
β-Mannosidase from Cellulomonas fimi: Immobilization Study and Application in the β-Mannoside Synthesis

 , , , ,
, , , , 
Abstract
:1. Introduction
2. Results and Discussion
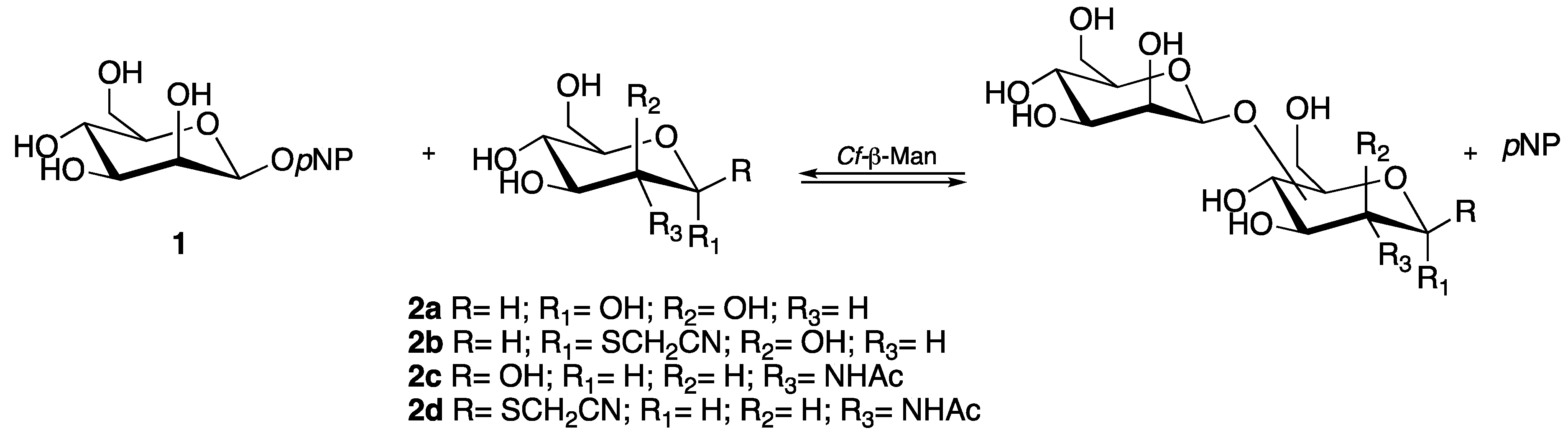
2.1. Transglycosylation: Screening of Acceptors
2.2. Transglycosylation: Reaction Conditions Optimization
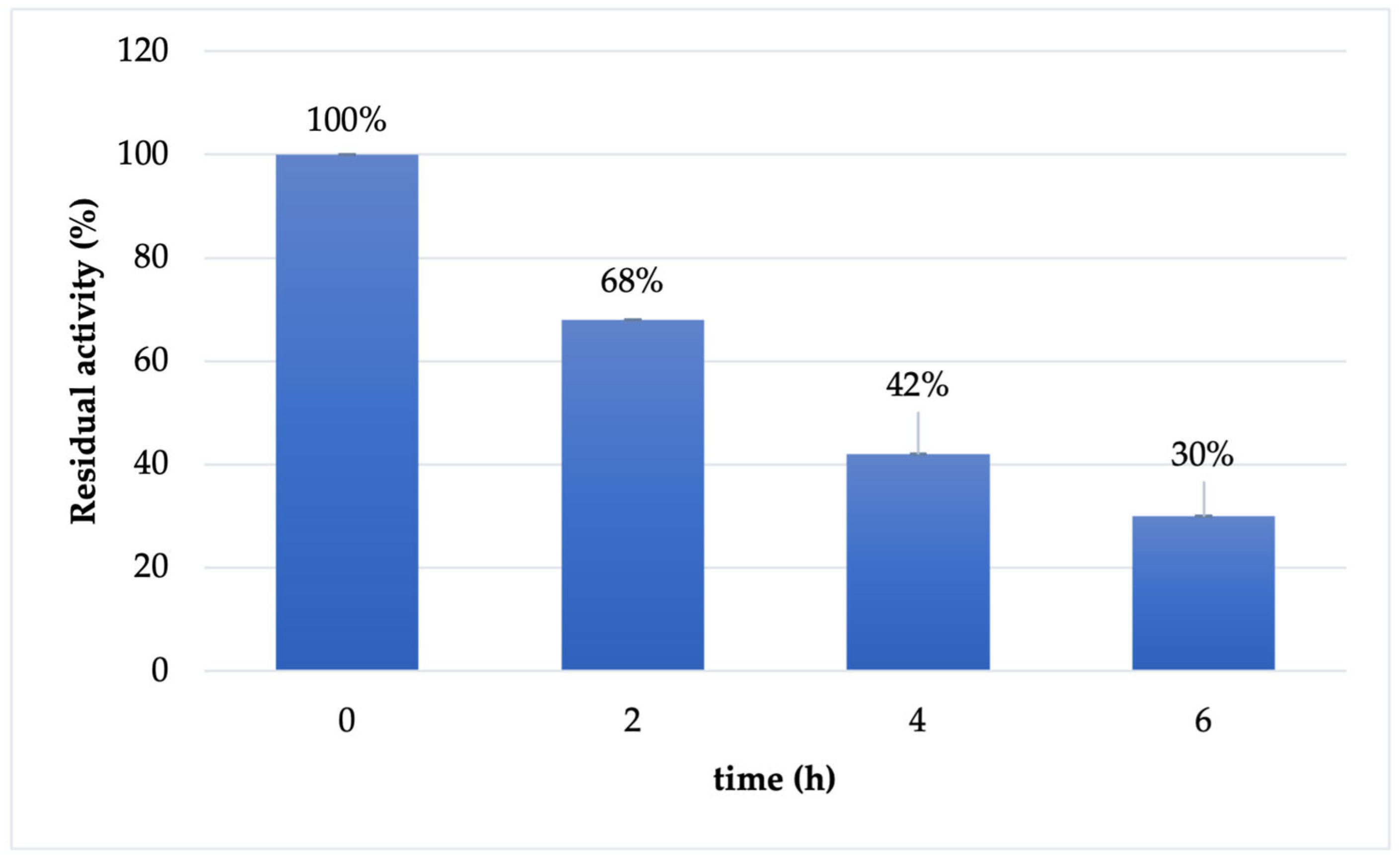
2.3. Cf-β-Man Stability
2.4. Immobilization of Cf-β-Man on Different Immobilization Carriers
2.5. Synthesis and Identification of Cyanomethyl β-d-Mannopyranosyl-(1→6)-2-Acetamido-2-Deoxy-1-thio-β-d-Glucopyranoside (3d)
3. Materials and Methods
3.1. Enzyme Characterization
3.1.1. Standard Activity Assay
3.1.2. Bradford Assay
3.2. Transglycosylation Reaction for the Synthesis of Mannose- and N-Acetylglucosamine-Based Disaccharides
3.2.1. Transglycosylation Reaction for the Synthesis of Mannose- and N-Acetylglucosamine-Based Disaccharides: Acceptor Screening
3.2.2. Transglycosylation Reaction for the Synthesis of Cyanomethyl β-d-Mannopyranosyl-(1→6)-2-Acetamido-2-Deoxy-1-thio-β-d-Glucopyranoside (3d): Reaction Conditions Screening
3.3. Cf-β-Man Stability
3.4. Enzyme Immobilization
3.4.1. Immobilization of Cf-β-Man on Glyoxyl-Sepabeads® (GLX-Sepabeads)
3.4.2. Immobilization of Cf-β-Man on Glyoxyl-Agarose (GLX-AG)
3.4.3. Immobilization of Cf-β-Man on Furan-2,5-Dicarbaldehyde-ReliZyme® (DFF-ReliZyme)
3.4.4. Immobilization of Cf-β-Man on Furan-2,5-Dicarbaldehyde-Ethylendiamine-Agarose (DFF-EDA-AG)
3.4.5. Immobilization of Cf-β-Man on Cyanogen Bromide Agarose (CNBr-AG)
3.4.6. Immobilization of Cf-β-Man on Sepabeads-Polyethylenimine (Sepabeads-PEI)
3.4.7. Immobilization of Cf-β-Man on Cobalt-Iminodiacetic Acid-Agarose (Co2+-IDA-AG)
3.5. Synthesis of Cyanomethyl β-d-Mannopyranosyl-(1→6)-2-Acetamido-2-Deoxy-1-thio-β-d-Glucopyranoside (3d) and Identification
3.5.1. Transglycosylation Reaction
3.5.2. Acetylation Reaction
3.5.3. Scale-Up and Identification
