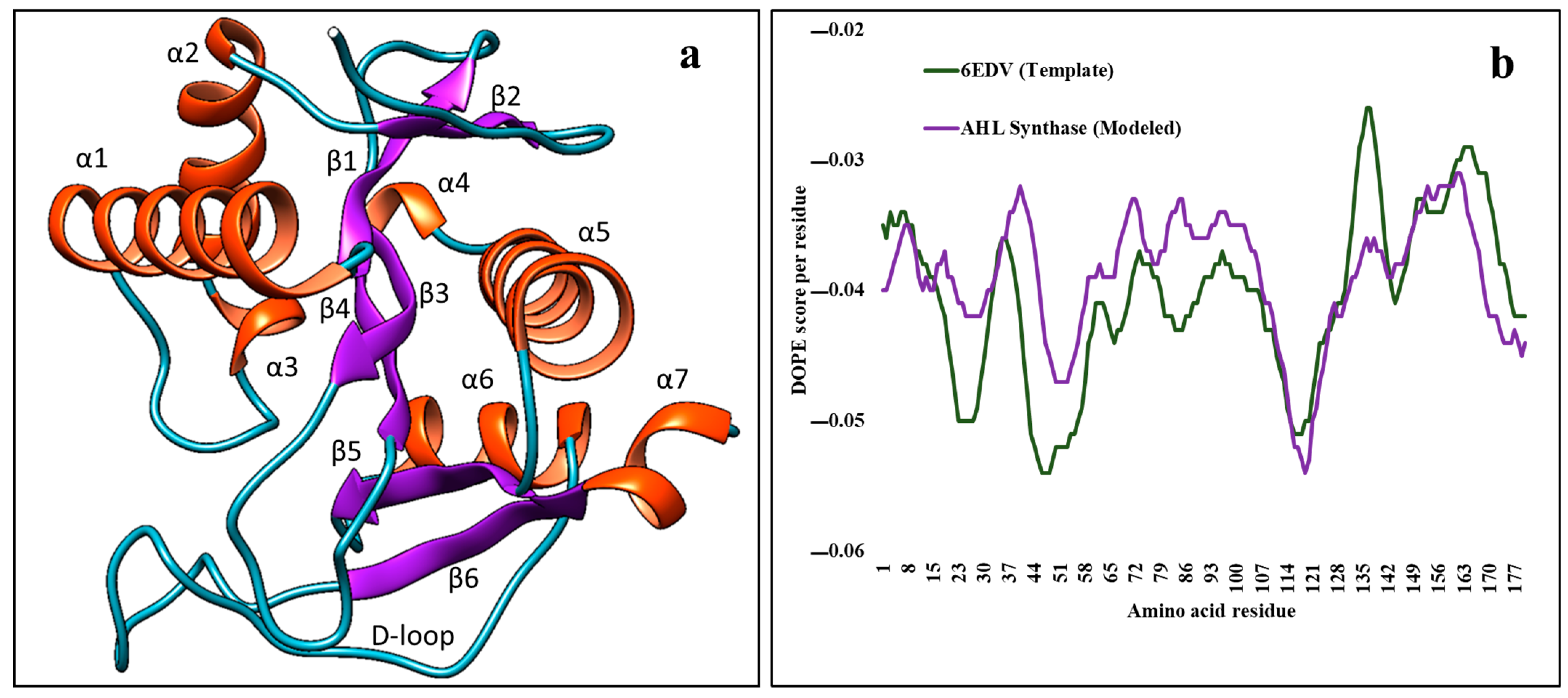
2.2. Modeling of AHL Synthase of Desulfovibrio vulgaris Hildenborough
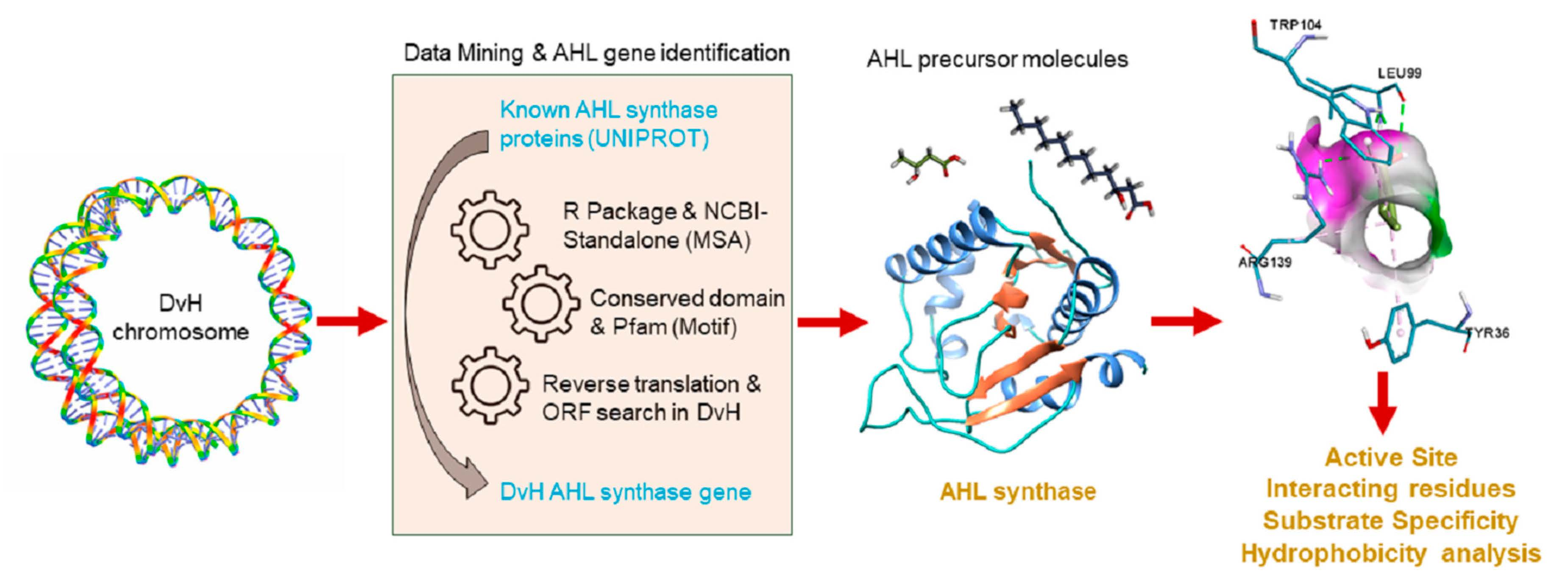
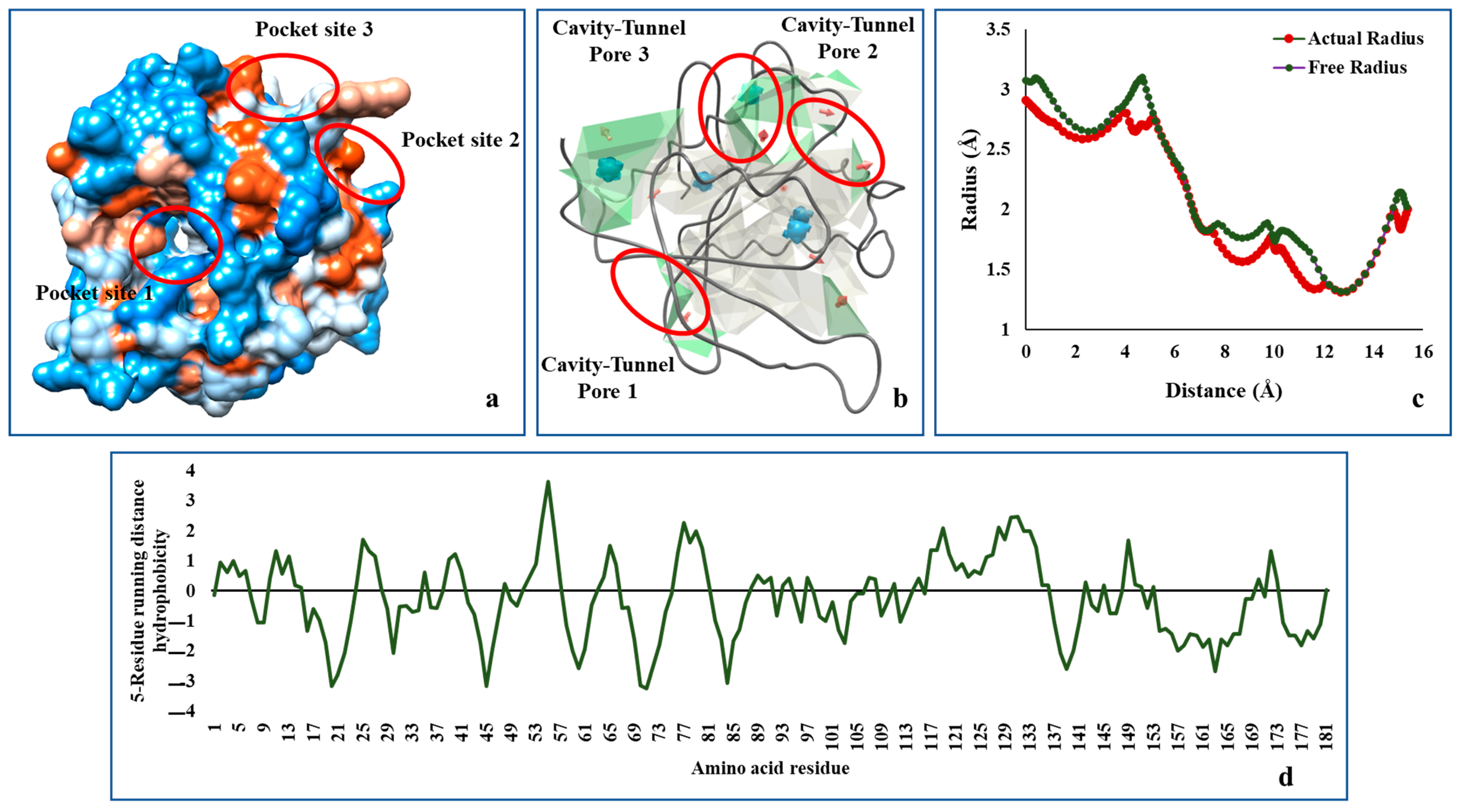
2.3. Analysis of Modelled AHL Synthase Structure with Others Present in Protein Data Bank
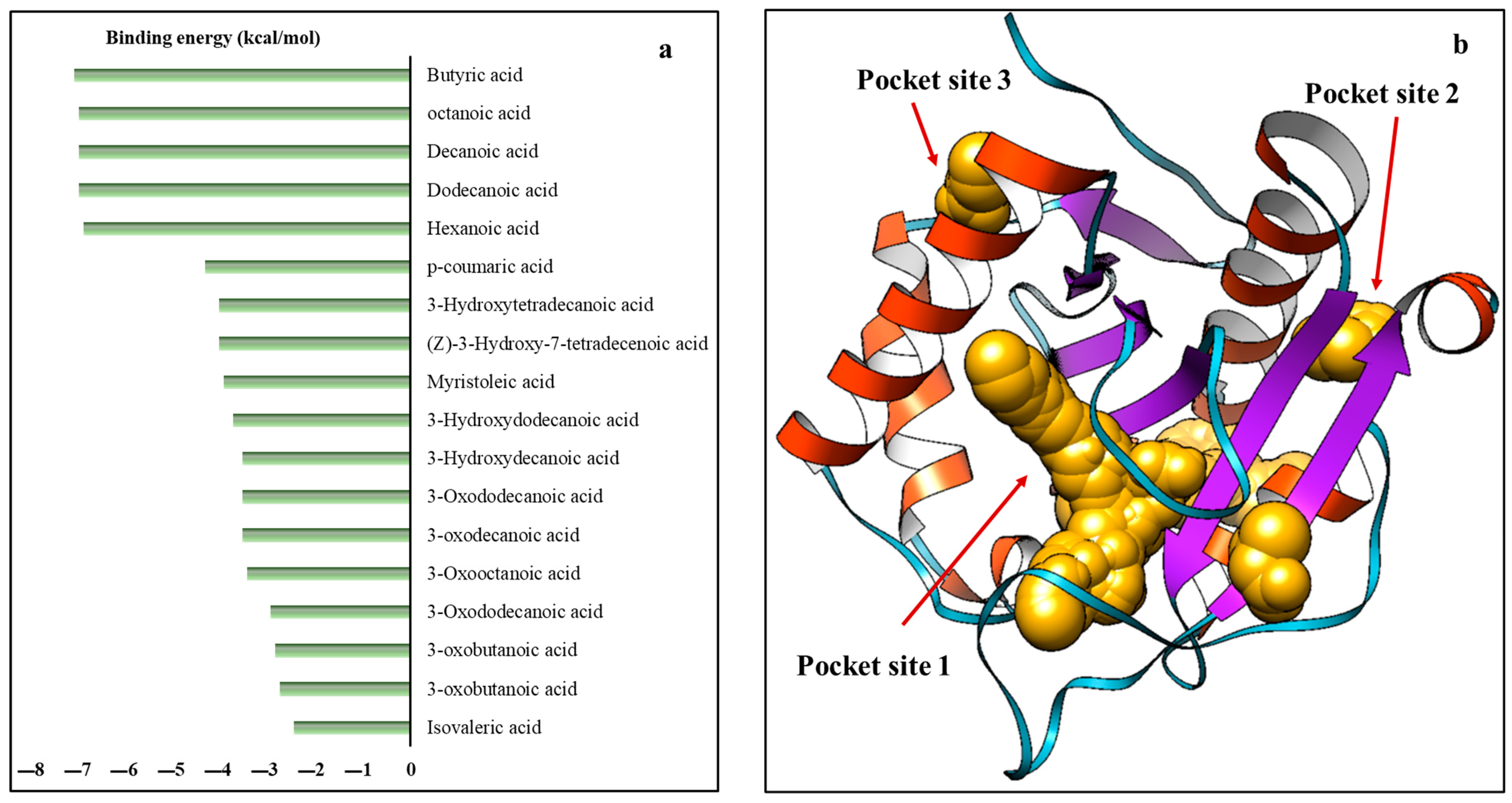
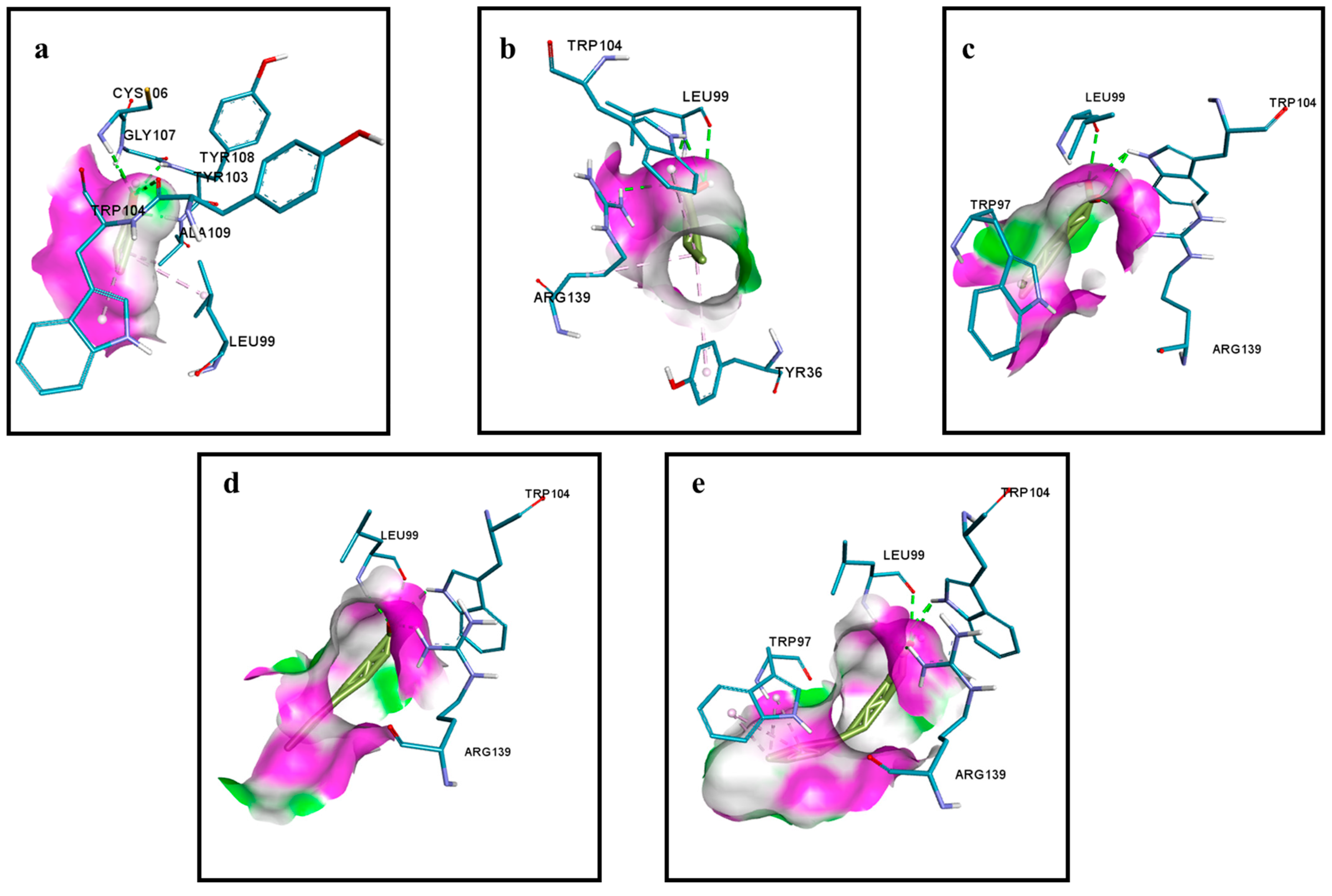
2.4. Interaction between AHL Synthase and ACP Bound Fatty Acyl Chain
3. Materials and Methods
3.1. Data Mining and Multiple Sequence Alignment to Find an AHL Synthase Gene in Desulfovibrio vulgaris Hildenborough
3.2. Modelling of AHL Synthase in Desulfovibrio vulgaris Hildenborough
3.3. Selection and Designing the Substrates of AHL Synthase
3.4. Interaction between the Substrates and Modelled AHL Synthase
3.5. Analysis of Hydropathy and Tunnels within the AHL Synthase of Desulfovibrio vulgaris Hildenborough
3.6. Superimposition between the Known and Modelled AHL Synthase
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Muyzer, G.; Stams, A. The Ecology and Biotechnology of Sulphate-Reducing Bacteria. Nat. Rev. Microbiol. 2008, 6, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.; Bardiau, M.; Brennan, R.; Burgess, H.; Caplin, J.; Ray, S.; Urios, T. Accelerated Low Water Corrosion: The Microbial Sulfur Cycle in Microcosm. NPJ Mater. Degrad. 2019, 3, 37. [Google Scholar] [CrossRef]
- Ning, J.; Zheng, Y.; Brown, B.; Young, D.; Nesic, S. The Role of Pyrite in Localized H2s Corrosion of Mild Steel. In Proceedings of the CORROSION 2017, New Orleans, LA, USA, 26–30 March 2017. [Google Scholar]
