
Electrocatalytic Oxygen Reduction to Hydrogen Peroxide on Graphdiyne-Based Single-Atom Catalysts: First-Principles Studies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
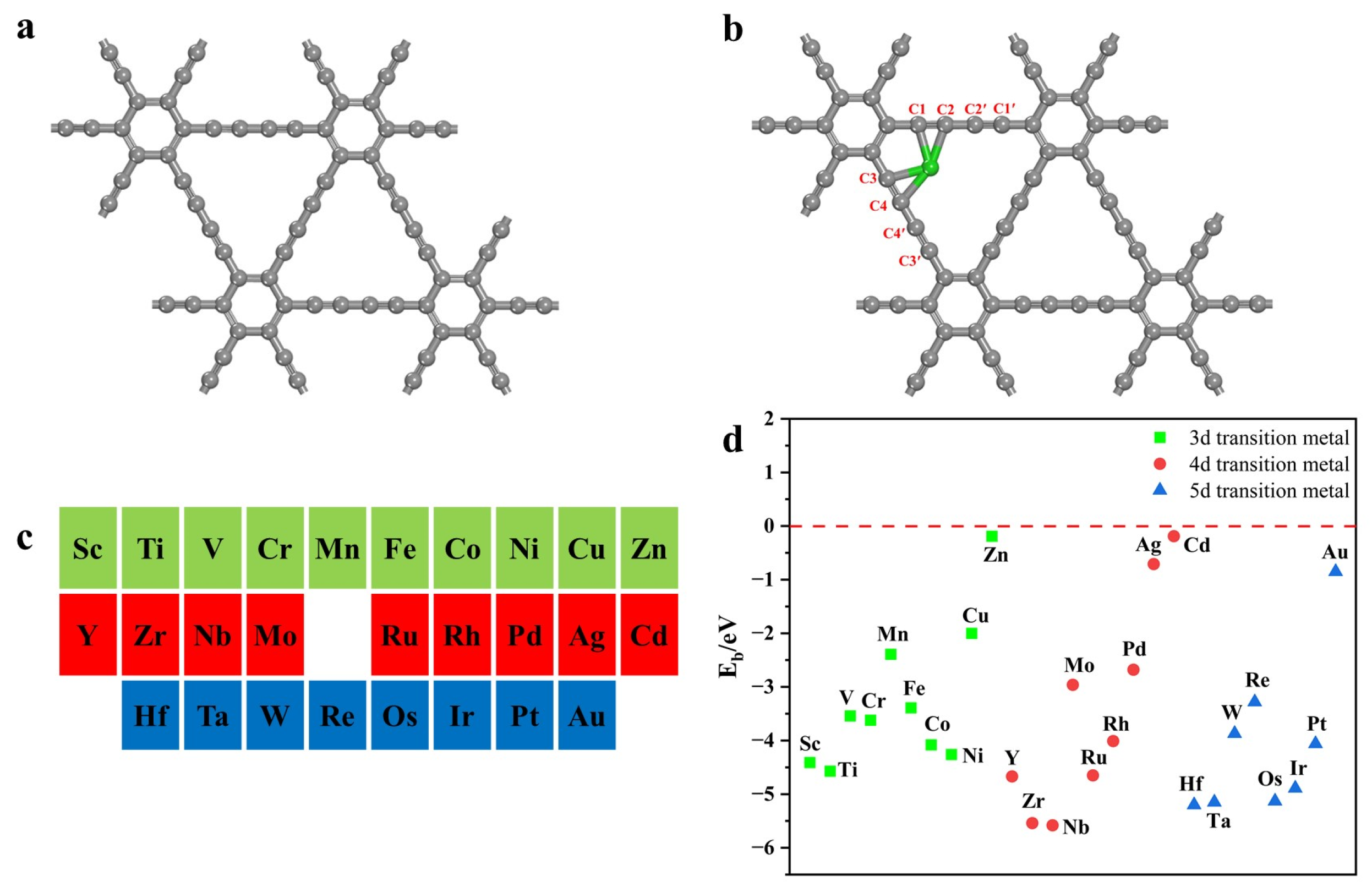
2.1. Catalyst Structure and Stability
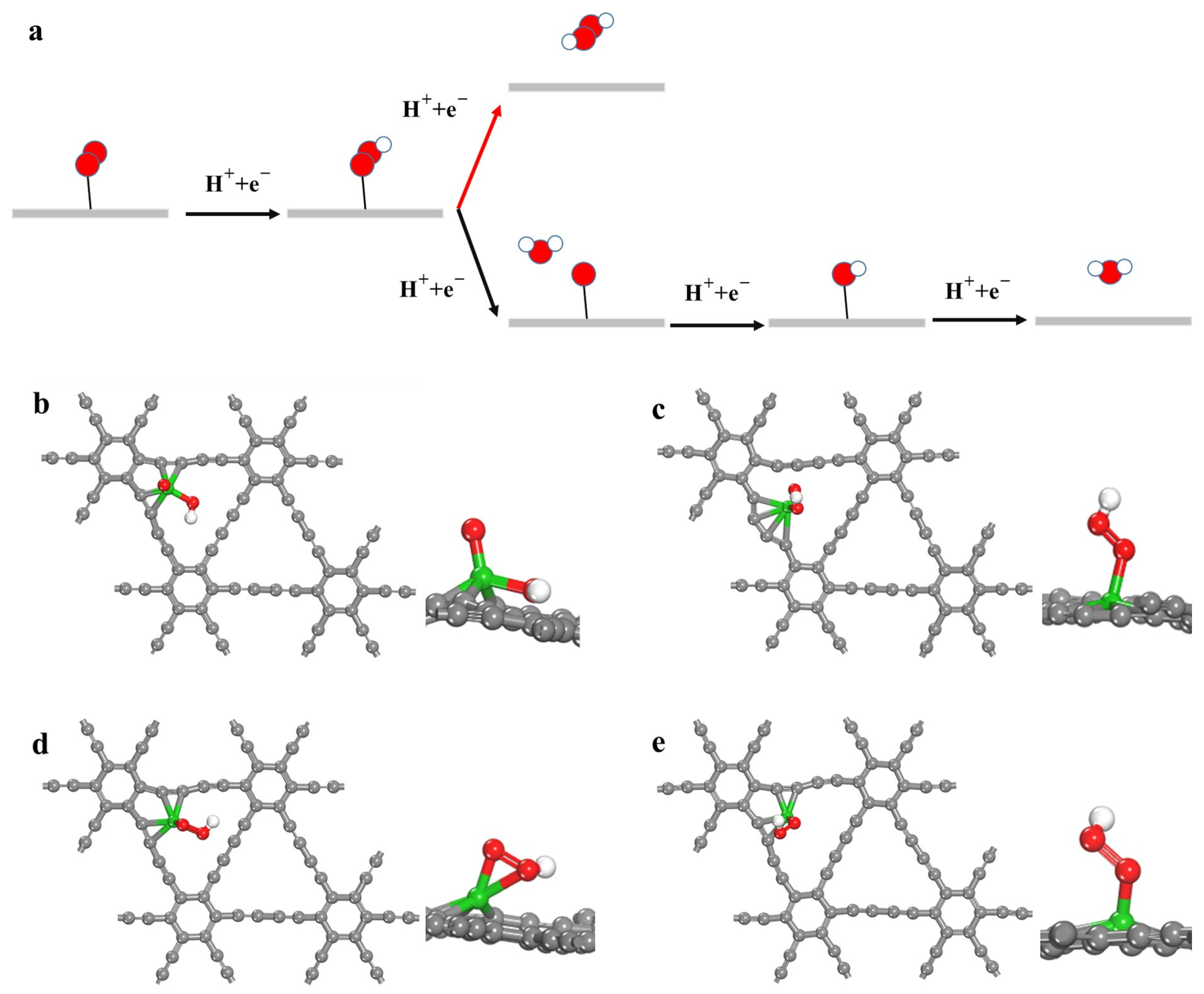
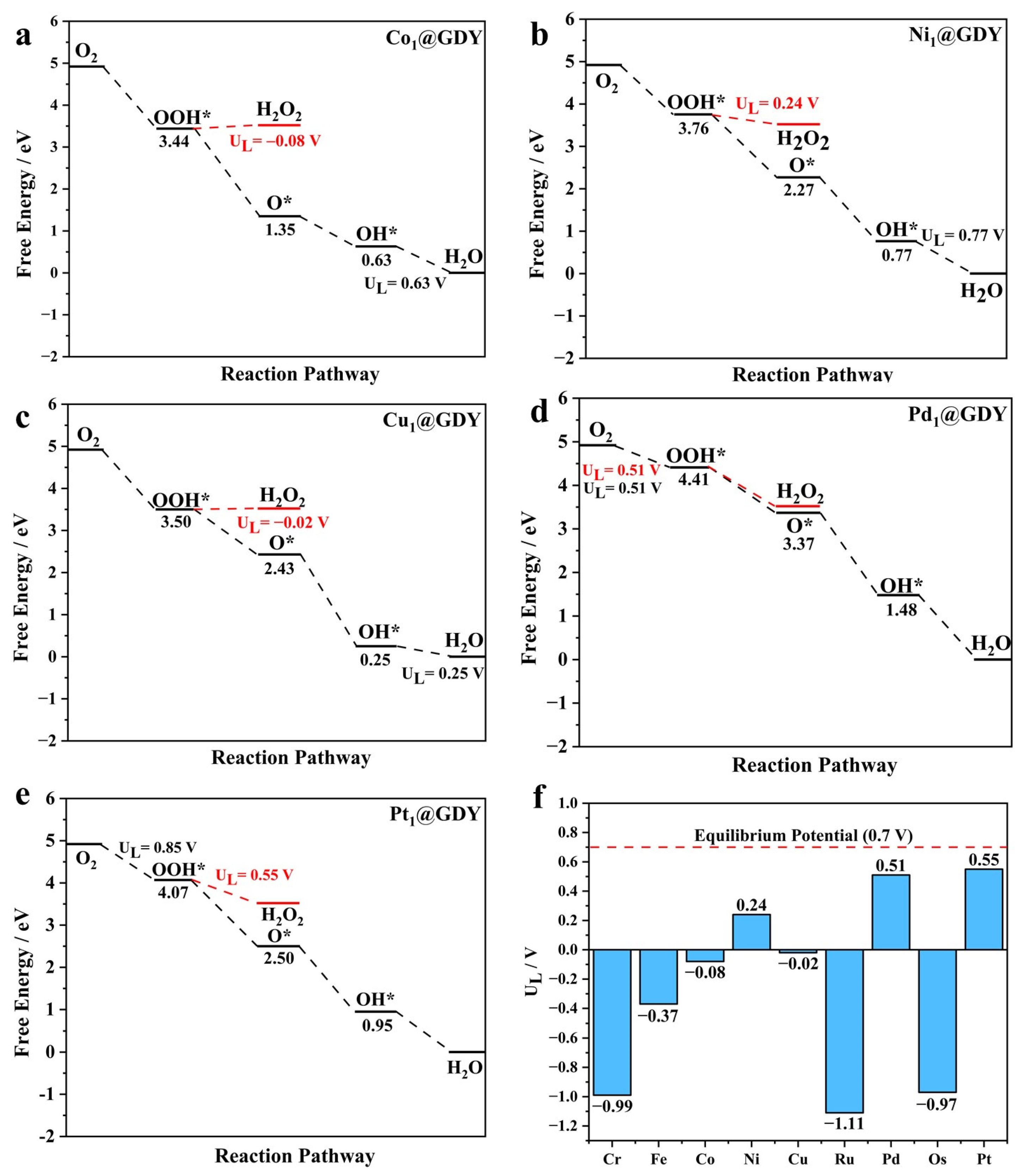
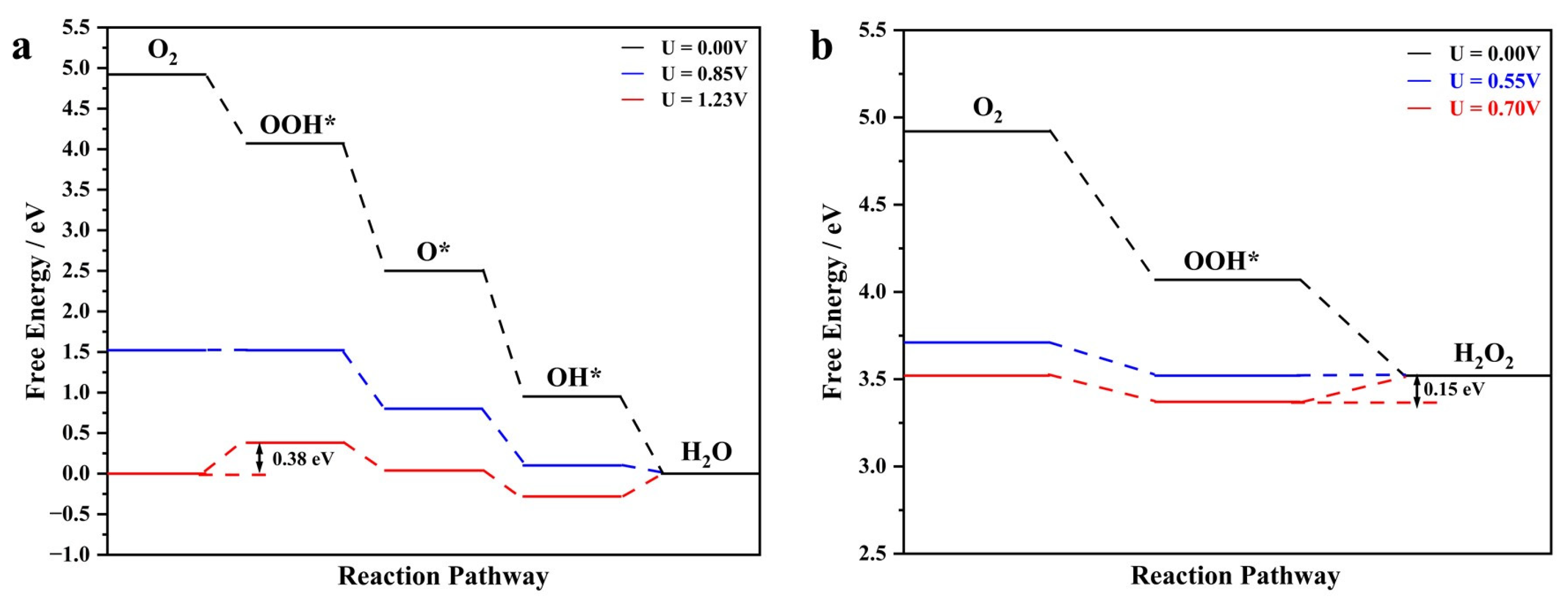
2.2. Catalyst Performance
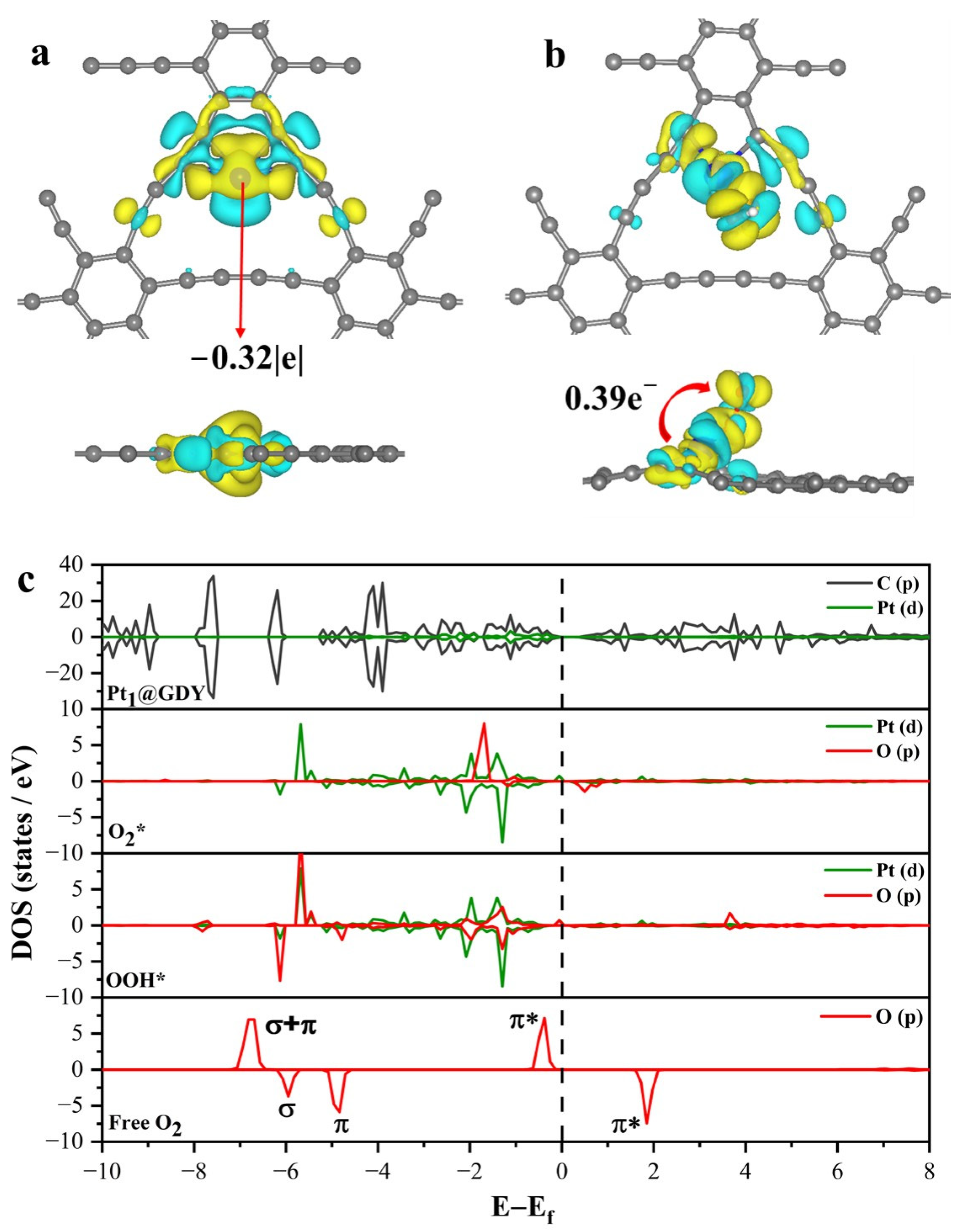
2.3. Origin of the 2e− ORR Activity
3. Materials and Methods
Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Melchionna, M.; Fornasiero, P.; Prato, M. The Rise of Hydrogen Peroxide as the Main Product by Metal-Free Catalysis in Oxygen Reductions. Adv. Mater. 2019, 31, e1802920. [Google Scholar] [CrossRef]
- Rozendal, R.A.; Leone, E.; Keller, J.; Rabaey, K. Efficient hydrogen peroxide generation from organic matter in a bioelectrochemical system. Electrochem. Commun. 2009, 11, 1752–1755. [Google Scholar] [CrossRef]
- Kosaka, K.; Yamada, H.; Shishida, K.; Echigo, S.; Minear, R.A.; Tsuno, H.; Matsui, S. Evaluation of the treatment performance of a multistage ozone/hydrogen peroxide process by decomposition by-products. Water Res. 2001, 35, 3587–3594. [Google Scholar] [CrossRef]
