
Characterization of Equilibrium Catalysts from the Fluid Catalytic Cracking Process of Atmospheric Residue

 ,
, 
Abstract
:1. Introduction
2. Results and Discussion
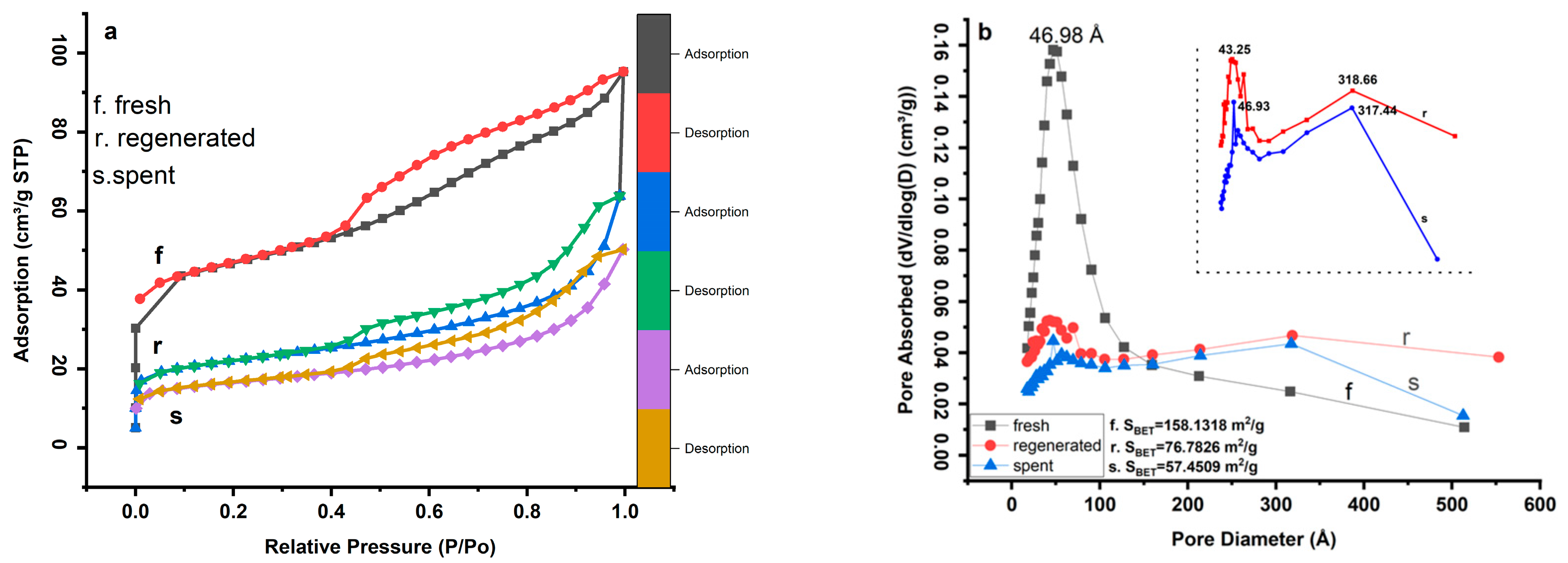
2.1. N2 Isotherm and Pore Size Distribution Analysis
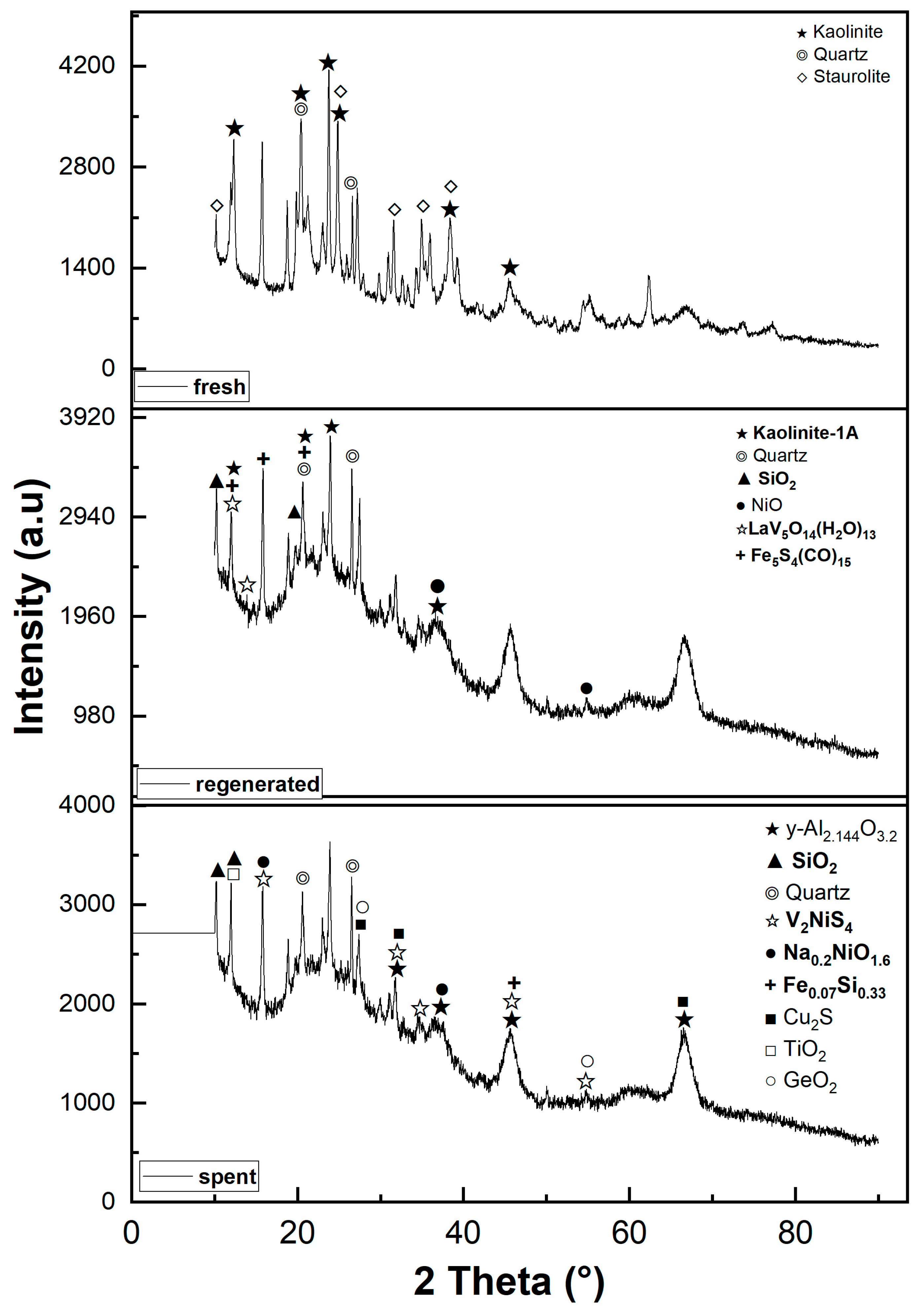
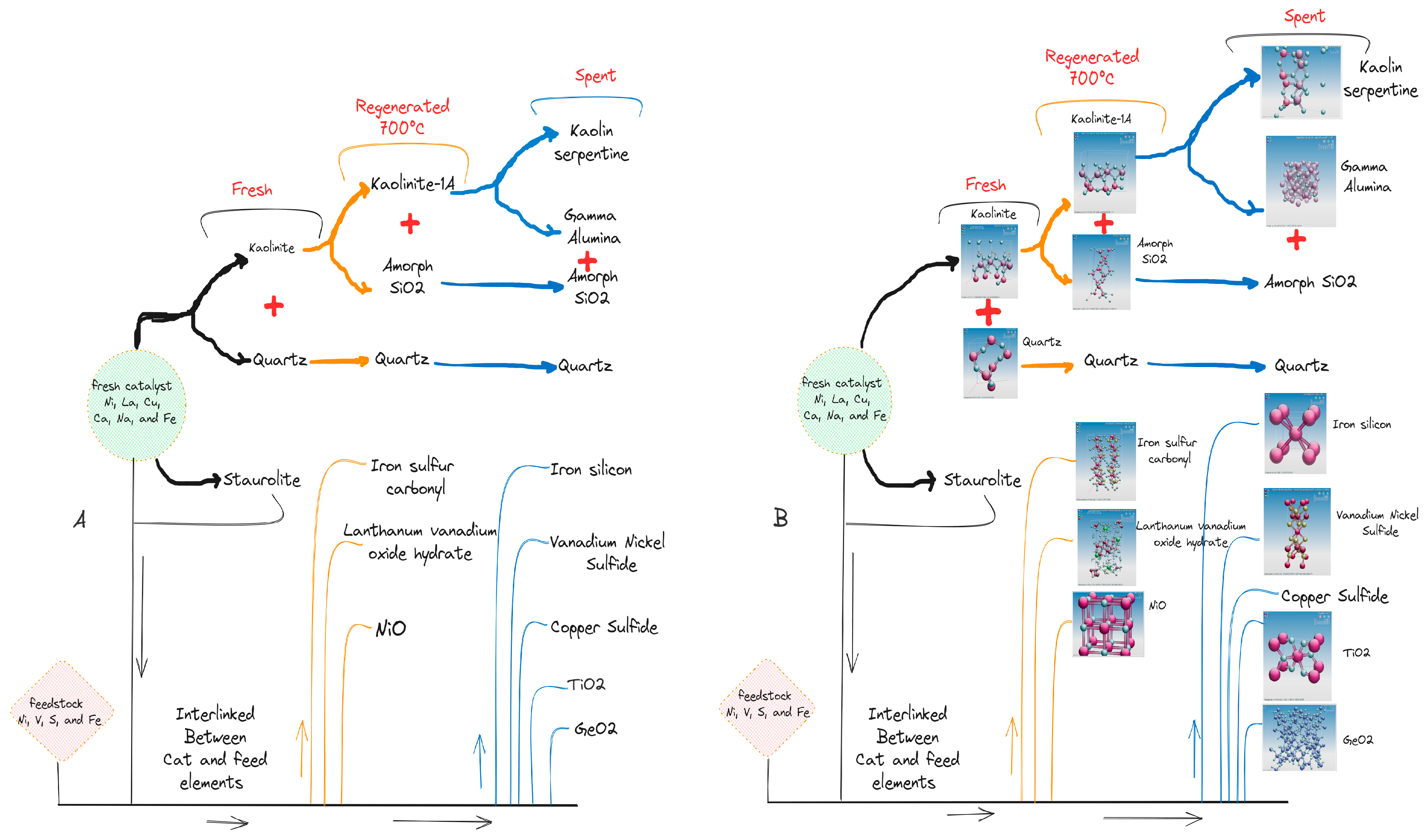
2.2. Crystallography Analysis
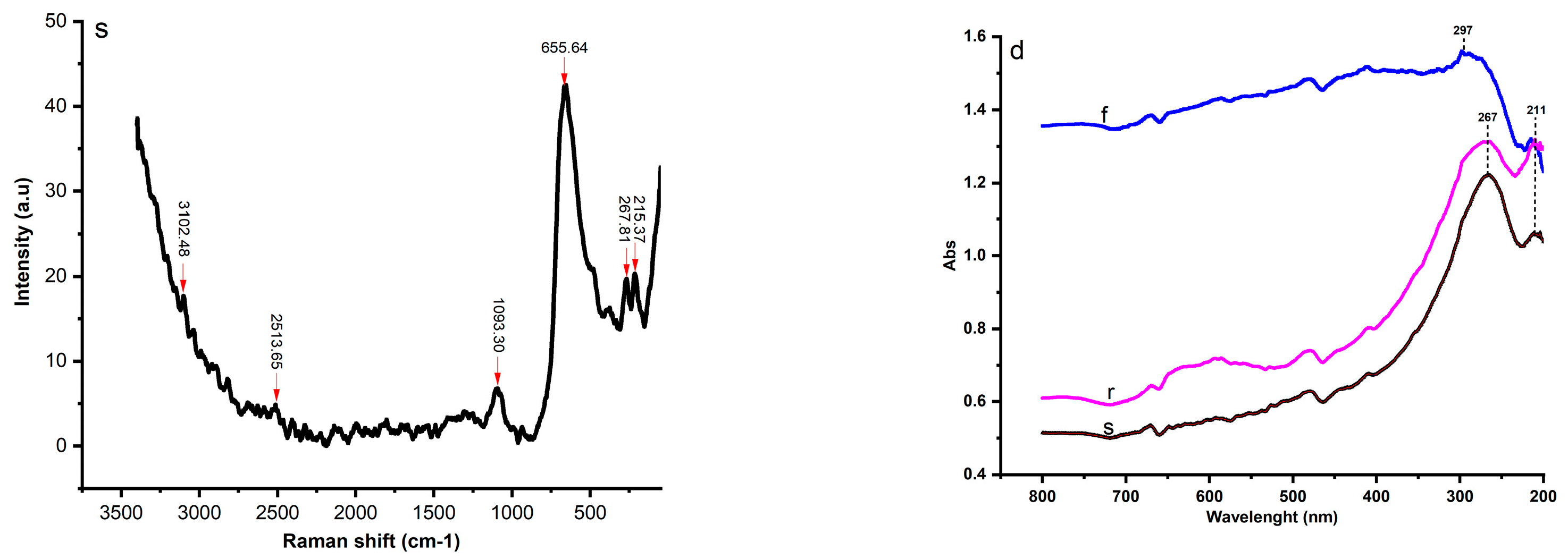
2.3. UV-Visible Near-Infrared Spectra and Raman Spectra Analysis
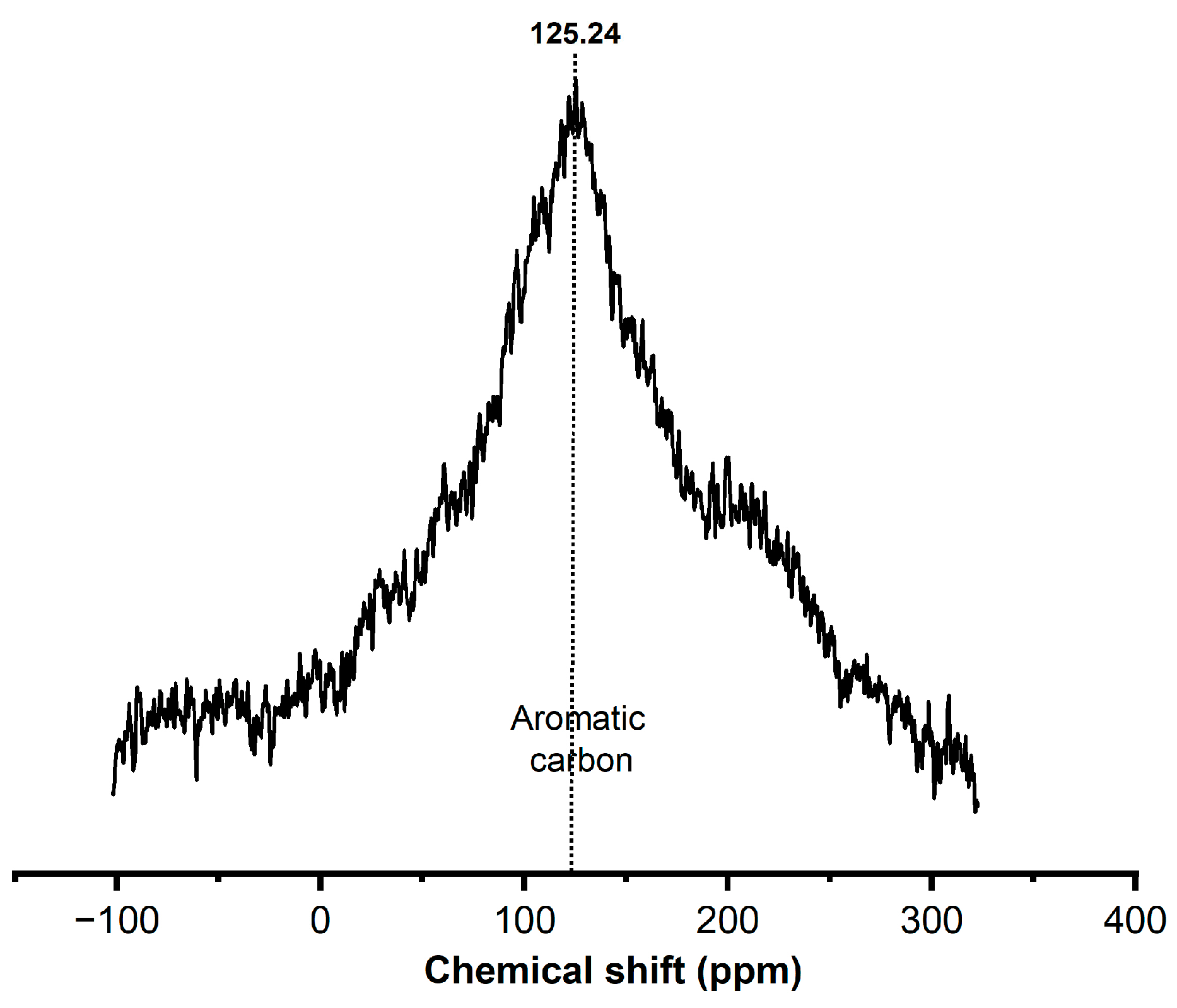
2.4. Solid-State LECO Carbon Analyzer and NMR 13C Studies
2.5. Catalyst Morphology Changes during the FCC Process
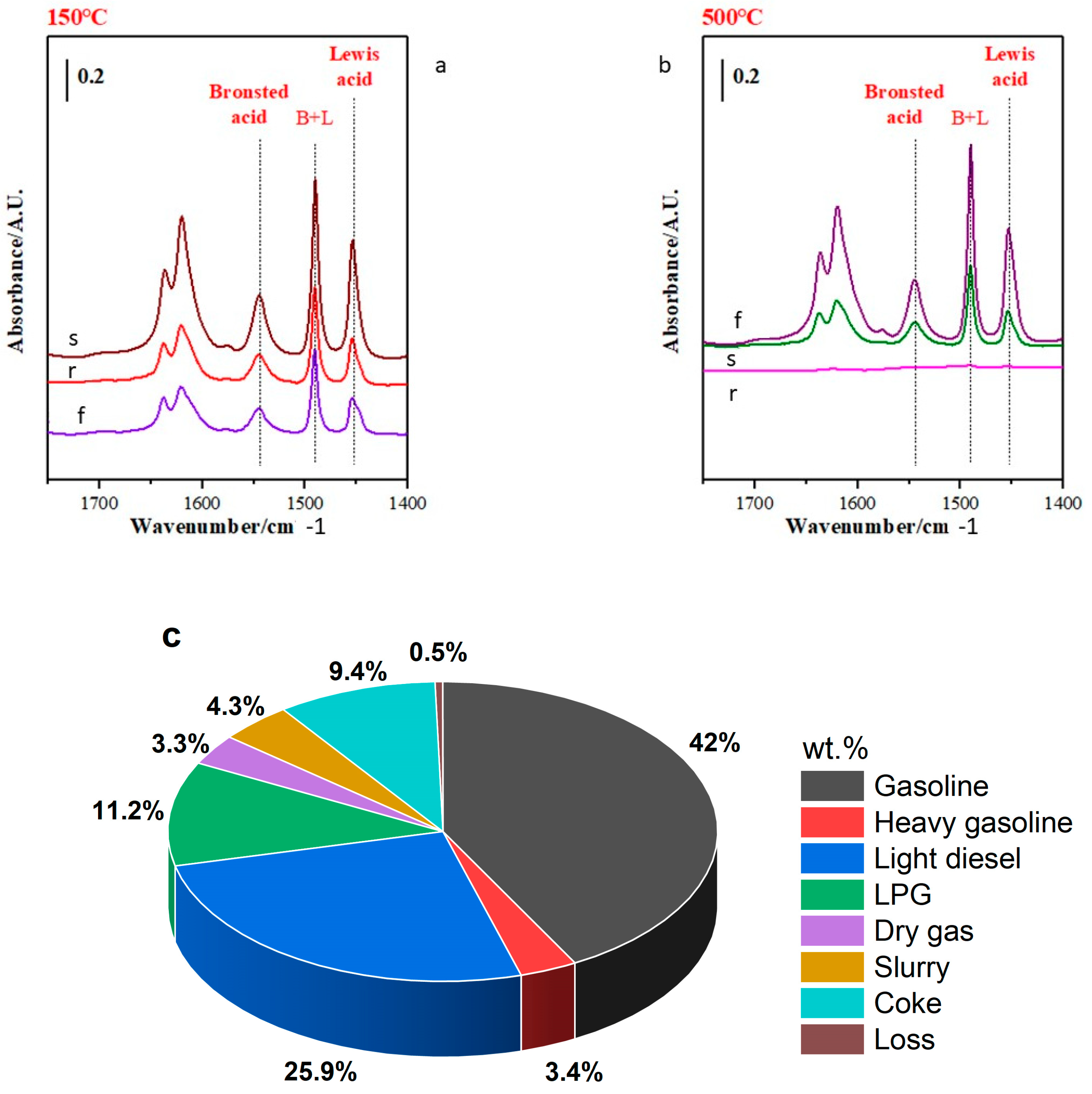
2.6. Catalyst Acidity Evolution during the FCC Process
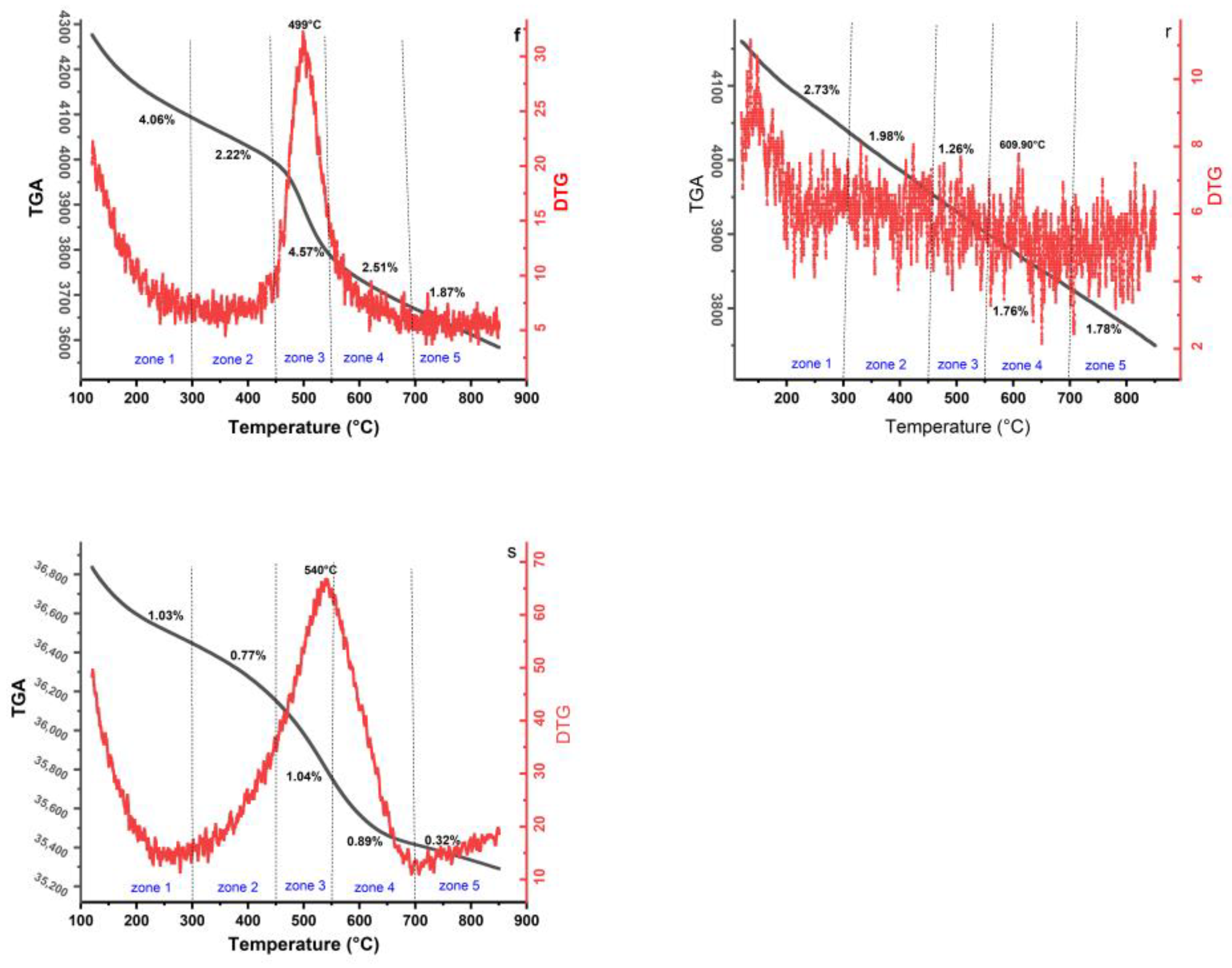
2.7. Thermal Analysis
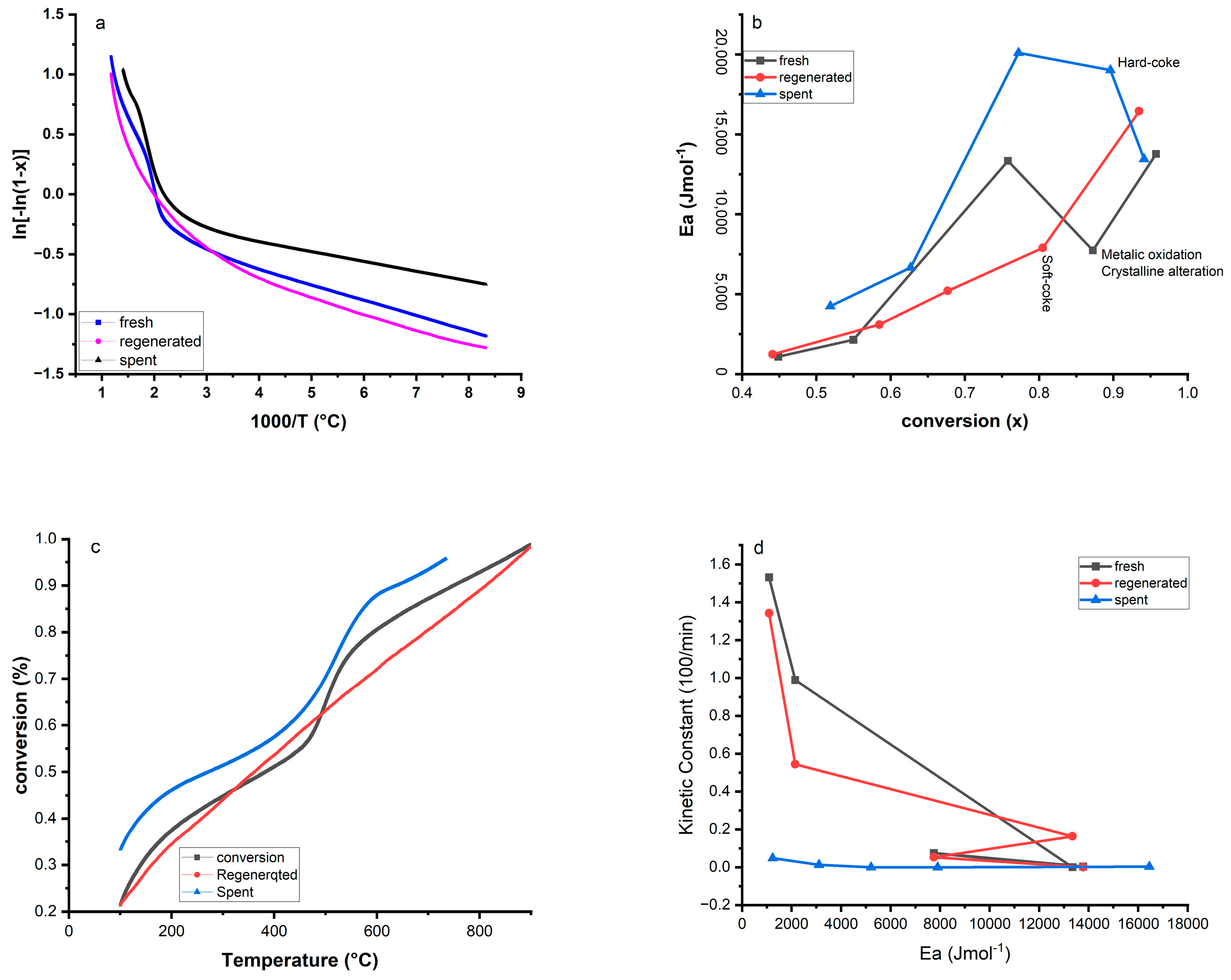
2.8. Kinetics and Thermodynamics Parameters Calculation Using TGA
3. Material and Methods
3.1. Sampling of Catalyst
3.2. Cracking Reaction and Regeneration Conditions
3.3. Catalyst Characterization
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, Y.S.; Lu, X.; Owen, R.E.; Manos, G.; Xu, R.; Wang, F.R.; Maskell, W.C.; Shearing, P.R.; Brett, D.J.L. Fine structural changes of fluid catalytic catalysts and characterization of coke formed resulting from heavy oil devolatilization. Appl. Catal. B. 2020, 263, 118329. [Google Scholar] [CrossRef]
- Jiménez-García, G.; Aguilar-López, R.; Maya-Yescas, R. The fluidized-bed catalytic cracking unit building its future environment. Fuel 2011, 90, 3531–3541. [Google Scholar] [CrossRef]
- Corma, A.; Martínez, A. Zeolites in refining and petrochemistry. Stud. Surf. Sci. Catal. 2005, 157, 337–366. [Google Scholar] [CrossRef]
- Krumeich, F.; Ihli, J.; Shu, Y.; Cheng, W.C.; Van Bokhoven, J.A. Structural Changes in Deactivated Fluid Catalytic Cracking Catalysts Determined by Electron Microscopy. ACS Catal. 2018, 8, 4591–4599. [Google Scholar] [CrossRef]
- Velthoen, M.E.Z.; Paioni, A.L.; Teune, I.E.; Baldus, M.; Weckhuysen, B.M. Matrix Effects in a Fluid Catalytic Cracking Catalyst Particle: Influence on Structure, Acidity, and Accessibility. Chem. A Eur. J. 2020, 26, 11995–12009. [Google Scholar] [CrossRef]