- Kato, S. Microbial Extracellular Electron Transfer and Its Relevance to Iron Corrosion. Microb. Biotechnol. 2016, 9, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Beech, I.B.; Gaylarde, C.C. Recent Advances in the Study of Biocorrosion: An Overview. Rev. Microbiol. 1999, 30, 117–190. [Google Scholar] [CrossRef]
- García-Contreras, R.; Nuñez-López, L.; Jasso-Chávez, R.; Kwan, B.W.; A Belmont, J.; Rangel-Vega, A.; Maeda, T.; Wood, T.K. Quorum Sensing Enhancement of the Stress Response Promotes Resistance to Quorum Quenching and Prevents Social Cheating. ISME J. 2015, 9, 115–125. [Google Scholar] [CrossRef]
- Rutherford, S.T.; Bassler, B.L. Bacterial Quorum Sensing: Its Role in Virulence and Possibilities for Its Control. Cold Spring Harb. Perspect. Med. 2012, 2, a012427. [Google Scholar] [CrossRef] [PubMed]
- Solano, C.; Echeverz, M.; Lasa, I. Biofilm Dispersion and Quorum Sensing. Curr. Opin. Microbiol. 2014, 18, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Verbeke, F.; De Craemer, S.; Debunne, N.; Janssens, Y.; Wynendaele, E.; Van de Wiele, C.; De Spiegeleer, B. Peptides as Quorum Sensing Molecules: Measurement Techniques and Obtained Levels in Vitro and in Vivo. Front. Neurosci. 2017, 11, 183. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Chen, Y.P.; Norman, R.S.; Decho, A.W. Rapid Screening of Quorum-Sensing Signal N-Acyl Homoserine Lactones by an in Vitro Cell-Free Assay. Appl. Environ. Microbiol. 2008, 74, 3667–3671. [Google Scholar] [CrossRef]