3.6. Calibration Curve of Cyanomethyl (2′,3′,4′,6′-Tetra-O-Acetyl-β-d-Mannopyranosyl)-(1→6)-2-Acetamido-3,4-di-O-Acetyl-2-Deoxy-1-thio-β-d-Glucopyranoside (4d)
3.7. Analytical Methods
3.7.1. LC-MS
3.7.2. HPLC
3.8. Building Block Synthesis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Varki, A. Biological Roles of Glycans. Glycobiology 2017, 27, 3–49. [Google Scholar] [CrossRef] [PubMed]
- Shivatare, S.S.; Shivatare, V.S.; Wong, C.-H. Glycoconjugates: Synthesis, Functional Studies, and Therapeutic Developments. Chem. Rev. 2022, 122, 15603–15671. [Google Scholar] [CrossRef] [PubMed]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in Health and Disease. Nat. Rev. Nephrol. 2019, 15, 347–366. [Google Scholar] [CrossRef] [PubMed]
- Gridley, J.J.; Osborn, H.M.I. Recent Advances in the Construction of β-D-Mannose and β-D-Mannosamine Linkages. J. Chem. Soc. Perkin Transform. 2000, 10, 1471–1491. [Google Scholar] [CrossRef]
- El Ashry, E.S.H.; Rashed, N.; Ibrahim, E.S.I. Strategies of Synthetic Methodologies for Constructing β-Mannosidic Linkage. Curr. Org. Synth. 2005, 2, 175–213. [Google Scholar] [CrossRef]
- Malgas, S.; van Dyk, J.S.; Pletschke, B.I. A Review of the Enzymatic Hydrolysis of Mannans and Synergistic Interactions between β-Mannanase, β-Mannosidase and α-Galactosidase. World J. Microbiol. Biotechnol. 2015, 31, 1167–1175. [Google Scholar] [CrossRef] [PubMed]
- Karav, S.; Cohen, J.L.; Barile, D.; Nobrega de Moura Bell, J.M.L. Recent Advances in Immobilization Strategies for Glycosidases. Biotechnol. Prog. 2017, 33, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Sadaqat, B.; Sha, C.; Ahmad Dar, M.; Dhanavade, M.J.; Sonawane, K.D.; Mohamed, H.; Shao, W.; Song, Y. Modifying Thermostability and Reusability of Hyperthermophilic Mannanase by Immobilization on Glutaraldehyde Cross-Linked Chitosan Beads. Biomolecules 2022, 12, 999. [Google Scholar] [CrossRef]