- Almas, Q.; Naeem, M.A.; Baldanza, M.A.S.; Solomon, J.; Kenvin, J.C.; Müller, C.R.; Da Silva, V.T.; Jones, C.W.; Sievers, C. Transformations of FCC catalysts and carbonaceous deposits during repeated reaction-regeneration cycles. Catal. Sci. Technol. 2019, 9, 6977–6992. [Google Scholar] [CrossRef]
- Xie, Y.; Zhang, Y.; He, L.; Jia, C.Q.; Yao, Q.; Sun, M.; Ma, X. Anti-deactivation of zeolite catalysts for residue fluid catalytic cracking. Appl. Catal. A Gen. 2023, 657, 119159. [Google Scholar] [CrossRef]
- Etim, U.J.; Bai, P.; Liu, X.; Subhan, F.; Ullah, R.; Yan, Z. Vanadium and nickel deposition on FCC catalyst: Influence of residual catalyst acidity on catalytic products. Microporous Mesoporous Mater. 2019, 273, 276–285. [Google Scholar] [CrossRef]
- Yan, Z.; Fan, Y.; Bi, X.; Lu, C. Dynamic behaviors of feed jets and catalyst particles in FCC feed injection zone. Chem. Eng. Sci. 2018, 189, 380–393. [Google Scholar] [CrossRef]
- Liu, Y.; Meirer, F.; Krest, C.M.; Webb, S.; Weckhuysen, B.M. Relating structure and composition with accessibility of a single catalyst particle using correlative 3-dimensional micro-spectroscopy. Nat. Commun. 2016, 7, 12634. [Google Scholar] [CrossRef]
- Meirer, F.; Morris, D.T.; Kalirai, S.; Liu, Y.; Andrews, J.C.; Weckhuysen, B.M. Mapping metals incorporation of a whole single catalyst particle using element specific X-ray nanotomography. J. Am. Chem. Soc. 2015, 137, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Ihli, J.; Jacob, R.R.; Holler, M.; Guizar-Sicairos, M.; Diaz, A.; Da Silva, J.C.; Sanchez, D.F.; Krumeich, F.; Grolimund, D.; Taddei, M.; et al. A three-dimensional view of structural changes caused by deactivation of fluid catalytic cracking catalysts. Nat. Commun. 2017, 8, 809. [Google Scholar] [CrossRef] [PubMed]
- Wallenstein, D.; Farmer, D.; Knoell, J.; Fougret, C.M.; Brandt, S. Progress in the deactivation of metals contaminated FCC catalysts by a novel catalyst metallation method. Appl. Catal. A Gen. 2013, 462–463, 91–99. [Google Scholar] [CrossRef]
- Liu, C.; Deng, Y.; Pan, Y.; Zheng, S.; Gao, X. Interactions between heavy metals and clay matrix in fluid catalytic cracking catalysts. Appl. Catal. A Gen. 2004, 257, 145–150. [Google Scholar] [CrossRef]