- Scarascia, G.; Wang, T.; Hong, P.-Y. Quorum Sensing and the Use of Quorum Quenchers as Natural Biocides to Inhibit Sulfate-Reducing Bacteria. Antibiotics 2016, 5, 39. [Google Scholar] [CrossRef] [Green Version]
- Decho, A.W.; Norman, R.S.; Visscher, P.T. Quorum Sensing in Natural Environments: Emerging Views from Microbial Mats. Trends Microbiol. 2010, 18, 73–80. [Google Scholar] [CrossRef]
- Decho, A.W.; Visscher, P.T.; Ferry, J.; Kawaguchi, T.; He, L.; Przekop, K.M.; Norman, R.S.; Reid, R.P. Autoinducers Extracted from Microbial Mats Reveal a Surprising Diversity of N-Acylhomoserine Lactones (Ahls) and Abundance Changes That May Relate to Diel Ph. Environ. Microbiol. 2009, 11, 409–420. [Google Scholar] [CrossRef]
- Smith, J.L.; Fratamico, P.M.; Gunther, N.W., IV. Shiga Toxin-Producing Escherichia Coli. Adv. Appl. Microbiol. 2014, 86, 145–197. [Google Scholar]
- van Keulen, G.; Dyson, P.J. Production of Specialized Metabolites by Streptomyces Coelicolor A3 (2). Adv. Appl. Microbiol. 2014, 89, 217–266. [Google Scholar]
- Dirix, G.; Monsieurs, P.; Dombrecht, B.; Daniels, R.; Marchal, K.; Vanderleyden, J.; Michiels, J. Peptide Signal Molecules and Bacteriocins in Gram-Negative Bacteria: A Genome-Wide in Silico Screening for Peptides Containing a Double-Glycine Leader Sequence and Their Cognate Transporters. Peptides 2004, 25, 1425–1440. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, N.A.; Barnard, A.M.; Slater, H.; Simpson, N.J.; Salmond, G.P. Quorum-Sensing in Gram-Negative Bacteria. FEMS Microbiol. Rev. 2001, 25, 365–404. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.K.; Thakur, P.; Saxena, P.; Rauniyar, S.; Gopalakrishnan, V.; Singh, R.N.; Gadhamshetty, V.; Gnimpieba, E.Z.; Jasthi, B.K.; Sani, R.K. Gene Sets and Mechanisms of Sulfate-Reducing Bacteria Biofilm Formation and Quorum Sensing with Impact on Corrosion. Front. Microbiol. 2021, 12, 3120. [Google Scholar] [CrossRef] [PubMed]
- Vance, R.E.; Zhu, J.; Mekalanos, J.J. A Constitutively Active Variant of the Quorum-Sensing Regulator Luxo Affects Protease Production and Biofilm Formation in Vibrio Cholerae. Infect. Immun. 2003, 71, 2571–2576. [Google Scholar] [CrossRef] [PubMed]
- Lenz, D.H.; Mok, K.C.; Lilley, B.N.; Kulkarni, R.V.; Wingreen, N.S.; Bassler, B.L. The Small Rna Chaperone Hfq and Multiple Small Rnas Control Quorum Sensing in Vibrio Harveyi and Vibrio Cholerae. Cell 2004, 118, 69–82. [Google Scholar] [CrossRef]
- Park, S.; Prévost, K.; Heideman, E.M.; Carrier, M.-C.; Azam, M.S.; A Reyer, M.; Liu, W.; Massé, E.; Fei, J. Dynamic Interactions between the Rna Chaperone Hfq, Small Regulatory Rnas, and Mrnas in Live Bacterial Cells. eLife 2021, 10, e64207. [Google Scholar] [CrossRef]
- Lorenz, N.; Shin, J.Y.; Jung, K. Activity, Abundance, and Localization of Quorum Sensing Receptors in Vibrio Harveyi. Front. Microbiol. 2017, 8, 634. [Google Scholar] [CrossRef]
- Li, Z.; Nair, S.K. Quorum Sensing: How Bacteria Can Coordinate Activity and Synchronize Their Response to External Signals? Protein Sci. 2012, 21, 1403–1417. [Google Scholar] [CrossRef] [PubMed]
