1. Introduction
The ability to site-selectively modify multifunctional substrates, bearing the same functional groups in several structurally different positions, is of considerable importance in synthetic organic as well as in medicinal chemistry [
1,
2,
3,
4,
5]. In particular, the selective functionalization of groups abundantly present in natural products, such as hydroxyls, could be particularly valuable for the design of new pharmaceuticals and SAR studies [
6,
7,
8,
9,
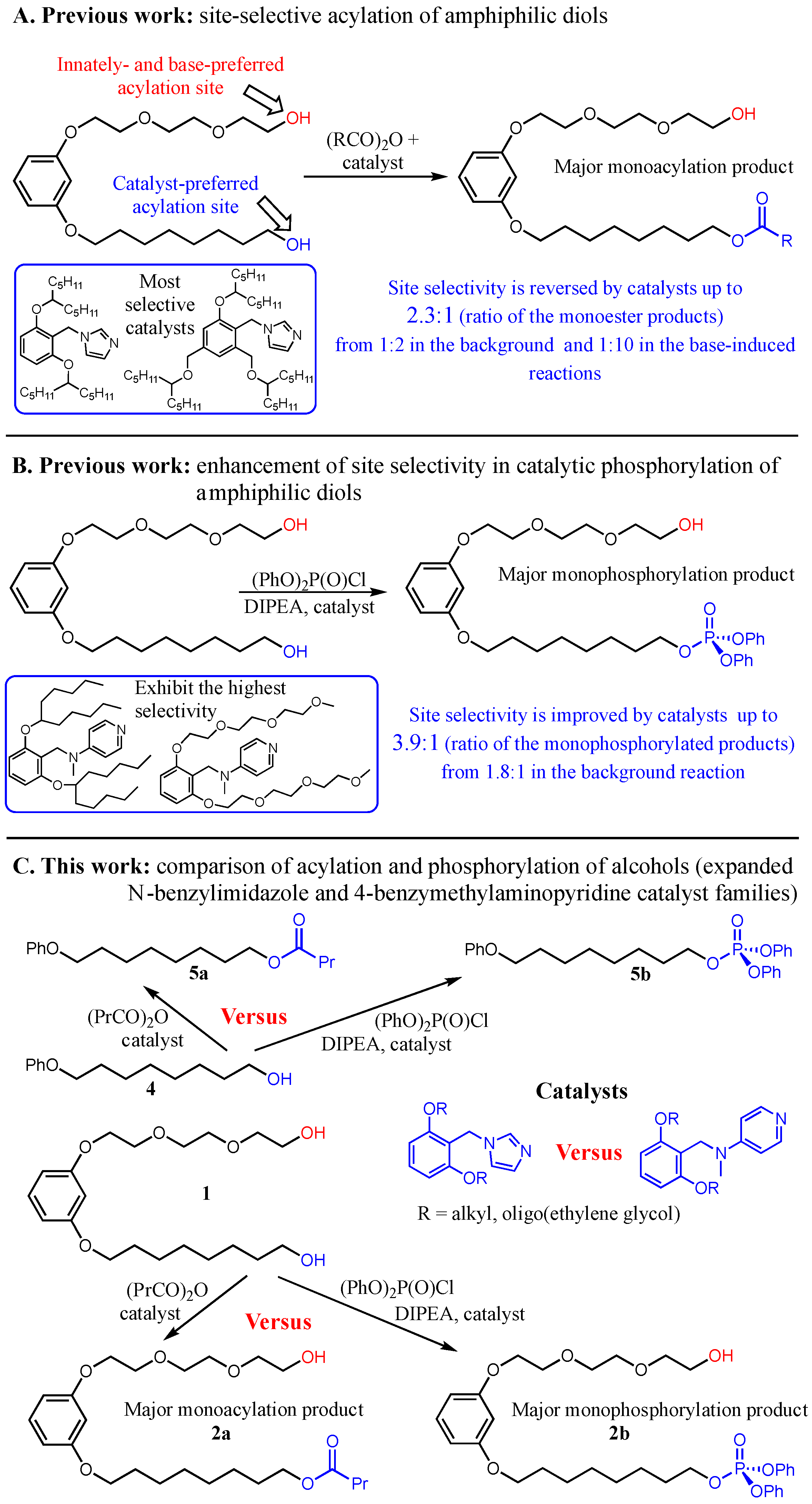
10]. While investigating the site-selective derivatization of amphiphilic diols, we recently reported that a family of catalysts, incorporating an N-benzylimidazole nucleophilic core and equipped with extensive outer-sphere hydrocarbon tails, was able to override the inherent preference of such substrates to undergo acylation of the alcohol at the polar site, promoting instead the reaction at the apolar site (
Scheme 1A) [