- Pala-Rosas, I.; Contreras, J.L.; Salmones, J.; López-Medina, R.; Angeles-Beltrán, D.; Zeifert, B.; Navarrete-Bolaños, J.; González-Hernández, N.N. Effects of the Acidic and Textural Properties of Y-Type Zeolites on the Synthesis of Pyridine and 3-Picoline from Acrolein and Ammonia. Catalysts 2023, 13, 652. [Google Scholar] [CrossRef]
- Zhang, H.; Shen, Z.; Gong, J.; Liu, H. Influences of regeneration atmospheres on structural transformation and renderability of FCC catalyst. Chin. J. Chem. Eng. 2023, 63, 71–80. [Google Scholar] [CrossRef]
- Duarte, L.; Garzón, L.; Baldovino-Medrano, V.G. An analysis of the physicochemical properties of spent catalysts from an industrial hydrotreating unit. Catal. Today 2019, 338, 100–107. [Google Scholar] [CrossRef]
- Vogt, E.T.C.; Fu, D.; Weckhuysen, B.M. Carbon Deposit Analysis in Catalyst Deactivation, Regeneration, and Rejuvenation. Angew. Chem. Int. Ed. 2023, 62, e202300319. [Google Scholar] [CrossRef]
- Wang, B.; Li, N.; Zhang, Q.; Li, C.; Yang, C.; Shan, H. Studies on the preliminary cracking: The reasons why matrix catalytic function is indispensable for the catalytic cracking of feed with large molecular size. J. Energ. Chem. 2016, 25, 641–653. [Google Scholar] [CrossRef]
- Hernández-Beltrán, F.; López-Salinas, E.; García-de-León, R.; Mogica-Martínez, E.; Moreno-Mayorga, J.C.; González-Serrano, R. Study on the deactivation-aging patterns of fluid cracking catalysts in industrial units. Stud. Surf. Sci. Catal. 2001, 134, 87–106. [Google Scholar] [CrossRef]
- Ruiz-Morales, Y.; Miranda-Olvera, A.D.; Portales-Martínez, B.; Domínguez, J.M. Determination of13C NMR Chemical Shift Structural Ranges for Polycyclic Aromatic Hydrocarbons (PAHs) and PAHs in Asphaltenes: An Experimental and Theoretical Density Functional Theory Study. Energ. Fuels 2019, 33, 7950–7970. [Google Scholar] [CrossRef]
- Behera, B.; Ray, S.S. Structural changes of FCC catalyst from fresh to regeneration stages and associated coke in a FCC refining unit: A multinuclear solid state NMR approach. Catal. Today 2009, 141, 195–204. [Google Scholar] [CrossRef]
- Wright, A.F.; Bailey, J.S. Organic carbon, total carbon, and total nitrogen determinations in soils of variable calcium carbonate contents using a Leco CN-2000 dry combustion analyzer. Commun. Soil Sci. Plant Anal. 2011, 32, 3243–3258. [Google Scholar] [CrossRef]