- Churchill, M.E.A.; Chen, L. Structural Basis of Acyl-Homoserine Lactone-Dependent Signaling. Chem. Rev. 2011, 111, 68–85. [Google Scholar] [CrossRef] [PubMed]
- Passador, L.; Cook, J.M.; Gambello, M.J.; Rust, L.; Iglewski, B.H. Expression of Pseudomonas Aeruginosa Virulence Genes Requires Cell-to-Cell Communication. Science 1993, 260, 1127–1130. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.; Li, P.-L.; Zhang, L.; Piper, K.R.; Cook, D.M.; Tate, M.E.; Farrand, S.K. Trai, a Luxi Homologue, Is Responsible for Production of Conjugation Factor, the Ti Plasmid N-Acylhomoserine Lactone Autoinducer. Proc. Natl. Acad. Sci. USA 1994, 91, 4639–4643. [Google Scholar] [CrossRef]
- Minna, P.; Flego, D.; Heikinheimo, R.; Palva, E.T. A Small Diffusible Signal Molecule Is Responsible for the Global Control of Virulence and Exoenzyme Production in the Plant Pathogen Erwinia Carotovora. EMBO J. 1993, 12, 2467–2476. [Google Scholar]
- Hanzelka, B.L.; Parsek, M.R.; Val, D.L.; Dunlap, P.V.; John, E.C.; Greenberg, E.P. Acylhomoserine Lactone Synthase Activity of the Vibrio Fischeri Ains Protein. J. Bacteriol. 1999, 181, 5766–5770. [Google Scholar] [CrossRef] [PubMed]
- Beck von Bodman, S.U.S.A.N.N.; Farrand, S.K. Capsular Polysaccharide Biosynthesis and Pathogenicity in Erwinia Stewartii Require Induction by an N-Acylhomoserine Lactone Autoinducer. J. Bacteriol. 1995, 177, 5000–5008. [Google Scholar] [CrossRef] [PubMed]
- Knight, T.F.; Papadakis, N.; Vibrio Fischeri Lux Operon Sali Digest. GENBANK AF170104 (Strain MJ-1); 1999. Available online: http://www.ncbi.nlm.nih.gov/nuccore/5726577 (accessed on 29 November 2022).