- Yamanaka, I.; Murayama, T. Neutral H2O2 synthesis by electrolysis of water and O2. Angew. Chem. Int. Ed. Engl. 2008, 47, 1900–1902. [Google Scholar] [CrossRef]
- Ksibi, M. Chemical oxidation with hydrogen peroxide for domestic wastewater treatment. Chem. Eng. J. 2006, 119, 161–165. [Google Scholar] [CrossRef]
- Zhang, X.L.; Su, X.; Zheng, Y.R.; Hu, S.J.; Shi, L.; Gao, F.Y.; Yang, P.P.; Niu, Z.Z.; Wu, Z.Z.; Qin, S.; et al. Strongly Coupled Cobalt Diselenide Monolayers for Selective Electrocatalytic Oxygen Reduction to H2O2 under Acidic Conditions. Angew. Chem. Int. Ed. Engl. 2021, 60, 26922–26931. [Google Scholar] [CrossRef]
- Campos-Martin, J.M.; Blanco-Brieva, G.; Fierro, J.L. Hydrogen peroxide synthesis: An outlook beyond the anthraquinone process. Angew. Chem. Int. Ed. Engl. 2006, 45, 6962–6984. [Google Scholar] [CrossRef]
- Jirkovsky, J.S.; Panas, I.; Ahlberg, E.; Halasa, M.; Romani, S.; Schiffrin, D.J. Single atom hot-spots at Au-Pd nanoalloys for electrocatalytic H2O2 production. J. Am. Chem. Soc. 2011, 133, 19432–19441. [Google Scholar] [CrossRef]
- Zheng, Z.; Ng, Y.H.; Wang, D.W.; Amal, R. Epitaxial Growth of Au-Pt-Ni Nanorods for Direct High Selectivity H2O2 Production. Adv. Mater. 2016, 28, 9949–9955. [Google Scholar] [CrossRef]