- Baldovino-Medrano, V.G.; Niño-Celis, V.; Isaacs-Giraldo, R.; Refugio, E. Systematic analysis of the nitrogen adsorption-desorption isotherms recorded for a series of microporous-mesoporous amorphous aluminosilicates using classical methods. J. Chem. Eng. Data 2023, 68, 2512–2528. [Google Scholar] [CrossRef]
- Gueu, S.; Finqueneisel, G.; Zimny, T.; Bartier, D.; Yao, B.K. Physicochemical characterization of three natural clays used as adsorbent for the humic acid removal from aqueous solution. Adsorpt. Sci. Technol. 2019, 37, 77–94. [Google Scholar] [CrossRef]
- Wrabetz, S.; Yang, X.; Tzolova-Müller, G.; Schlögl, R.; Jentoft, F.C. Characterization of Catalysts in Their Active State by Adsorption Microcalorimetry: Experimental Design and Application to Sulfated Zirconia. J. Catal. 2010, 269, 351–358. [Google Scholar] [CrossRef]
- Silva, J.M.R.; de Morais Araújo, A.M.; da Costa Evangelista, J.P.; da Silva, D.R.; Gondim, A.D.; de Araujo, A.S. Evaluation of the kinetic and thermodynamic parameters in catalytic pyrolysis process of sunflower oil using Al-MCM-41 and zeolite H-ZSM-5. Fuel 2023, 333, 126225. [Google Scholar] [CrossRef]
- Patidar, K.; Singathia, A.; Vashishtha, M.; Sangal, V.K.; Upadhyaya, S. Investigation of kinetic and thermodynamic parameters approaches to non-isothermal pyrolysis of mustard stalk using model-free and master plots methods. Mater. Sci. Energ. Technol. 2022, 5, 6–14. [Google Scholar] [CrossRef]
- Vasudev, V.; Ku, X.; Lin, J. Kinetic study and pyrolysis characteristics of algal and lignocellulosic biomasses. Bioresour. Technol. 2019, 288, 121496. [Google Scholar] [CrossRef]
- Cui, W.; Zhu, D.; Tan, J.; Chen, N.; Fan, D.; Wang, J.; Han, J.; Wang, L.; Tian, P.; Liu, Z. Synthesis of mesoporous high-silica zeolite Y and their catalytic cracking performance. Chin. J. Catal. 2022, 43, 1945–1954. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Alothman, Z.A. A review: Fundamental aspects of silicate mesoporous materials. Materials 2012, 5, 2874–2902. [Google Scholar] [CrossRef]