11]. We also found that a catalyst of a family closely related to the said imidazole-based catalysts, which bears peripheral oligoether rather than hydrocarbon tails, imposes enhanced apolar site-favoring selectivity in a related phosphorylation of amphiphilic diols [
12]. Moreover, the extension of this study revealed that a family of catalysts of a similar design, with a 4-aminopyridine active core decorated with extensive outer-sphere appendages [
12,
13], exhibited both remarkable activities and superior site selectivities favoring the apolar site in phosphorylation of such diols, regardless of the nature of the appendages (
Scheme 1B) [
12]. Intrigued by these findings, we sought to apply both expanded families of the catalysts (Im and BMAP families, respectively) for acylation of the model diol amphiphile, while comparing the performances of both types (activity- and selectivity-wise) between themselves in this transformation, and to their respective performances in the related phosphorylation catalysis (
Scheme 1C).
2. Results and Discussion
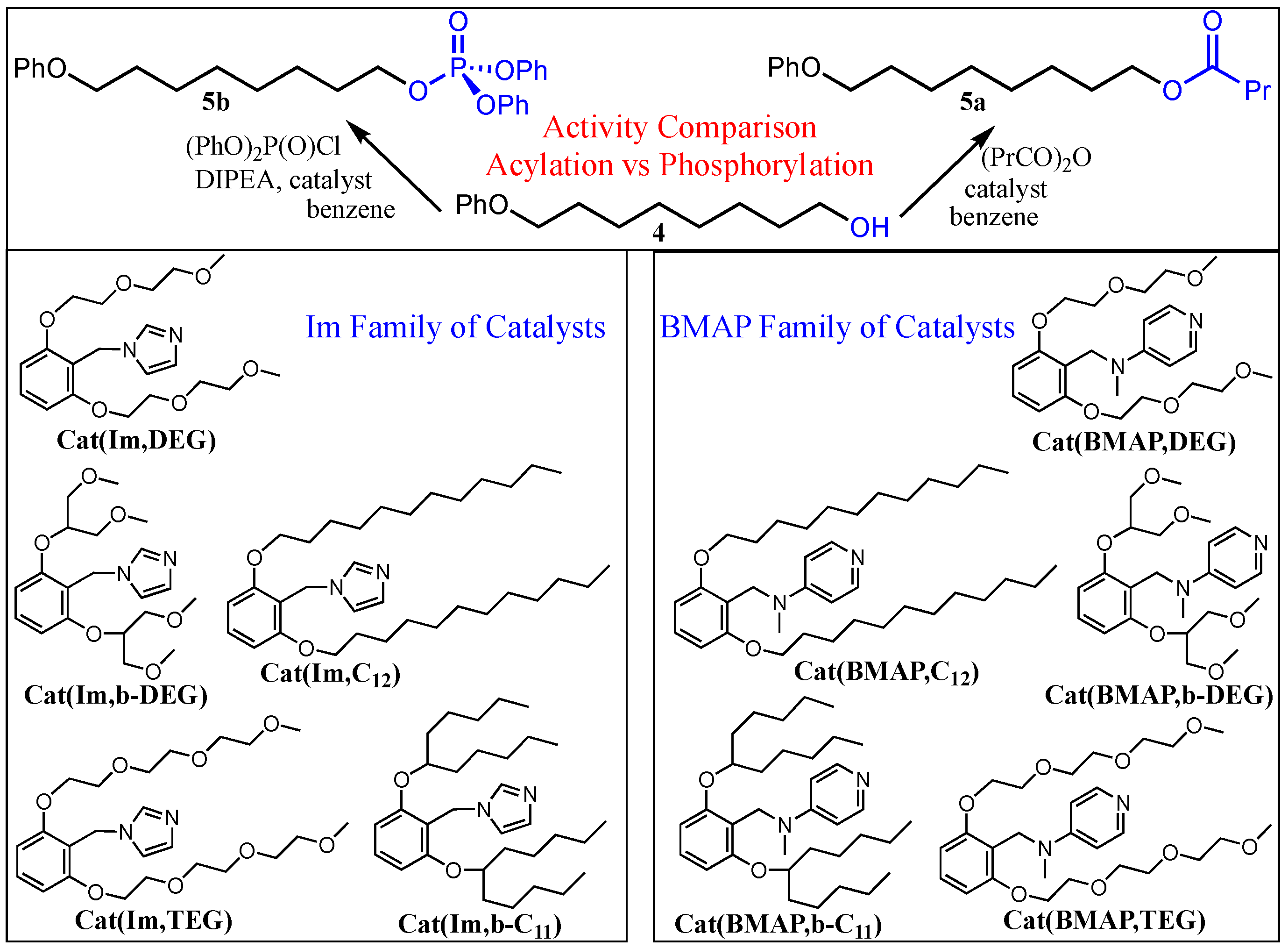
