
Exploring the Methane to Methanol Oxidation over Iron and Copper Sites in Metal–Organic Frameworks

Abstract
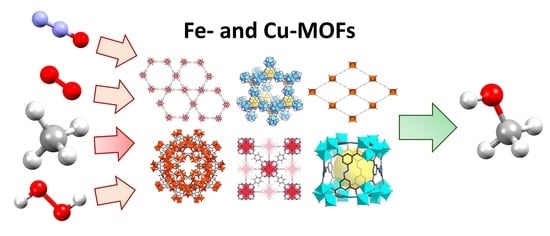
:
1. Introduction
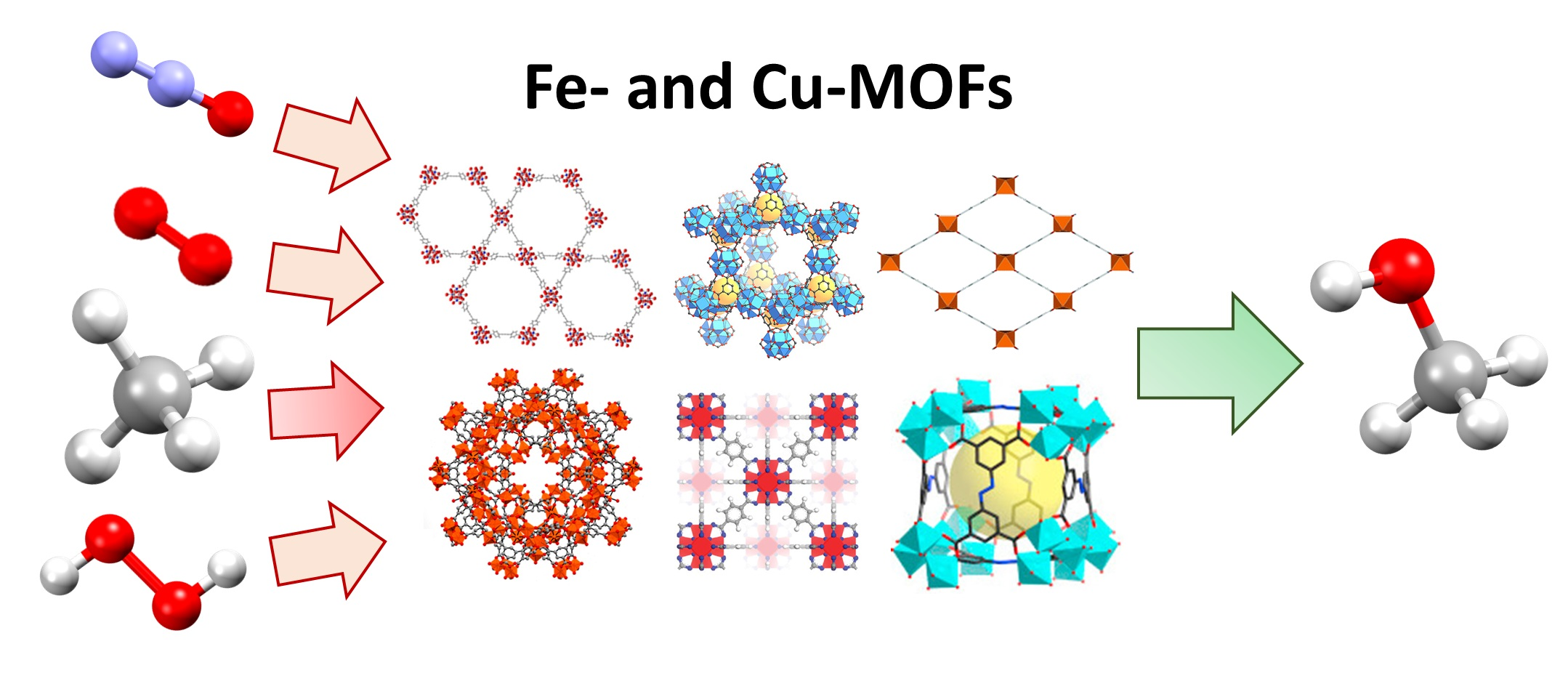
2. Metal–Organic Frameworks