- Hua, D.; Zhou, Z. Deactivation of Mesoporous Titanosilicate-supported WO3 Catalyst for Metathesis of Butene to Propene by Coke. J. Thermodyn. Catal. 2016, 7, 1–5. [Google Scholar] [CrossRef]
- Alabdullah, M.A.; Shoinkhorova, T.; Dikhtiarenko, A.; Ould-Chikh, S.; Rodriguez-Gomez, A.; Chung, S.H.; Alahmadi, A.O.; Hita, I.; Pairis, S.; Hazemann, J.L.; et al. Understanding catalyst deactivation during the direct cracking of crude oil. Catal. Sci. Technol. 2022, 12, 5657–5670. [Google Scholar] [CrossRef]
- Lu, G.; Lu, X.; Liu, P. Reactivation of spent FCC catalyst by mixed acid leaching for efficient catalytic cracking. J. Ind. Eng. Chem. 2020, 92, 236–242. [Google Scholar] [CrossRef]
- Palmay, P.; Medina, C.; Donoso, C.; Barzallo, D.; Bruno, J.C. Catalytic pyrolysis of recycled polypropylene using a regenerated FCC catalyst. Clean Technol. Env. Policy 2022, 25, 1539–1549. [Google Scholar] [CrossRef]
- Mathieu, Y.; Corma, A.; Echard, M.; Bories, M. Single and combined Fluidized Catalytic Cracking (FCC) catalyst deactivation by iron and calcium metal–organic contaminants. Appl. Catal. A Gen. 2014, 469, 451–465. [Google Scholar] [CrossRef]
- Cora, I.; Dódony, I.; Pekker, P. Electron crystallographic study of a kaolinite single crystal. Appl. Clay Sci. 2014, 90, 6–10. [Google Scholar] [CrossRef]
- Wright, A.F.; Lehmann, M.S. The structure of quartz at 25 and 590 °C determined by neutron diffraction. J. Solid State Chem. 1981, 36, 371–380. [Google Scholar] [CrossRef]
- Wang, Y.X.; Gies, H.; Marler, B.; Müller, U. Synthesis and crystal structure of zeolite RUB-41 obtained as calcination product of a layered precursor: A systematic approach to a new synthesis route. Chem. Mater. 2005, 17, 43–49. [Google Scholar] [CrossRef]
- Peng, X.-H.; Li, Y.-Z.; Cai, L.-X.; Wang, L.-F.; Wu, J.-G. A novel polyoxometalate supramolecular compound: [La(H2O)9] 2 [V10O28]·8H2O. Acta Crystallogr. Sect. E Struct. Rep. Online 2002, 58, i111–i113. [Google Scholar] [CrossRef]
- Koide, S. Electrical Properties of LixNi(1−x)O Single Crystals. J. Physical. Soc. Jpn. 1965, 20, 123–132. [Google Scholar] [CrossRef]
- Wei, C.H.; Dahl, L.F. The Molecular Structure of a Tricyclic Complex, [SFe(CO)3]2. Inorg Chem. 1965, 4, 1–11. [Google Scholar] [CrossRef]