In order to assess the activity of the catalysts in both transformations, they were applied in the acylation and phosphorylation of a truncated substrate
4, imitating the apolar site of the model diol amphiphile, using butyric anhydride and diphenylphosphoryl chloride, respectively (
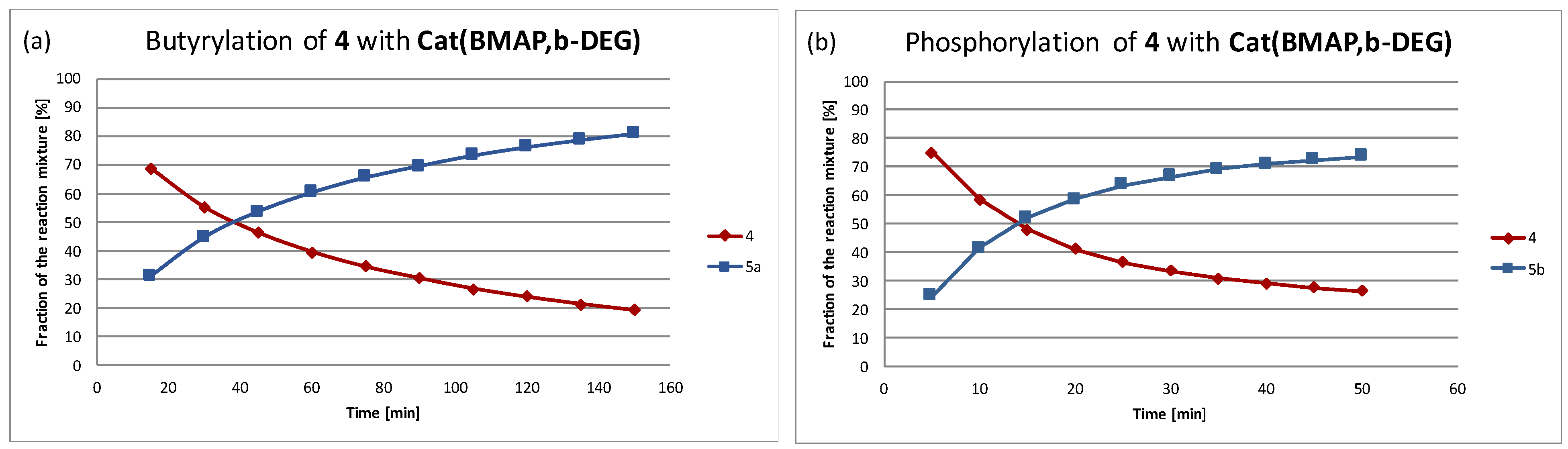
Scheme 2). The reactions were monitored using HPLC (see, for instance,
Figure 1, as well as
Supplementary Materials), demonstrating the clean conversion of
4 to
5a or
5b, respectively, and the apparent initial reaction rates, as well as the yields of the reactions after 45 min were summarized in
Table 1 (the Im series) and
Table 2 (the BMAP series). While the majority of the experiments were carried out with the 0.02 M initial substrate concentration, the acylation with the catalysts of the Im family was conducted with 0.2 M initial concentration, since acylation is the slower reaction of the two and the imidazole-based catalysts are of lower activity compared to the BMAP-derived species.