as Catalytic Platforms for the MTM Reaction

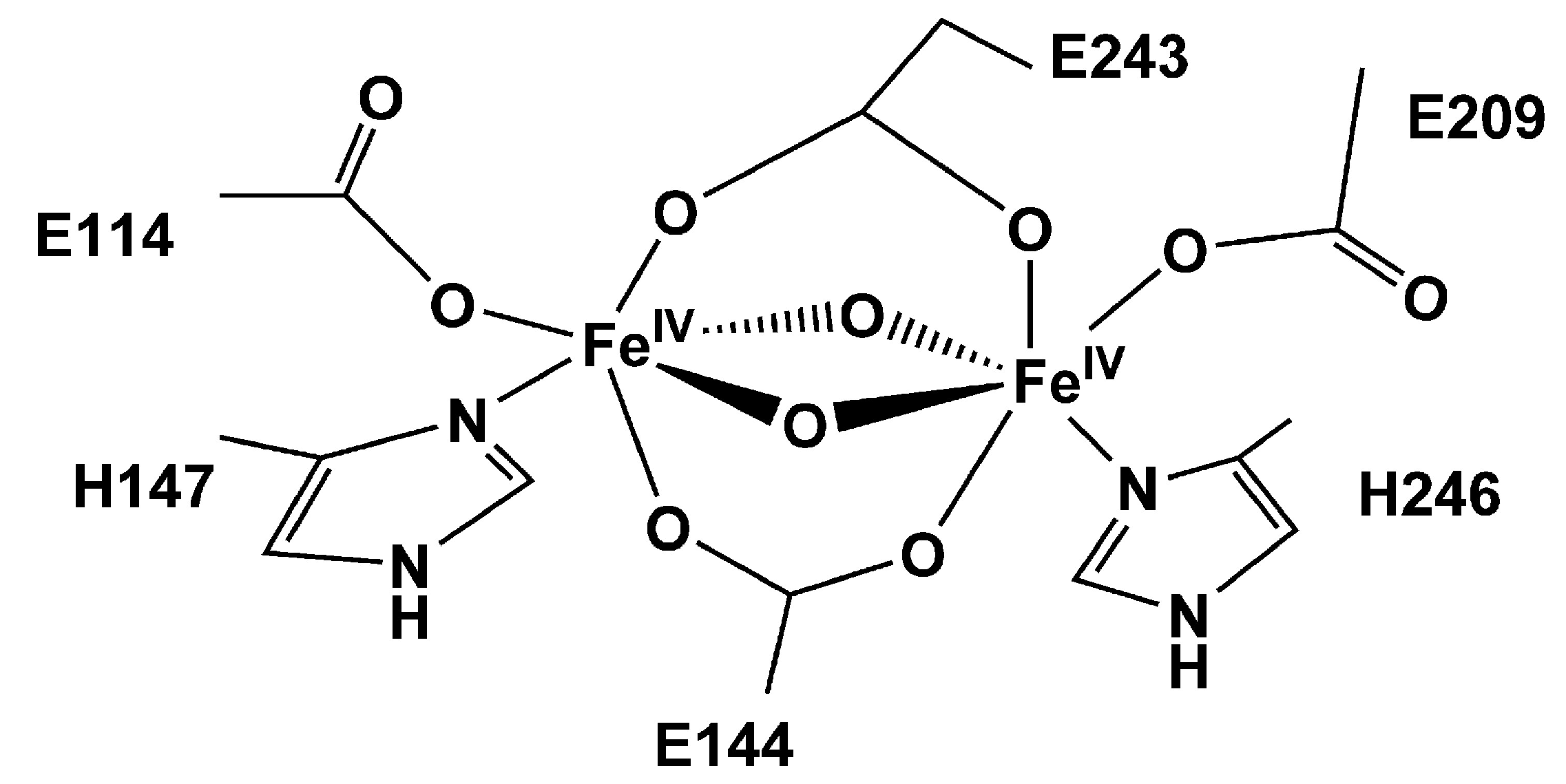
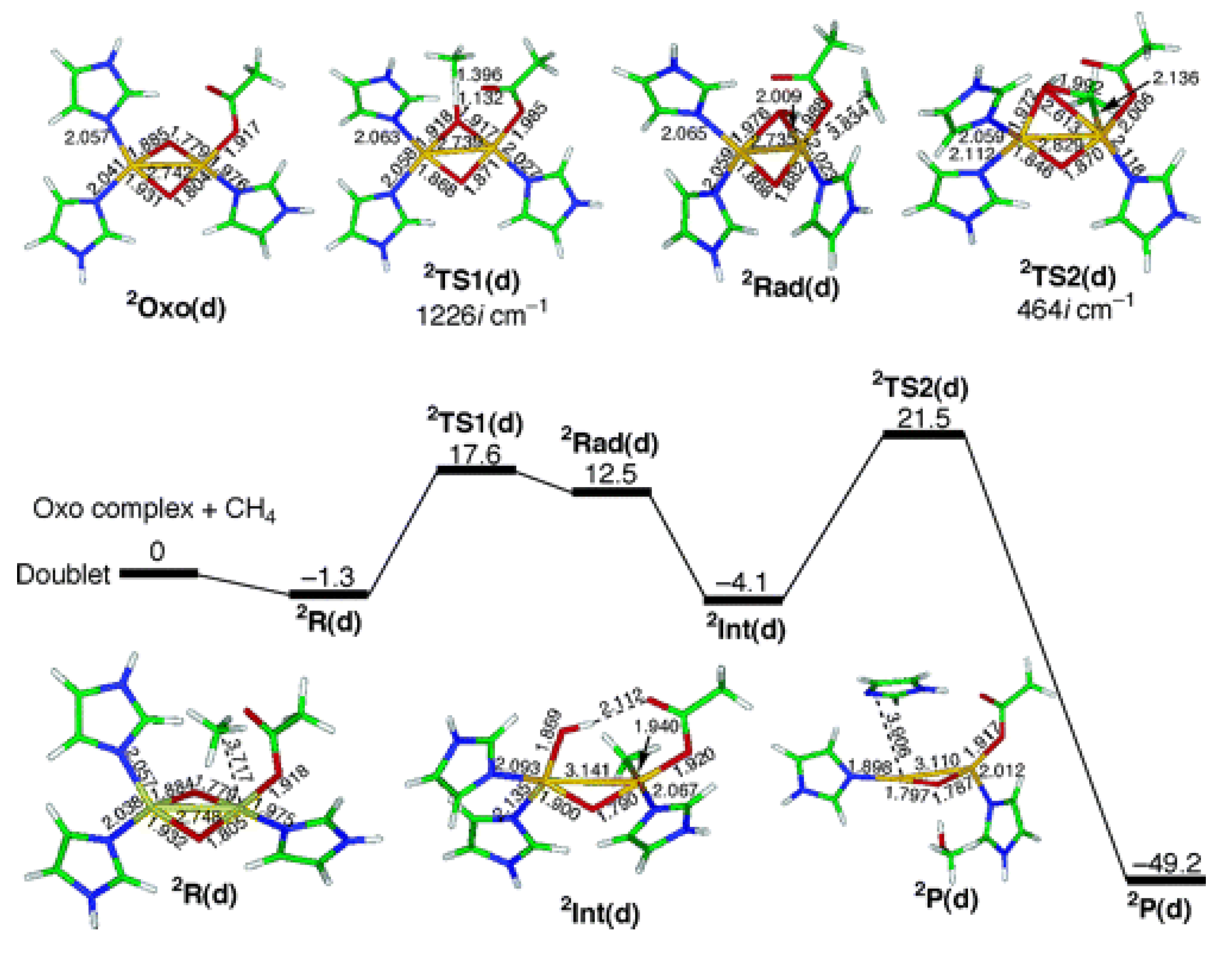