- Jiang, Y.; Ni, P.; Chen, C.; Lu, Y.; Yang, P.; Kong, B.; Fisher, A.; Wang, X. Selective Electrochemical H2O2 Production through Two-Electron Oxygen Electrochemistry. Adv. Energy Mater. 2018, 8, 1801909. [Google Scholar] [CrossRef]
- Gervasini, A.; Carniti, P.; Desmedt, F.; Miquel, P. Liquid Phase Direct Synthesis of H2O2: Activity and Selectivity of Pd-Dispersed Phase on Acidic Niobia-Silica Supports. ACS Catal. 2017, 7, 4741–4752. [Google Scholar] [CrossRef]
- Arrigo, R.; Schuster, M.E.; Abate, S.; Giorgianni, G.; Centi, G.; Perathoner, S.; Wrabetz, S.; Pfeifer, V.; Antonietti, M.; Schlögl, R. Pd Supported on Carbon Nitride Boosts the Direct Hydrogen Peroxide Synthesis. ACS Catal. 2016, 6, 6959–6966. [Google Scholar] [CrossRef]
- Edwards, J.K.; Ntainjua, E.; Carley, A.F.; Herzing, A.A.; Kiely, C.J.; Hutchings, G.J. Direct synthesis of H2O2 from H2 and O2 over gold, palladium, and gold-palladium catalysts supported on acid-pretreated TiO2. Angew. Chem. Int. Ed. Engl. 2009, 48, 8512–8515. [Google Scholar] [CrossRef]
- Li, F.; Shao, Q.; Hu, M.; Chen, Y.; Huang, X. Hollow Pd–Sn Nanocrystals for Efficient Direct H2O2 Synthesis: The Critical Role of Sn on Structure Evolution and Catalytic Performance. ACS Catal. 2018, 8, 3418–3423. [Google Scholar] [CrossRef]