- Flavier, A.B.; Ganova-Raeva, L.M.; Schell, M.A.; Denny, T.P. Hierarchical Autoinduction in Ralstonia Solanacearum: Control of Acyl-Homoserine Lactone Production by a Novel Autoregulatory System Responsive to 3-Hydroxypalmitic Acid Methyl Ester. J. Bacteriol. 1997, 179, 7089–7097. [Google Scholar] [CrossRef]
- Christensen, Q.H.; Brecht, R.M.; Dudekula, D.; Greenberg, E.P.; Nagarajan, R. Evolution of Acyl-Substrate Recognition by a Family of Acyl-Homoserine Lactone Synthases. PLoS ONE 2014, 9, e112464. [Google Scholar] [CrossRef]
- Lindemann, A.; Pessi, G.; Schaefer, A.L.; Mattmann, M.E.; Christensen, Q.H.; Kessler, A.; Hennecke, H.; Blackwell, H.E.; Greenberg, E.P.; Harwood, C.S. Isovaleryl-Homoserine Lactone, an Unusual Branched-Chain Quorum-Sensing Signal from the Soybean Symbiont Bradyrhizobium Japonicum. Proc. Natl. Acad. Sci. USA 2011, 108, 16765–16770. [Google Scholar] [CrossRef] [PubMed]
- Gotschlich, A.; Huber, B.; Geisenberger, O.; Tögl, A.; Steidle, A.; Riedel, K.; Hill, P.; Tümmler, B.; Vandamme, P.; Middleton, B. Synthesis of Multiple N-Acylhomoserine Lactones Is Wide-Spread among the Members of the Burkholderia Cepacia Complex. Syst. Appl. Microbiol. 2001, 24, 1–14. [Google Scholar] [CrossRef]
- Cheng, F.; Ma, A.; Zhuang, G.; Fray, R.G. Exogenous N-Acyl-Homoserine Lactones Enhance the Expression of Flagella of Pseudomonas Syringae and Activate Defence Responses in Plants. Mol. Plant Pathol. 2018, 19, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Ochsner, U.A.; Reiser, J. Autoinducer-Mediated Regulation of Rhamnolipid Biosurfactant Synthesis in Pseudomonas Aeruginosa. Proc. Natl. Acad. Sci. USA 1995, 92, 6424–6428. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.H.; Choi, C.H. Role of Luxir Homologue Anoir in Acinetobacter Nosocomialis and the Effect of Virstatin on the Expression of Anor Gene. J. Microbiol. Biotechnol. 2015, 25, 1390–1400. [Google Scholar] [CrossRef] [PubMed]
- Chalupowicz, L.; Manulis-Sasson, S.; Itkin, M.; Sacher, A.; Sessa, G.; Barash, I. Quorum-Sensing System Affects Gall Development Incited by Pantoea Agglomerans Pv. Gypsophilae. Mol. Plant Microbe Interact. 2008, 21, 1094–1105. [Google Scholar] [CrossRef]
- Bassler, B.L.; Wright, M.; Showalter, R.E.; Silverman, M.R. Intercellular Signalling in Vibrio Harveyi: Sequence and Function of Genes Regulating Expression of Luminescence. Mol. Microbiol. 1993, 9, 773–786. [Google Scholar] [CrossRef]
- Throup, J.P.; Camara, M.; Briggs, G.S.; Winson, M.K.; Chhabra, S.R.; Bycroft, B.W.; Williams, P.; Stewart, G.S. Characterisation of the Yeni/Yenr Locus from Yersinia Enterocolitica Mediating the Synthesis of Two N-Acylhomoserine Lactone Signal Molecules. Mol. Microbiol. 1995, 17, 345–356. [Google Scholar] [CrossRef]
- Rosemeyer, V.; Michiels, J.; Verreth, C.; Vanderleyden, J. Luxi-and Luxr-Homologous Genes of Rhizobium Etli Cnpaf512 Contribute to Synthesis of Autoinducer Molecules and Nodulation of Phaseolus Vulgaris. J. Bacteriol. 1998, 180, 815–821. [Google Scholar] [CrossRef]
- Gamage, A.M.; Shui, G.; Wenk, M.R.; Chua, K.L. N-Octanoylhomoserine Lactone Signalling Mediated by the Bpsi–Bpsr Quorum Sensing System Plays a Major Role in Biofilm Formation of Burkholderia Pseudomallei. Microbiology 2011, 157, 1176–1186. [Google Scholar] [CrossRef]
- Niu, C.; Clemmer, K.M.; Bonomo, R.A.; Rather, P.N. Isolation and Characterization of an Autoinducer Synthase from Acinetobacter Baumannii. J. Bacteriol. 2008, 190, 3386–3392. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.Y.; Bian, H.S.; Tan, T.M.C.; Mattmann, M.E.; Geske, G.D.; Igarashi, J.; Hatano, T.; Suga, H.; Blackwell, H.E.; Chua, K.L. Control of Quorum Sensing by a Burkholderia Pseudomallei Multidrug Efflux Pump. J. Bacteriol. 2007, 189, 4320–4324. [Google Scholar] [CrossRef] [PubMed]
- Milton, D.L.; Chalker, V.J.; Kirke, D.; Hardman, A.; Cámara, M.; Williams, P. The Luxm Homologue Vanm from Vibrio Anguillarum Directs the Synthesis of N-(3-Hydroxyhexanoyl) Homoserine Lactone and N-Hexanoylhomoserine Lactone. J. Bacteriol. 2001, 183, 3537–3547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, W.T.; Minogue, T.D.; Val, D.L.; Von Bodman, S.B.; Churchill, M.E. Structural Basis and Specificity of Acyl-Homoserine Lactone Signal Production in Bacterial Quorum Sensing. Mol. Cell 2002, 9, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Calatrava-Morales, N.; McIntosh, M.; Soto, M.J. Regulation Mediated by N-Acyl Homoserine Lactone Quorum Sensing Signals in the Rhizobium-Legume Symbiosis. Genes 2018, 9, 263. [Google Scholar] [CrossRef] [PubMed]
- Malott, R.J. Quorum-Sensing in Burkolderia Cenocepacia and Burkolderia Vietnamiensis. 2007. Available online: https://library-archives.canada.ca/eng/services/services-libraries/theses/Pages/item.aspx?idNumber=436330118 (accessed on 29 November 2022).