- Zhou, R.-S.; Snyder, R.L. Structures and transformation mechanisms of the η, γ and θ transition aluminas. Acta Crystallogr. Sect. B: Struct. Sci. 1991, 47, 617–630. [Google Scholar] [CrossRef]
- Foster, M.D.; Friedrichs, O.D.; Bell, R.G.; Paz, F.A.A.; Klinowski, J. Chemical evaluation of hypothetical uninodal zeolites. J. Am. Chem. Soc. 2004, 126, 9769–9775. [Google Scholar] [CrossRef]
- Haiyan, L.; Liyuan, C.; Baoying, W.; Yu, F.; Gang, S.; Xaojun, B. In-situ Synthesis and Catalytic Properties of ZSM-5/Rectorite Composites as Propylene Boosting Additive in Fluid Catalytic Cracking Process. Chin. J. Chem. Eng. 2012, 20, 158–166. [Google Scholar]
- Komarneni, S.; Katsuki, H.; Furuta, S. Novel honeycomb structure: A microporous ZSM-5 and macroporous mullite composite. J. Mater. Chem. 1998, 8, 2327–2329. [Google Scholar] [CrossRef]
- Wicks, F.J. Status of the reference X-ray powder-diffraction patterns for the serpentine minerals in the PDF database—1997. Powder Diffr. 2000, 15, 42–50. [Google Scholar] [CrossRef]
- Hu, Q.; Yong, Q.; He, L.; Gu, Y.; Zeng, J. Structural evolution of kaolinite in muddy intercalation under microwave heating. Mater. Res. Express 2021, 8, 105503. [Google Scholar] [CrossRef]
- Nong, L.; Yang, X.; Zeng, L.; Liu, J. Qualitative and quantitative phase analyses of Pingguo bauxite mineral using X-ray powder diffraction and the Rietveld method. Powder Diffr. 2007, 22, 300–302. [Google Scholar] [CrossRef]
- Murugesan, T.; Ramesh, S.; Gopalakrishnan, J.; Rao, C.N.R. Ternary vanadium sulfides. J. Solid State Chem. 1982, 44, 119–125. [Google Scholar] [CrossRef]
- Kools, F.X.N.M.; Koster, A.S.; Rieck, G.D. The structures of potassium, rubidium, and caesium molybdate and tungstate. Acta Cryst. B 1970, 26, 1974–1977. [Google Scholar] [CrossRef]
- Frolov, A.A.; Krentsis, R.P.; Sidorenko, F.A.; Gel’d, P.V. Some physical properties of Co2Si and Ni2Si in the 10–350 °k temperature range. Sov. Phys. J. 1972, 15, 418–420. [Google Scholar] [CrossRef]
- Meagher, E.P.; Lager, G.A. Polyhedral Thermal Expansion in the TiO2 Polymorphs; Refinement of the Crystal Structures of Rutile and Brookite at High Temperature, Can Mineral. 1979. Available online: https://rruff-2.geo.arizona.edu/uploads/CM17_77.pdf (accessed on 4 October 2023).