- Panwar, D.; Kaira, G.S.; Kapoor, M. Cross-Linked Enzyme Aggregates (CLEAs) and Magnetic Nanocomposite Grafted CLEAs of GH26 Endo-β-1,4-Mannanase: Improved Activity, Stability and Reusability. Int. J. Biol. Macromol. 2017, 105, 1289–1299. [Google Scholar] [CrossRef]
- Dhiman, S.; Srivastava, B.; Singh, G.; Khatri, M.; Arya, S.K. Immobilization of Mannanase on Sodium Alginate-Grafted-β-Cyclodextrin: An Easy and Cost Effective Approach for the Improvement of Enzyme Properties. Int. J. Biol. Macromol. 2020, 156, 1347–1358. [Google Scholar] [CrossRef]
- Behera, S.; Dev, M.J.; Singhal, R.S. Cross-Linked β-Mannanase Aggregates: Preparation, Characterization, and Application for Producing Partially Hydrolyzed Guar Gum. Appl. Biochem. Biotechnol. 2022, 194, 1981–2004. [Google Scholar] [CrossRef] [PubMed]
- El-Naggar, M.Y.; El-Assar, S.A.; Youssef, A.S.; El-Sersy, N.A.; Beltagy, E.A. Extracellular β-Mannanase Production by the Immobilization of the Locally Isolated Aspergillus niger. Int. J. Agric. Biol. 2006, 8, 57–62. [Google Scholar]
- Hoyos, P.; Bavaro, T.; Perona, A.; Rumbero, A.; Tengattini, S.; Terreni, M.; Hernáiz, M.J. Highly Efficient and Sustainable Synthesis of Neoglycoproteins Using Galactosidases. ACS Sustain. Chem. Eng. 2020, 8, 6282–6292. [Google Scholar] [CrossRef]
- Benítez-Mateos, A.I.; Contente, M.L. Agarose vs. Methacrylate as Material Supports for Enzyme Immobilization and Continuous Processing. Catalysts 2021, 11, 814. [Google Scholar] [CrossRef]
- Zucca, P.; Fernandez-Lafuente, R.; Sanjust, E. Agarose and Its Derivatives as Supports for Enzyme Immobilization. Molecules 2016, 21, 1577. [Google Scholar] [CrossRef] [PubMed]
- Danielli, C.; van Langen, L.; Boes, D.; Asaro, F.; Anselmi, S.; Provenza, F.; Renzi, M.; Gardossi, L. 2,5-Furandicarboxaldehyde as a Bio-Based Crosslinking Agent Replacing Glutaraldehyde for Covalent Enzyme Immobilization. RSC Adv. 2022, 12, 35676. [Google Scholar] [CrossRef] [PubMed]