- Sun, Y.; Sinev, I.; Ju, W.; Bergmann, A.; Dresp, S.; Kühl, S.; Spöri, C.; Schmies, H.; Wang, H.; Bernsmeier, D.; et al. Efficient Electrochemical Hydrogen Peroxide Production from Molecular Oxygen on Nitrogen-Doped Mesoporous Carbon Catalysts. ACS Catal. 2018, 8, 2844–2856. [Google Scholar] [CrossRef]
- Han, L.; Sun, Y.; Li, S.; Cheng, C.; Halbig, C.E.; Feicht, P.; Hübner, J.L.; Strasser, P.; Eigler, S. In-Plane Carbon Lattice-Defect Regulating Electrochemical Oxygen Reduction to Hydrogen Peroxide Production over Nitrogen-Doped Graphene. ACS Catal. 2019, 9, 1283–1288. [Google Scholar] [CrossRef]
- Iglesias, D.; Giuliani, A.; Melchionna, M.; Marchesan, S.; Criado, A.; Nasi, L.; Bevilacqua, M.; Tavagnacco, C.; Vizza, F.; Prato, M.; et al. N-Doped Graphitized Carbon Nanohorns as a Forefront Electrocatalyst in Highly Selective O2 Reduction to H2O2. Chem 2018, 4, 106–123. [Google Scholar] [CrossRef]
- Sun, Y.; Silvioli, L.; Sahraie, N.R.; Ju, W.; Li, J.; Zitolo, A.; Li, S.; Bagger, A.; Arnarson, L.; Wang, X.; et al. Activity-Selectivity Trends in the Electrochemical Production of Hydrogen Peroxide over Single-Site Metal-Nitrogen-Carbon Catalysts. J. Am. Chem. Soc. 2019, 141, 12372–12381. [Google Scholar] [CrossRef]