The results demonstrated that, as expected and was already demonstrated for the phosphorylation reaction [
12,
13], also in the acylation reaction, the BMAP-type catalysts were much more active than the imidazole-type analogues. Furthermore, it is evident that, both in acylation and in phosphorylation catalysis and for both catalytic families, the ortho-alkoxy groups on the benzyl substituent of the nitrogen augment the catalyst activity. It seems that the augmenting effect of these groups is much stronger for the phosphorylation reaction, compared to the acylation, and somewhat stronger in the case of the BMAP family of catalysts, compared to the Im family. On the other hand, while the effect of these groups derived from oligoethers is visibly stronger than that of those derived from alkanes, this difference is more pronounced in the case of the Im series. Finally, the comparison between the two reactions within the series of experiments with BMAP-type catalysts reveals that, as already abovementioned, the phosphorylation reaction is faster than the acylation reaction under the indicated conditions. It was observed that, in the case of phosphorylation, lack of the auxiliary base (DIPEA) prevents the reaction progress almost completely [
12], while in acylation, the influence of such base on the rate of the transformation is marginal [
11]. However, it is possible that the lack of the auxiliary base in the acylation reaction mixture, as appears in this study, slows somewhat this transformation. On the other hand, larger excess of the modifying agent (butyric anhydride) applied in the acylation should counterbalance the lack-of-base effect.
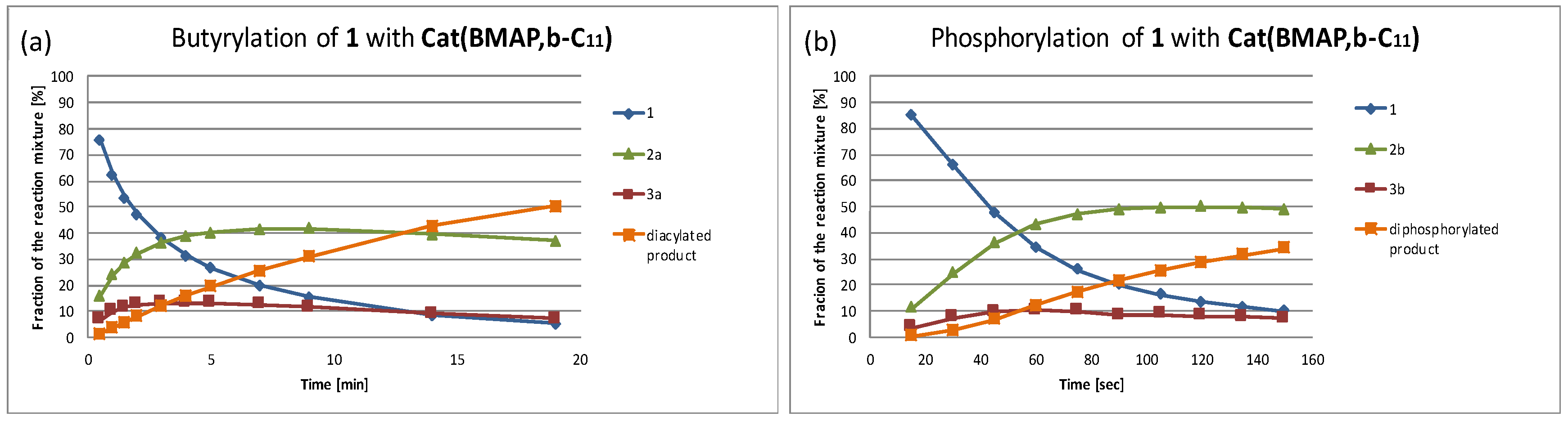
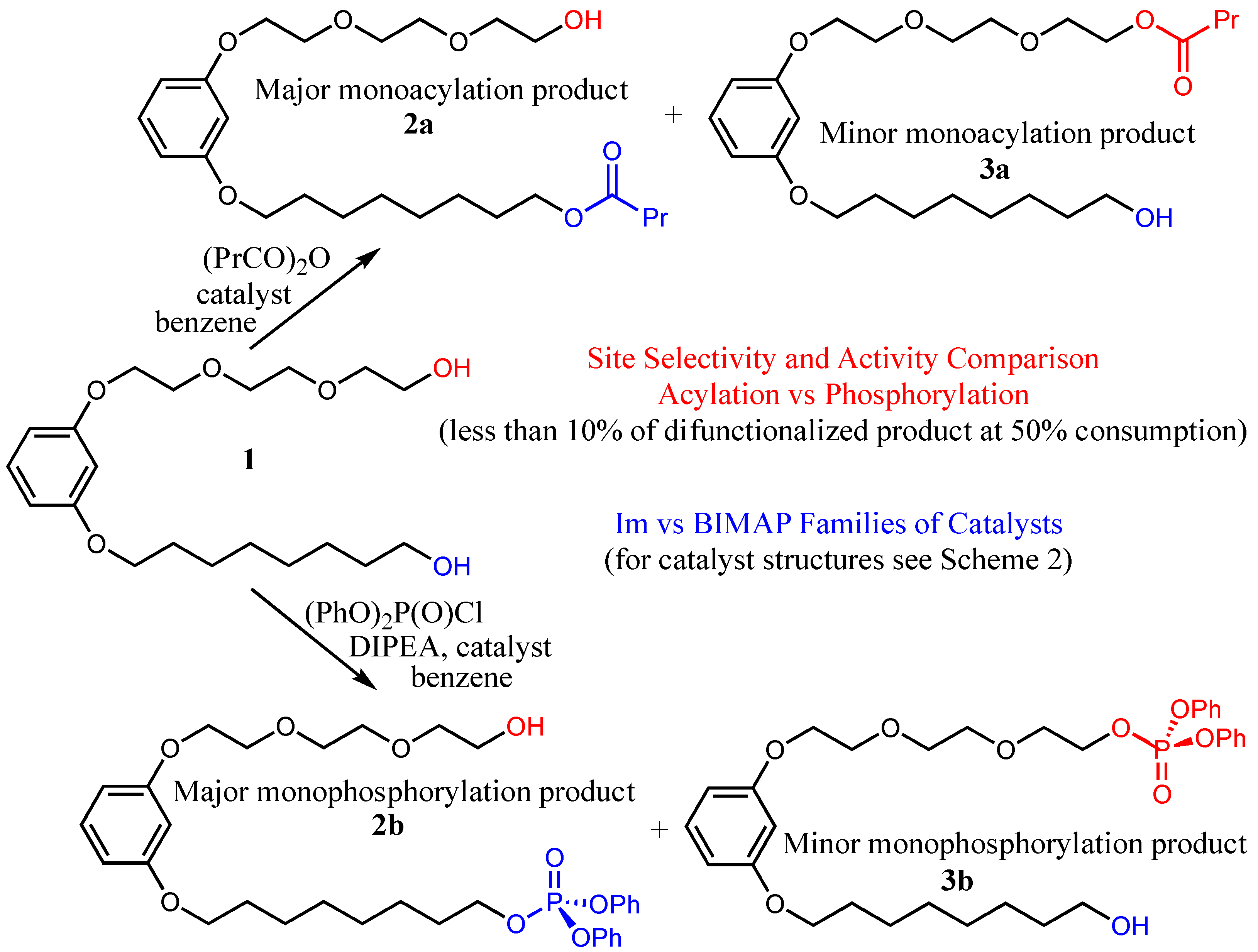
Following the comparison of the reactions with the simple primary alcohol, we turned to comparing the two catalytic processes with model amphiphilic diol
1 (
Scheme 3). The phosphorylation of this substrate using the catalysts of both families was recently reported [