- Bubb, W.A. NMR Spectroscopy in the Study of Carbohydrates: Characterizing the Structural Complexity. Concepts Magn. Reson. 2003, 19, 1–19. [Google Scholar] [CrossRef]
- Tanzi, L.; Robescu, M.S.; Marzatico, S.; Recca, T.; Zhang, Y.; Terreni, M.; Bavaro, T. Developing a Library of Mannose-Based Mono- and Disaccharides: A General Chemoenzymatic Approach to Monohydroxylated Building Blocks. Molecules 2020, 25, 5764. [Google Scholar] [CrossRef]
- Nashiru, O.; Zechel, D.L.; Stoll, D.; Mohammadzadeh, T.; Warren, R.A.J.; Withers, S.G. β-Mannosynthase: Synthesis of β-Mannosides with a Mutant β-Mannosidase. Angew. Chem. Int. Ed. 2001, 40, 417–420. [Google Scholar] [CrossRef]
- Semproli, R.; Robescu, M.S.; Sangiorgio, S.; Pargoletti, E.; Bavaro, T.; Rabuffetti, M.; Cappelletti, G.; Speranza, G.; Ubiali, D. From Lactose to Alkyl Galactoside Fatty Acid Esters as Non-ionic Biosurfactants: A Two-step Enzymatic Approach to Cheese Whey Valorization. ChemPlusChem 2023, 88, e202200331. [Google Scholar] [CrossRef]
- Bruni, M.; Robescu, M.S.; Ubiali, D.; Marrubini, G.; Vanna, R.; Morasso, C.; Benucci, I.; Speranza, G.; Bavaro, T. Immobilization of γ-Glutamyl Transpeptidase from Equine Kidney for the Synthesis of Kokumi Compounds. ChemCatChem 2020, 12, 210–218. [Google Scholar] [CrossRef]
- Robescu, M.S.; Serra, I.; Terreni, M.; Ubiali, D.; Bavaro, T. A Multi-Enzymatic Cascade Reaction for the Synthesis of Vidarabine 5′-Monophosphate. Catalysts 2020, 10, 60. [Google Scholar] [CrossRef]
- Bavaro, T.; Filice, M.; Temporini, C.; Tengattini, S.; Serra, I.; Morelli, C.F.; Massolini, G.; Terreni, M. Chemoenzymatic Synthesis of Neoglycoproteins Driven by the Assessment of Protein Surface Reactivity. RSC Adv. 2014, 4, 56455–56466. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Carrier | Experimental Conditions | Immobilization Yield (Protein) (%) a | Immobilization Yield (Activity) (%) b | Activity Recovery (%) c | Efficiency (%) d | Activity (IU/g) | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | Additive (1% w/v) | Time (h) | T (°C) | |||||||