- Gao, J.; Yang, H.B.; Huang, X.; Hung, S.-F.; Cai, W.; Jia, C.; Miao, S.; Chen, H.M.; Yang, X.; Huang, Y.; et al. Enabling Direct H2O2 Production in Acidic Media through Rational Design of Transition Metal Single Atom Catalyst. Chem 2020, 6, 658–674. [Google Scholar] [CrossRef] [Green Version]
- Li, B.Q.; Zhao, C.X.; Liu, J.N.; Zhang, Q. Electrosynthesis of Hydrogen Peroxide Synergistically Catalyzed by Atomic Co-Nx-C Sites and Oxygen Functional Groups in Noble-Metal-Free Electrocatalysts. Adv. Mater. 2019, 31, e1808173. [Google Scholar] [CrossRef]
- Li, Y.; Xu, L.; Liu, H.; Li, Y. Graphdiyne and graphyne: From theoretical predictions to practical construction. Chem. Soc. Rev. 2014, 43, 2572–2586. [Google Scholar] [CrossRef]
- Jiao, Y.; Du, A.; Hankel, M.; Zhu, Z.; Rudolph, V.; Smith, S.C. Graphdiyne: A versatile nanomaterial for electronics and hydrogen purification. Chem. Commun. 2011, 47, 11843–11845. [Google Scholar] [CrossRef]
- He, J.; Ma, S.Y.; Zhou, P.; Zhang, C.X.; He, C.; Sun, L.Z. Magnetic Properties of Single Transition-Metal Atom Absorbed Graphdiyne and Graphyne Sheet from DFT+U Calculations. J. Phys. Chem. C 2012, 116, 26313–26321. [Google Scholar] [CrossRef] [Green Version]
- He, T.; Matta, S.K.; Will, G.; Du, A. Transition-Metal Single Atoms Anchored on Graphdiyne as High-Efficiency Electrocatalysts for Water Splitting and Oxygen Reduction. Small Methods 2019, 3, 1800419. [Google Scholar] [CrossRef]