- Kirwan, J.P.; Gould, T.A.; Schweizer, H.P.; Bearden, S.W.; Murphy, R.C.; Churchill, M.E. Quorum-Sensing Signal Synthesis by the Yersinia Pestis Acyl-Homoserine Lactone Synthase Yspi. J. Bacteriol. 2006, 188, 784–788. [Google Scholar] [CrossRef] [PubMed]
- Ortori, C.A.; Atkinson, S.; Chhabra, S.R.; Cámara, M.; Williams, P.; Barrett, D.A. Comprehensive Profiling of N-Acylhomoserine Lactones Produced by Yersinia Pseudotuberculosis Using Liquid Chromatography Coupled to Hybrid Quadrupole–Linear Ion Trap Mass Spectrometry. Anal. Bioanal. Chem. 2007, 387, 497–511. [Google Scholar] [CrossRef]
- Samanta, D.; Govil, T.; Saxena, P.; Gadhamshetty, V.; Krumholz, L.R.; Salem, D.R.; Sani, R.K. Enhancement of Methane Catalysis Rates in Methylosinus Trichosporium Ob3b. Biomolecules 2022, 12, 560. [Google Scholar] [CrossRef]
- Puskas, A.; Greenberg, E.P.; Kaplan, S.; Schaefer, A.L. A Quorum-Sensing System in the Free-Living Photosynthetic Bacterium Rhodobacter Sphaeroides. J. Bacteriol. 1997, 179, 7530–7537. [Google Scholar] [CrossRef]
- Hoang, T.T.; Ma, Y.; Stern, R.J.; McNeil, M.R.; Schweizer, H.P. Construction and Use of Low-Copy Number T7 Expression Vectors for Purification of Problem Proteins: Purification of Mycobacterium Tuberculosis Rmld and Pseudomonas Aeruginosa Lasi and Rhli Proteins, and Functional Analysis of Purified Rhli. Gene 1999, 237, 361–371. [Google Scholar] [CrossRef]
- Gould, T.A.; Schweizer, H.P.; Churchill, M.E.A. Structure of the Pseudomonas Aeruginosa Acyl-Homoserinelactone Synthase Lasi. Mol. Microbiol. 2004, 53, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Moré, M.I.; Finger, L.D.; Stryker, J.L.; Fuqua, C.; Eberhard, A.; Winans, S.C. Winans. Enzymatic Synthesis of a Quorum-Sensing Autoinducer through Use of Defined Substrates. Science 1996, 272, 1655–1658. [Google Scholar] [CrossRef]
- Shin, D.; Gorgulla, C.; Boursier, M.E.; Rexrode, N.; Brown, E.C.; Arthanari, H.; Blackwell, H.E.; Nagarajan, R. N-Acyl Homoserine Lactone Analog Modulators of the Pseudomonas Aeruginosa Rhll Quorum Sensing Signal Synthase. ACS Chem. Biol. 2019, 14, 2305–2314. [Google Scholar]
- Dong, S.-H.; Frane, N.D.; Christensen, Q.H.; Greenberg, E.P.; Nagarajan, R.; Nair, S.K. Molecular Basis for the Substrate Specificity of Quorum Signal Synthases. Proc. Natl. Acad. Sci. USA 2017, 114, 9092–9097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, J.; Goo, E.; Yu, S.; Choi, O.; Lee, J.; Kim, J.; Kim, H.; Igarashi, J.; Suga, H.; Moon, J.S.; et al. Small-Molecule Inhibitor Binding to an N-Acyl-Homoserine Lactone Synthase. Proc. Natl. Acad. Sci. USA 2011, 108, 12089–12094. [Google Scholar] [CrossRef] [PubMed]
- Uniprot: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [CrossRef]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. Interproscan: Protein Domains Identifier. Nucleic Acids Res. 2005, 33, W116–W120. [Google Scholar] [CrossRef]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The Protein Families Database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. Emboss: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The Rast Server: Rapid Annotations Using Subsystems Technology. BMC Genom. 2008, 9, 1–15. [Google Scholar] [CrossRef]
- Wright, E.S. Decipher: Harnessing Local Sequence Context to Improve Protein Multiple Sequence Alignment. BMC Bioinform. 2015, 16, 322. [Google Scholar] [CrossRef] [PubMed]
- Fiser, A.; Šali, A. Modeller: Generation and Refinement of Homology-Based Protein Structure Models. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2003; pp. 461–491. [Google Scholar]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. Ncbi Blast: A Better Web Interface. Nucleic Acids Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Mark, A.E. Refinement of Homology-Based Protein Structures by Molecular Dynamics Simulation Techniques. Protein Sci. 2004, 13, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.R.; Heo, L.; Seok, C. Effective Protein Model Structure Refinement by Loop Modeling and Overall Relaxation. Proteins 2016, 84, 293–301. [Google Scholar] [CrossRef]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable Molecular Dynamics on Cpu and Gpu Architectures with Namd. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]
- Pedretti, A.; Mazzolari, A.; Gervasoni, S.; Fumagalli, L.; Vistoli, G. The Vega Suite of Programs: An Versatile Platform for Cheminformatics and Drug Design Projects. Bioinformatics 2021, 37, 1174–1175. [Google Scholar] [CrossRef]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. Molprobity: More and Better Reference Data for Improved All-Atom Structure Validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
- Cooley, M.A.; Whittall, C.; Rolph, M.S. Pseudomonas Signal Molecule 3-Oxo-C12-Homoserine Lactone Interferes with Binding of Rosiglitazone to Human Pparγ. Microbes Infect. 2010, 12, 231–237. [Google Scholar] [CrossRef]
- Parsek, M.R.; Greenberg, E.P. Acyl-Homoserine Lactone Quorum Sensing in Gram-Negative Bacteria: A Signaling Mechanism Involved in Associations with Higher Organisms. Proc. Natl. Acad. Sci. USA 2000, 97, 8789–8793. [Google Scholar] [CrossRef]