12], with the results being reproduced in the two subsequent tables, and thus only the acylation study with both families of catalysts was carried out [
14]. The reactions were monitored, using HPLC (see, for instance,
Figure 2, as well as
Supplementary Materials), and the apparent initial reaction rates, as well as the half-life times of the substrate and the ratio between the monofunctionalized products, were summarized in
Table 3 (the Im series) and
Table 4 (the BMAP series). These results confirm the trends deduced from
Table 1 and
Table 2 regarding the activity of the catalysts; i.e., (1) the BMAP-type catalysts are orders of magnitude more active than the Im-type ones; (2) the di-ortho-alkoxy substituted benzyls almost always augment the activity of the catalysts, compared to the reference catalysts without such substituents; (3) the said augmentation is substantially stronger in the phosphorylation reaction; and (4) this augmentation is more significant for the BMAP family of the catalysts, while the differences between the oligoether-derived alkoxy substituents and the alkane-derived ones are stronger within the Im family (particularly in the phosphorylation reaction).
Regarding the site selectivity of the transformations, the more important of the investigated catalyst characteristics, profound effects of the catalyst structure on this parameter were observed. The effects were notably different for the two examined reactions. In the case of phosphorylation, the ortho-alkoxy substituents on the benzyl moiety increased the tendency of the first functionalization to occur at the apolar site (thus forming the product 2b), compared to substituent-less reference catalysts BnIm and BMAP, almost regardless of the nature of this substituents. While in the case of imidazole-based catalysts, better selectivity was obtained with the catalysts with oligoether-derived substituents and slightly lower with the hydrocarbon-derived appendages, in the case of BMAP-including catalysts both the hydrocarbon- and tri(ethylene glycol)-derived substituents on the benzyl moiety imparted superior (almost equivalent) selectivities. In the case of acylation, however, for both catalytic series, the presence of hydrocarbon-derived alkoxy groups on the benzylic moiety of the catalyst improved the apolar site-favoring selectivity compared to the substituent-less reference catalysts (though only modestly), while the presence of oligoether derived substituents did not affect or was even detrimental to this critical parameter. To our disappointment, in acylation experiments, the most selective catalyst of the BMAP family, Cat(BMAP,b-C11), was only marginally better, selectivity-wise, than its analogue of the Im family or even the benchmark DMAP catalyst.
Up to this stage of the research, the comparison between the two reactions regarding the discussed catalytic families revealed that while the new catalytic blueprint (a nucleophilic core with a benzyl substituent decorated with two ortho-positioned large alkoxy appendages) was highly beneficial for conducting site-selective catalytic phosphorylation of a diol amphiphile, it was less fruitful for the similar acylation catalysis. However, at this point, and while seeking ways to achieve a better site selectivity in the acylation of the model diol, we encountered improvement in the
2a:
3a ratio upon the addition of the reaction byproduct, butyric acid, prior to the beginning of the acylation experiment. Although butyric acid addition caused a substantial decrease in the reaction rate, optimization experiments conducted with DMAP revealed that a consistent improvement in site selectivity is observed with the increase in the number of equivalents of the additive, up to six equivalents. Further increase was counterproductive since it slowed the reaction even more without affecting the selectivity. The butyric acid additive imposed a similar effect also with all the catalysts of the BMAP series (
Table 5). Under the optimized conditions, a
2a:
3a ratio of 2.9:1 was achieved with DMAP, while our best catalyst,
Cat(BMAP,b-C11), induced the highest, thus far, 3.2:1 ratio at 50% consumption.