- Etim, U.J.; Xu, B.; Bai, P.; Ullah, R.; Subhan, F.; Yan, Z. Role of nickel on vanadium poisoned FCC catalyst: A study of physiochemical properties. J. Energ. Chem. 2016, 25, 667–676. [Google Scholar] [CrossRef]
- Wang, Y.J.; Wang, C.; Li, L.L.; Chen, Y.; He, C.H.; Zheng, L. Assessment of ecotoxicity of spent fluid catalytic cracking (FCC) refinery catalysts on Raphidocelis subcapitata and predictive models for toxicity. Ecotoxicol. Environ. Saf. 2021, 222, 112466. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Liu, X.; Madon, R.J. Pathways for Y zeolite destruction: The role of sodium and vanadium. J. Catal. 2002, 207, 237–246. [Google Scholar] [CrossRef]
- Argyle, M.D.; Bartholomew, C.H. Heterogeneous catalyst deactivation and regeneration: A review. Catalysts 2015, 5, 145–269. [Google Scholar] [CrossRef]
- Zakariyaou, M.S.Y.; Hua, Y.; Makaou, O.A.D. A Four-Lump Kinetic Model for Atmospheric Residue Conversion in the Fluid Catalytic Cracking Unit: Effect of the Inlet Gas Oil Temperature and Catalyst-Oil Weight Ratio on the Catalytic Reaction Behavior. E. Afr. Sch. J. Eng. Comput. Sci. 2023, 6, 39–42. [Google Scholar] [CrossRef]
- Jonsson, R.; Ho, P.H.; Wang, A.; Skoglundh, M.; Olsson, L. The impact of lanthanum and zeolite structure on hydrocarbon storage. Catalysts 2021, 11, 635. [Google Scholar] [CrossRef]
- Hallac, B.B.; Brown, J.C.; Stavitski, E.; Harrison, R.G.; Argyle, M.D. In situ UV-visible assessment of iron-based high-temperature water-gas shift catalysts promoted with Lanthana: An extent of reduction study. Catalysts 2018, 8, 63. [Google Scholar] [CrossRef]
- Alcaraz, L.; Rodríguez-Largo, O.; Álvarez-Montes, M.; López, F.A.; Baudín, C. Effect of lanthanum content on physicochemical properties and thermal evolution of spent and beneficiated spent FCC catalysts. Ceram. Int. 2022, 48, 17691–17702. [Google Scholar] [CrossRef]
- Haddadnezhad, M.N.; Babaei, A.; Molaei, M.J.; Ataie, A. Synthesis and characterization of lanthanum nickelate nanoparticles with Rudllesden-Popper crystal structure for cathode materials of solid oxide fuel cells. J. Ultrafine Grained Nanostruct. Mater. 2020, 53, 98–109. [Google Scholar] [CrossRef]
- Zhou, J.; Zhao, J.; Zhang, J.; Zhang, T.; Ye, M.; Liu, Z. Regeneration of catalysts deactivated by coke deposition: A review. Chin. J. Catal. 2020, 41, 1048–1061. [Google Scholar] [CrossRef]
- Tsai, Y.L.; Huang, E.; Li, Y.H.; Hung, H.T.; Jiang, J.H.; Liu, T.C.; Fang, J.N.; Chen, H.F. Raman spectroscopic characteristics of zeolite group minerals. Minerals 2021, 11, 167. [Google Scholar] [CrossRef]
- Yu, J.; Guo, Q.; Ding, L.; Gong, Y.; Yu, G. Studying effects of solid structure evolution on gasification reactivity of coal chars by in-situ Raman spectroscopy. Fuel 2020, 270, 117603. [Google Scholar] [CrossRef]
- Sadezky, A.; Muckenhuber, H.; Grothe, H.; Niessner, R.; Pöschl, U. Raman microspectroscopy of soot and related carbonaceous materials: Spectral analysis and structural information. Carbon 2005, 43, 1731–1742. [Google Scholar] [CrossRef]
- Kukade, S.; Kumar, P.; Rao, P.V.C.; Choudary, N.V. Comparative study of attrition measurements of commercial FCC catalysts by ASTM fluidized bed and jet cup test methods. Powder Technol. 2016, 301, 472–477. [Google Scholar] [CrossRef]
- Zhang, X.; Han, Y.; Li, D.; Zhang, Z.; Ma, X. Study on attrition of spherical-shaped Mo/HZSM-5 catalyst for methane dehydro-aromatization in a gas–solid fluidized bed. Chin. J. Chem. Eng. 2021, 38, 172–183. [Google Scholar] [CrossRef]
- Bemrose, C.R.; Bridgwater, J. A review of attrition and attrition test methods. Powder Technol. 1987, 49, 97–126. [Google Scholar] [CrossRef]
- Hao, J.; Zhao, Y.; Ye, M.; Liu, Z. Influence of Temperature on Fluidized-Bed Catalyst Attrition Behavior. Chem. Eng. Technol. 2016, 39, 927–934. [Google Scholar] [CrossRef]
- Miandad, R.; Barakat, M.A.; Rehan, M.; Aburiazaiza, A.S.; Ismail, I.M.I.; Nizami, A.S. Plastic waste to liquid oil through catalytic pyrolysis using natural and synthetic zeolite catalysts. Waste Manag. 2017, 69, 66–78. [Google Scholar] [CrossRef]
- Meisel, S.L. Catalytic Cracking: Catalysis. Chemistry, and Kinetics. Wojciechowski, B.W. and Corma, A. Marcel Dekker, 1986, 236 pp. $65.00 (U.S. and Canada), $66.00 (all other countries). AIChE J. 1987, 33, 1581. [Google Scholar] [CrossRef]
- Feng, R.; Bai, P.; Liu, S.; Zhang, P.; Liu, X.; Yan, Z.; Zhang, Z.; Gao, X. The application of mesoporous alumina with rich Brönsted acidic sites in FCC catalysts. Appl. Petrochem. Res. 2014, 4, 367–372. [Google Scholar] [CrossRef]
- Chester, A.W. Studies on the Metal Poisoning and Metal Resistance of Zeolitic Cracking Catalysts. Ind. Eng. Chem. Res. 1987, 26, 863–869. [Google Scholar] [CrossRef]
- Xu, J.; Zhou, W.; Wang, J.; Li, Z.; Ma, J. Characterization and analysis of carbon deposited during the dry reforming of methane over Ni/La2O3/Al2O3 catalysts. Cuihua Xuebao/Chin. J. Catal. 2009, 30, 1076–1084. [Google Scholar] [CrossRef]
- Luo, J.Z.; Yu, Z.L.; Ng, C.F.; Au, C.T. CO2/CH4 reforming over Ni-La2O3/5A: An investigation on carbon deposition and reaction steps. J. Catal. 2000, 194, 198–210. [Google Scholar] [CrossRef]
- Zhang, Z.; Verykios, X.E. Carbon dioxide reforming of methane to synthesis gas over Ni/La2O3 catalysts. Appl. Catal. A Gen. 1996, 138, 109–133. [Google Scholar] [CrossRef]
- Ramukutty, S.; Ramachandran, E. Reaction Rate Models for the Thermal Decomposition of Ibuprofen Crystals. J. Cryst. Process Technol. 2014, 4, 71–78. [Google Scholar] [CrossRef]
- Arjmandi, R.; Hassan, A.; Haafiz, M.K.M.; Zakaria, Z.; Islam, M.S. Effect of hydrolysed cellulose nanowhiskers on properties of montmorillonite/polylactic acid nanocomposites. Int. J. Biol. Macromol. 2016, 82, 998–1010. [Google Scholar] [CrossRef]
- Trache, D. Comments on “thermal degradation behavior of hypochlorite-oxidized starch nanocrystals under different oxidized levels”. Carbohydr Polym. 2016, 151, 535–537. [Google Scholar] [CrossRef]
- Farrukh, M.A.; Butt, K.M.; Chong, K.K.; Chang, W.S. Photoluminescence emission behavior on the reduced band gap of Fe doping in CeO2-SiO2 nanocomposite and photophysical properties. J. Saudi Chem. Soc. 2019, 23, 561–575. [Google Scholar] [CrossRef]