| 1 | GLX-Sepabeads | 50 mM NaHCO3 pH 10 | Man | 6 | 4 | 100 | 89.5 | 13.8 | 15.5 | 0.94 |
| 2 | GLX-Sepabeads | 50 mM KH2PO4 pH 8 | - | 3 | 25 | 100 | 93.2 | 15.0 | 16.0 | 0.96 |
| 3 | GLX-AG | 50 mM NaHCO3 pH 10 | Man | 24 | 4 | 77.4 | 48.8 | <5 | 9.6 | 0.08 |
| 4 | DFF- Sepabeads | 50 mM NaHCO3 pH 10 | Man | 24 | 4 | 51.3 | 76.5 | <5 | <5 | 0.06 |
| 5 | DFF-EDA-AG | 50 mM NaHCO3 pH 10 | Man | 24 | 4 | 88.8 | 74.0 | <5 | <5 | 0.06 |
| 6 | CNBr-AG | 50 mM KH2PO4 pH 7.5 | Man | 2 | 4 | 100 | 100 | 70.6 | 70.6 | 1.75 |
| 7 | Sepabeads-PEI | 5 mM KH2PO4 pH 7.5 | - | 1 | 25 | 97.4 | 94.8 | 5.2 | 5.5 | 0.26 |
| 8 | Sepabeads-PEI | 5 mM KH2PO4 pH 7.5 | Suc | 2 | 4 | 100 | 95.3 | 33.5 | 35.0 | 1.74 |
| 9 | Sepabeads-PEI + Dx ox. 10% | 5 mM KH2PO4 pH 7.5 | Suc | 2 | 4 | 60 | 92.5 | 13.5 | 14.6 | 0.63 |
| 10 | IDA-Co2+-AG | 5 mM KH2PO4 pH 7.5 | Man | 4 | 4 | 100 | 82.2 | 72.8 | 88.5 | 3.10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robescu, M.S.; Tengattini, S.; Rabuffetti, M.; Speranza, G.; Terreni, M.; Bavaro, T. β-Mannosidase from Cellulomonas fimi: Immobilization Study and Application in the β-Mannoside Synthesis. Catalysts 2023, 13, 1399. https://doi.org/10.3390/catal13111399
Robescu MS, Tengattini S, Rabuffetti M, Speranza G, Terreni M, Bavaro T. β-Mannosidase from Cellulomonas fimi: Immobilization Study and Application in the β-Mannoside Synthesis. Catalysts. 2023; 13(11):1399. https://doi.org/10.3390/catal13111399
Chicago/Turabian StyleRobescu, Marina S., Sara Tengattini, Marco Rabuffetti, Giovanna Speranza, Marco Terreni, and Teodora Bavaro. 2023. "β-Mannosidase from Cellulomonas fimi: Immobilization Study and Application in the β-Mannoside Synthesis" Catalysts 13, no. 11: 1399. https://doi.org/10.3390/catal13111399