3. Materials and Methods
General information: All reactions, requiring anhydrous conditions, were conducted under an atmosphere of nitrogen in oven-dried glassware in dry solvents. Dry benzene, toluene, and DMF were purchased at highest available purity and used as received. Chloroform, ethyl acetate, hexanes, and methanol, as well as HPLC grade water, methanol, and acetonitrile, were purchased and used as received. THF was dried and distilled over sodium metal with benzophenone as the indicator. DCM was dried and distilled over calcium hydride. All reagents were purchased at the highest available purity and used as received. Thin layer chromatography (TLC) was performed on silica gel plates Merck 60 F254, and the compounds were visualized by irradiation with UV light or by KMnO4. Flash column chromatography was carried out using flash-grade silica gel (particle size 0.040–0.063 mm).
1H NMR (400 MHz), 13C NMR (100 MHz) and 31P NMR (162 MHz) spectra were recorded on Bruker AVANCE-400 spectrometers, in CDCl3 with residual CHCl3 (1H, 7.26 ppm) and CDCl3 (13C, 77.16 ppm) as internal standards, or 85% H3PO4 (31P, 0.0 ppm) as an external standard. MS analyses were conducted on Waters SYNAPT or Waters XEVO G2-XSTOF instruments (ESI ionization method, TOF detection method) or Waters Autospec instrument (EI ionization method, magnetic sector detection method). HPLC experiments were carried out using Apollo C18 5u column on a Hitachi Elite LaChrome instrument, equipped with a diode array UV/Vis detector, with acetonitrile and water as the eluting solvents.
The synthesis of the catalysts (except for commercially available DMAP and BnIm) followed the recently disclosed routes [
12,
13,
14].
General procedure for the butyrylation reaction of the model alcohol substrate: To the solution of alcohol substrate
4 (0.044 g, 0.2 mmol, 1.0 equiv) in benzene (1 mL or 10 mL), the catalyst (0.005 mmol, 0.05 equiv),
N,
N-diisopropylethylamine (52 μL 0.3 mmol, 3 equiv) and, finally, butyric anhydride (65 μL 0.4 mmol, 4 equiv) were added. The solution was stirred at room temperature. During the reaction, aliquots were taken at constant intervals (30 μL or 300 μL each sample, according to the volume of the reaction), and quenched with methanol (0.5 mL or 1.0 mL, according to the volume of the reaction). Each sample was analyzed using HPLC to determine the ratio of the products and the degree of the conversion. The product of the reaction was fully characterized previously [
11].
General procedure for the butyrylation reaction of the model diol substrate: To the solution of diol model substrate
1 (0.037 g, 0.1 mmol, 1.0 equiv, 2.0 hydroxyl equiv) in benzene (1 mL), the catalyst (0.005 mmol, 0.05 equiv),
N,
N-diisopropylethylamine (52 μL 0.3 mmol, 3 equiv) and, finally, butyric anhydride (65 μL 0.4 mmol, 4 equiv) were added. The solution was stirred at room temperature. During the reaction, aliquots were taken at constant intervals (30 μL each sample), and quenched with methanol (0.5 mL). Each sample was analyzed using HPLC to determine the ratio of the products and the degree of the conversion. The products of the reaction were fully characterized previously [
11].
General procedure for the phosphorylation reaction of the model alcohol substrate: To the solution of alcohol substrate
4 (0.044 g, 0.2 mmol, 1.0 equiv) in benzene (10 mL), the catalyst (0.005 mmol, 0.025 equiv),
N,
N-diisopropylethylamine (44 μL, 0.25 mmol, 1.25 equiv) and, finally, diphenyl chlorophosphate (51.8 μL, 0.25 mmol, 1.25 equiv) were added. The solution was stirred at room temperature. During the reaction, aliquots were taken at constant intervals (300 μL), quenched with methanol (1 mL), and the obtained solution was stirred for 30 min. Each sample was analyzed using HPLC to determine the ratio of the products and the degree of the conversion. The product of the reaction was fully characterized previously [
13].