- Yu, J.; Cao, C.; Jin, H.; Chen, W.; Shen, Q.; Li, P.; Zheng, L.; He, F.; Song, W.; Li, Y. Uniform single atomic Cu1-C4 sites anchored in graphdiyne for hydroxylation of benzene to phenol. Natl. Sci. Rev. 2022, 9, nwac018. [Google Scholar] [CrossRef]
- Kulkarni, A.; Siahrostami, S.; Patel, A.; Norskov, J.K. Understanding Catalytic Activity Trends in the Oxygen Reduction Reaction. Chem. Rev. 2018, 118, 2302–2312. [Google Scholar] [CrossRef]
- Ye, C.-W.; Xu, L. Recent advances in the design of a high performance metal–nitrogen–carbon catalyst for the oxygen reduction reaction. J. Mater. Chem. A 2021, 9, 22218–22247. [Google Scholar] [CrossRef]
- Viswanathan, V.; Hansen, H.A.; Rossmeisl, J.; Norskov, J.K. Unifying the 2e- and 4e- Reduction of Oxygen on Metal Surfaces. J. Phys. Chem. Lett. 2012, 3, 2948–2951. [Google Scholar] [CrossRef] [Green Version]
- Siahrostami, S.; Verdaguer-Casadevall, A.; Karamad, M.; Deiana, D.; Malacrida, P.; Wickman, B.; Escudero-Escribano, M.; Paoli, E.A.; Frydendal, R.; Hansen, T.W.; et al. Enabling direct H2O2 production through rational electrocatalyst design. Nat. Mater. 2013, 12, 1137–1143. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.; Fang, Q.; Xia, Q.; Hu, A.; Sun, F.; Zhang, W.; Yu, Y.; Zhuang, G.; Jiang, J.; Wang, J. Dual effect of the coordination field and sulphuric acid on the properties of a single-atom catalyst in the electrosynthesis of H2O2. Phys. Chem. Chem. Phys. 2021, 23, 21338–21349. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Blochl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Valdés, Á.; Qu, Z.W.; Kroes, G.J.; Rossmeisl, J.; Nørskov, J.K. Oxidation and Photo-Oxidation of Water on TiO2 Surface. J. Phys. Chem. C 2008, 112, 9872–9879. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lian, K.; Wan, Q.; Jiang, R.; Lin, S. Electrocatalytic Oxygen Reduction to Hydrogen Peroxide on Graphdiyne-Based Single-Atom Catalysts: First-Principles Studies. Catalysts 2023, 13, 307. https://doi.org/10.3390/catal13020307
Lian K, Wan Q, Jiang R, Lin S. Electrocatalytic Oxygen Reduction to Hydrogen Peroxide on Graphdiyne-Based Single-Atom Catalysts: First-Principles Studies. Catalysts. 2023; 13(2):307. https://doi.org/10.3390/catal13020307
Chicago/Turabian StyleLian, Kangkang, Qiang Wan, Rong Jiang, and Sen Lin. 2023. "Electrocatalytic Oxygen Reduction to Hydrogen Peroxide on Graphdiyne-Based Single-Atom Catalysts: First-Principles Studies" Catalysts 13, no. 2: 307. https://doi.org/10.3390/catal13020307