- Del Mar Graciani, M.; Rodríguez, A.; Muñoz, M.; Moyá, M.L. Micellar solutions of sulfobetaine surfactants in water-ethylene glycol mixtures: Surface tension, fluorescence, spectroscopic, conductometric, kinetic studies. Langmuir 2005, 21, 7161–7169. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, G.; Kumar, M.; Mishra, P.K.; Upadhyay, S.N. Kinetic analysis of the slow pyrolysis of paper wastes. Biomass Convers. Biorefin. 2023, 13, 3087–3100. [Google Scholar] [CrossRef]
- Verdeş, O.; Popa, A.; Borcănescu, S.; Suba, M.; Sasca, V. Thermogravimetry Applied for Investigation of Coke Formation in Ethanol Conversion over Heteropoly Tungstate Catalysts. Catalysts 2022, 12, 1059. [Google Scholar] [CrossRef]
- Saadatkhah, N.; Garcia, A.C.; Ackermann, S.; Leclerc, P.; Latifi, M.; Samih, S.; Patience, G.S.; Chaouki, J. Experimental methods in chemical engineering: Thermogravimetric analysis—TGA. Can. J. Chem. Eng. 2020, 98, 34–43. [Google Scholar] [CrossRef]
- Taynara, J.; Cunha, B.; Castro, V.M.; Da Silva, D.S.; De Aguiar Pontes, D.; Da Silva, V.L.; Antônio, L.; Pontes, M.; Santos, R.C. Coke Deposition on Cracking Catalysts Study by Thermogravimetric Analysis. Int. J. Eng. Res. Appl. 2020, 10, 43–47. [Google Scholar] [CrossRef]
- Marcilla, A.; Gómez-Siurana, A.; Odjo, A.O.; Navarro, R.; Berenguer, D. Characterization of vacuum gas oil-low density polyethylene blends by thermogravimetric analysis. Polym. Degrad. Stab. 2008, 93, 723–730. [Google Scholar] [CrossRef]
- Ochoa, A.; Ibarra, Á.; Bilbao, J.; Arandes, J.M.; Castaño, P. Assessment of thermogravimetric methods for calculating coke combustion-regeneration kinetics of deactivated catalyst. Chem. Eng. Sci. 2017, 171, 459–470. [Google Scholar] [CrossRef]
- Aguayo, A.T.; Gayubo, A.G.; Atutxa, A.; Valle, B.; Bilbao, J. Regeneration of a HZSM-5 zeolite catalyst deactivated in the transformation of aqueous ethanol into hydrocarbons. Catal. Today 2005, 107–108, 410–416. [Google Scholar] [CrossRef]
- Ran, C.; Liu, Y.; Siddiqui, A.R.; Siyal, A.A.; Mao, X.; Kang, Q.; Fu, J.; Ao, W.; Dai, J. Pyrolysis of textile dyeing sludge in fluidized bed: Analysis of products, and migration and distribution of heavy metals. J. Clean Prod. 2019, 241, 118308. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | SBET (m2g−1) | Sext (m2g−1) | Smicro (m2g−1) | Vtotal (cm3g−1) | Vmicro (cm3g−1) | Microporosity (%) |
|---|---|---|---|---|---|---|
| Fresh | 158.13 | 94.24 | 63.8825 | 0.147 | 0.028 | 40.39 |
| Regenerated | 76.78 | 38.55 | 38.2327 | 0.098 | 0.017 | 49.79 |
| Spent | 57.45 | 28.87 | 28.5726 | 0.077 | 0.012 | 49.73 |
| property | Fresh | Regenerated | Spent |
|---|---|---|---|
| Composition (wt.%) | |||
| CO2 | 9.64 | 3.56 | 9.58 |
| N | 0.27 | ||
| Na2O | 0.10 | 0.12 | 0.11 |
| MgO | 0.35 | 0.14 | 0.11 |
| Al2O3 | 50.71 | 50.28 | 47.04 |
| SiO2 | 34.65 | 39.04 | 37.35 |
| P2O5 | 0.51 | 0.54 | 0.35 |
| SO3 | 0.33 | 0.2 | 0.26 |
| K2O | 0.47 | 0.54 | 0.56 |
| CaO | 0.07 | 0.12 | 0.10 |
| TiO2 | 0.22 | 0.19 | 0.20 |
| V2O5 | 0.06 | 0.05 | |
| Fe2O3 | 0.44 | 0.55 | 0.50 |
| Co2O3 | 0.12 | 0.08 | |
| NiO | 2.05 | 1.52 | |
| CuO | 0.004 | ||
| ZnO | 0.003 | 0.013 | 0.008 |
| Ga2O3 | 0.012 | 0.012 | 0.010 |
| GeO2 | 0.004 | ||
| Rb2O | 0.002 | 0.003 | |
| Y2O3 | 0.66 | 0.14 | 0.08 |
| ZrO2 | 0.003 | 0.004 | |
| Sb2O3 | 0.05 | 0.02 | |
| La2O3 | 1.18 | 1.66 | 1.61 |
| CeO2 | 0.48 | 0.32 | |
| Samples | Pyridine, μmol g−1 | |||
|---|---|---|---|---|
| Brönsted Acid | Lewis Acid | |||
| 150 °C | 500 °C | 150 °C | 500 °C | |
| Fresh | 89 | 138 | 121 | 71 |
| Regenerated | 25 | 5 | 44 | 6 |
| Spent | 14 | 0 | 37 | 2 |
| Fresh Catalyst | Regenerated Catalyst | Spent Catalyst | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Zone | T °C | Ea Jmol−1 (×102) | Jmol−1 (×10−2) | Jmol−1K−1 (×105) | Jmol−1 (×106) | Fitting Equation | R2 | Ea Jmol−1 (×102) | Jmol−1 (×10−2) | Jmol−1K−1 (×105) | Jmol−1 (×106) | Fitting Equation | R2 | Ea Jmol−1 (×102) | Jmol−1 (×10−2) | Jmol−1K−1 (×105) | Jmol−1 (×106) | Fitting Equation | R2 |
| 1 | 100–300 | 10.85 | −3.03 | 51.73 | 5.34 | Y = −0.13065x − 0.0994 | 0.99 | 12.37 | −3.02 | 58.93 | 6.06 | Y = −0.1488x −0.0984 | 0.99 | 42.45 | −2.86 | 202.24 | 20.38 | Y = −0.5106x + 0.5913 | 0.98 |
| 2 | 300–450 | 21.44 | −3.00 | 128.89 | 13.10 | Y = −0.2579x + 0.318 | 0.98 | 31.01 | −2.94 | 186.42 | 18.85 | Y = −0.373x + 0.6749 | 0.99 | 66.63 | −2.84 | 400.58 | 40.26 | Y = −0.8015x + 1.0601 | 0.97 |
| 3 | 450–550 | 133.44 | −2.63 | 913.11 | 91.52 | Y = −1.605x + 3.2677 | 0.98 | 52.08 | −2.88 | 356.37 | 35.87 | Y = −0.6264x + 1.252 | 0.99 | 201.00 | −2.59 | 1380 | 138 | Y = −2.4176x + 3.296 | 0.98 |
| 4 | 550–700 | 77.43 | −2.82 | 626.40 | 62.91 | Y = −0.9313x + 2.0443 | 0.99 | 79.01 | −2.84 | 639.18 | 64.19 | Y = −0.9503x + 1.8307 | 0.99 | 190.16 | −2.65 | 1540 | 154 | Y = −2.2872x + 3.2081 | 0.97 |
| 5 | 700–850 | 137.75 | −2.72 | 1290 | 129 | Y = −1.6568x + 3.0534 | 0.98 | 164.49 | −2.69 | 1540 | 154 | Y = −1.9784x + 3.275 | 0.98 | 134.56 | −2.77 | 1260 | 126 | Y = −1.6185x + 2.4638 | 0.98 |
| Item | Value |
|---|---|
| Density (g/cm3) | 0.9215 |
| API | 22.05 |
| Aniline point (°C) | 52.9 |
| Residual carbon (wt.%) | 5.5 |
| Nickel (ppm) | 22.9 |
| Vanadium (ppm) | 0.3 |
| Iron (ppm) | 3.2 |
| Sulfur (wt.%) | 0.24 |
| Nitrogen (wt.%) | 0.17 |
| Average molecular weight (g/mol) | 462 |
| Distillation TBP (°C) | |
| 50% | 343.0 |
| 90% | 378.0 |
| 95% | 385.0 |
| SARA fractions (wt.%) a | |
| Aromatics | 18.34 |
| Asphaltenes | 7.10 |
| Saturates | 33.44 |
| NS0 b | 41.12 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zakariyaou, S.Y.; Ye, H.; Oumarou, A.D.M.; Abdoul Aziz, M.S.; Ke, S. Characterization of Equilibrium Catalysts from the Fluid Catalytic Cracking Process of Atmospheric Residue. Catalysts 2023, 13, 1483. https://doi.org/10.3390/catal13121483
Zakariyaou SY, Ye H, Oumarou ADM, Abdoul Aziz MS, Ke S. Characterization of Equilibrium Catalysts from the Fluid Catalytic Cracking Process of Atmospheric Residue. Catalysts. 2023; 13(12):1483. https://doi.org/10.3390/catal13121483
Chicago/Turabian StyleZakariyaou, Seybou Yacouba, Hua Ye, Abdoulaye Dan Makaou Oumarou, Mamane Souley Abdoul Aziz, and Shixian Ke. 2023. "Characterization of Equilibrium Catalysts from the Fluid Catalytic Cracking Process of Atmospheric Residue" Catalysts 13, no. 12: 1483. https://doi.org/10.3390/catal13121483