General procedure for the phosphorylation reaction of the model diol substrate: To the solution of model diol substrate
1 (0.037 g, 0.1 mmol, 1.0 equiv, 2.0 hydroxyl equiv) in benzene (1 mL), the catalyst (0.005 mmol, 0.05 equiv),
N,
N-diisopropylethylamine (43.5 μL, 0.25 mmol, 2.5 equiv) and, finally, diphenyl chlorophosphate (52 μL, 0.25 mmol, 2.5 equiv) were added. The solution was stirred at room temperature. During the reaction, aliquots were taken at constant intervals (300 μL each sample), quenched with methanol (1 mL), and the obtained solution was stirred for 30 min. Each sample was analyzed using HPLC to determine the ratio of the products and the degree of consumption. The products of the reaction were fully characterized previously [
12].
4. Conclusions
In conclusion, there are a number of similarities between the two reactions, when promoted by the Im- or BMAP-series catalysts. These are mainly related to the catalysts’ activity. BMAP-catalyzed reactions are substantially faster than those promoted by the catalysts of the imidazole series. Adding two alkoxy substituents in the ortho positions of the phenyl ring of the catalyst’s benzyl moiety generally augments the catalysis, particularly within the BMAP family. The influence of the nature of the alkoxy appendages is also similar between the reactions, with the oligo(ethylene glycol)-derived tails providing higher activity than the alkane-derived ones. This difference between the two types of alkoxy groups is stronger for the phosphorylation reaction and within the Im family of catalysts.
The selectivity aspect of the catalysis emphasizes, however, the differences between the two alcohol-modifying reactions. While, in the case of phosphorylation, both higher activity and extensive apolar outer sphere of the catalyst are associated with the higher site selectivity of the system in favor of the apolar site, in the case of acylation, increasing the activity of the catalyst is accompanied by deterioration in its selectivity, and only extensive apolar outer sphere appendages affect the apolar site-favoring selectivity enhancement. Accordingly, while the BMAP catalysts with hydrophobic tails demonstrate superior selectivity (compared with tailless analogues) in both reactions, their counterparts with oligoether tails, which display exceptionally high activity [
15], offer comparable selectivity in the phosphorylation reaction, but are inferior (selectivity-wise) in acylation, even compared with tailless series members. This difference may explain the substantially higher site-selectivity that we have been able to realize in the phosphorylation of the model diol amphiphile, compared with that in its acylation, with the best catalysts, such as
Cat(BMAP,b-C11). In the case of the former reaction, the increase in the catalyst activity, induced by the electron donation of the lipophilic alkoxy moieties on the benzyl substituent, works in concert with the lipophilicity of the envelope of the active site, generated by the very same groups, in favor of the modification of the alcohol at the apolar site. In the case of acylation, however, the said increase in the activity erodes the selectivity, which, on the other hand, is enhanced by the extensive apolar wrapping of the catalytic center (with both factors originating from the installment of the ortho-alkoxy groups on the benzyl moiety). Hence, the highest ratio between the monofunctionalized products achieved in the acylation reaction (2.5:1 for
Cat(BMAP,b-C11)) is substantially lower than the parallel ratios observed in the phosphorylation catalysis (above 3:1 for all the catalysts, we prepared, 3.9:1 for
Cat(BMAP,b-C11)). It is possible that the inhibition of the catalytic activity caused in the acylation reactions by the butyric acid additive reduces the influence of the activity on the selectivity, thus allowing the site selectivity increase (up to a 3.2:1 ratio between the monoester products), as we demonstrated in the last series of experiments.
The trends observed for the studied catalytic series in both reactions are likely to provide critical assistance in the future development of even more selective catalysts, though it is not necessarily that one particular catalyst will constitute the optimal choice for both reactions.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}