- Laue, B.E.; Jiang, Y.; Chhabra, S.R.; Jacob, S.; Stewart, G.S.; Hardman, A.; Downie, J.A.; O’Gara, F.; Williams, P. The Biocontrol Strain Pseudomonas Fluorescens F113 Produces the Rhizobium Small Bacteriocin, N-(3-Hydroxy-7-Cis-Tetradecenoyl) Homoserine Lactone, Via Hdts, a Putative Novel N-Acylhomoserine Lactone Synthase. Microbiology 2000, 146, 2469–2480. [Google Scholar] [CrossRef]
- Schaefer, A.L.; Greenberg, E.P.; Oliver, C.M.; Oda, Y.; Huang, J.J.; Bittan-Banin, G.; Peres, C.M.; Schmidt, S.; Juhaszova, K.; Sufrin, J.R. A New Class of Homoserine Lactone Quorum-Sensing Signals. Nature 2008, 454, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Papenfort, K.; Bassler, B.L. Quorum Sensing Signal–Response Systems in Gram-Negative Bacteria. Nat. Rev. Microbiol. 2016, 14, 576–588. [Google Scholar] [CrossRef]
- Stock, F.; Cirri, E.; Nuwanthi, S.G.L.I.; Stock, W.; Ueberschaar, N.; Mangelinckx, S.; Pohnert, G.; Vyverman, W. Sampling, Separation, and Quantification of N-Acyl Homoserine Lactones from Marine Intertidal Sediments. Limnol. Oceanogr. Methods 2021, 19, 145–157. [Google Scholar] [CrossRef]
- Vajedsamiei, J.; Melzner, F.; Raatz, M.; Kiko, R.; Khosravi, M.; Pansch, C. Simultaneous Recording of Filtration and Respiration in Marine Organisms in Response to Short-Term Environmental Variability. Limnol. Oceanogr. Methods 2021, 19, 196–209. [Google Scholar] [CrossRef]
- Biswa, P.; Doble, M. Production of Acylated Homoserine Lactone by Gram-Positive Bacteria Isolated from Marine Water. FEMS Microbiol. Lett. 2013, 343, 34–41. [Google Scholar] [CrossRef]
- Kaur, J.; Yogalakshmi, K.N. Degradation of N-Hexanoyl Homoserine Lactone with Quorum Quenching Bacteria Immobilised Magnetic Nanocomposite Beads. Environ. Technol. 2022, 43, 885–892. [Google Scholar] [CrossRef]
- Tabraiz, S.; Shamurad, B.; Petropoulos, E.; Charlton, A.; Mohiudin, O.; Danish Khan, M.; Ekwenna, E.; Sallis, P. Diversity of Acyl Homoserine Lactone Molecules in Anaerobic Membrane Bioreactors Treating Sewage at Psychrophilic Temperatures. Membranes 2020, 10, 320. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, J.; Marsili, M. A New Model for Calculating Atomic Charges in Molecules. Tetrahedron Lett. 1978, 19, 3181–3184. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. Autodock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Dallakyan, S.; Olson, A.J. Small-Molecule Library Screening by Docking with Pyrx. In Chemical Biology: Methods and Protocols; Hempel, J.E., Williams, C.H., Hong, C.C., Eds.; Springer: New York, NY, USA, 2015; pp. 243–250. [Google Scholar]
- Brooks, B.R.; Brooks, C.L., III; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, C.; Bartels, G.; Boresch, S.A.; et al. Charmm: The Biomolecular Simulation Program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Sehnal, D.; Svobodová Vařeková, R.; Berka, K.; Pravda, L.; Navrátilová, V.; Banáš, P.; Ionescu, C.-M.; Otyepka, M.; Koča, J. Mole 2.0: Advanced Approach for Analysis of Biomacromolecular Channels. J. Cheminform. 2013, 5, 39. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.-H.; Lam, N.; Nagarajan, R.; Nair, S.K. Structure-Guided Biochemical Analysis of Quorum Signal Synthase Specificities. ACS Chem. Biol. 2020, 15, 1497–1504. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fold Type | Modeled AHL | IRO5 (LasI) | 2P2F | 6WNS | IKZF (EsaI) |
|---|---|---|---|---|---|
| α1 RH | 🗸 | 🗸 | 🗸 | 🗸 | 🗸 |
| α2 RH | 🗸 | X | 🗸 | 🗸 | X |
| α3 RH | 🗸 | X | X | 🗸 | 🗸 |
| α4 RH | 🗸 | X | X | 🗸 | X |
| α5 RH | 🗸 | 🗸 | X | 🗸 | 🗸 |
| α6 RH | 🗸 | 🗸 | 🗸 | 🗸 | 🗸 |
| α7 RH | 🗸 | 🗸 | X | X | 🗸 |
| β1 AP(β2) | 🗸 | 🗸 | 🗸 | 🗸 | 🗸 |
| β2 AP(β1) | 🗸 | 🗸 | 🗸 | 🗸 | X |
| β3 AP(β4) | 🗸 | 🗸 | 🗸 | X | 🗸 |
| β4 AP(β3) | 🗸 | 🗸 | 🗸 | X | X |
| β5 AP(β6) | 🗸 | 🗸 | 🗸 | 🗸 | 🗸 |
| β6 AP(β5) | 🗸 | 🗸 | 🗸 | 🗸 | 🗸 |
| AHL Synthase Gene | Organism Name | PDB ID | Resolution (Å) | Crystallography Method | References |
| EsaI | Escherichia coli BL21 | 1KZF | 1.80 | X-ray diffraction | [46] |
| MesI | Mesorhizobium sp. ORS 3359 | 6WNS | 1.93 | X-ray diffraction | [88] |
| LasI | Escherichia coli | 1RO5 | 2.30 | X-ray diffraction | [54] |
| TofI | Burkholderia glumae | 3P2F | 2.30 | X-ray diffraction | [58] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tripathi, A.K.; Samanta, D.; Saxena, P.; Thakur, P.; Rauniyar, S.; Goh, K.M.; Sani, R.K. Identification of AHL Synthase in Desulfovibrio vulgaris Hildenborough Using an In-Silico Methodology. Catalysts 2023, 13, 364. https://doi.org/10.3390/catal13020364
Tripathi AK, Samanta D, Saxena P, Thakur P, Rauniyar S, Goh KM, Sani RK. Identification of AHL Synthase in Desulfovibrio vulgaris Hildenborough Using an In-Silico Methodology. Catalysts. 2023; 13(2):364. https://doi.org/10.3390/catal13020364
Chicago/Turabian StyleTripathi, Abhilash Kumar, Dipayan Samanta, Priya Saxena, Payal Thakur, Shailabh Rauniyar, Kian Mau Goh, and Rajesh Kumar Sani. 2023. "Identification of AHL Synthase in Desulfovibrio vulgaris Hildenborough Using an In-Silico Methodology" Catalysts 13, no. 2: 364. https://doi.org/10.3390/catal13020364