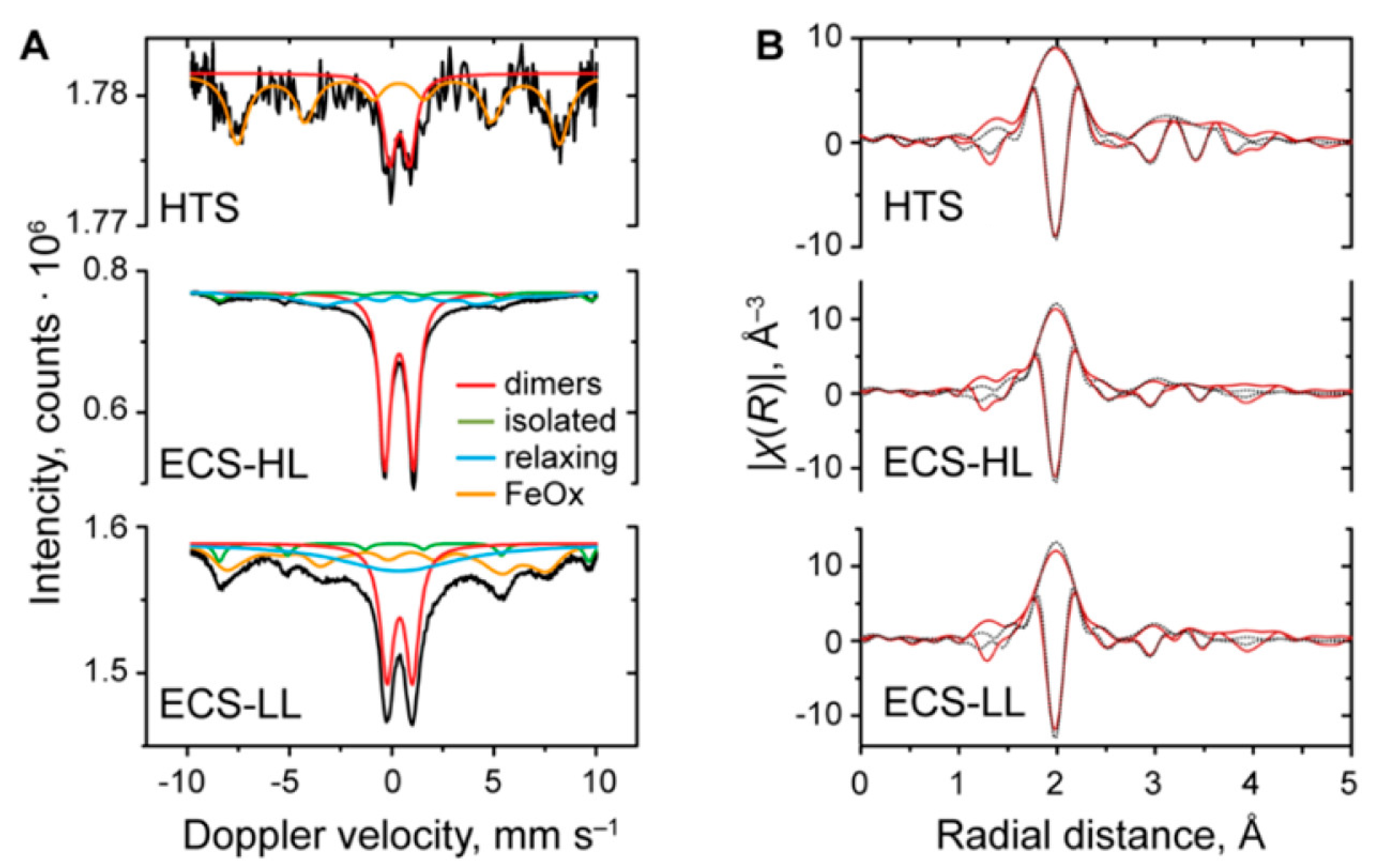
3. MOFs Exhibiting Iron Active Sites for the MTM Conversion
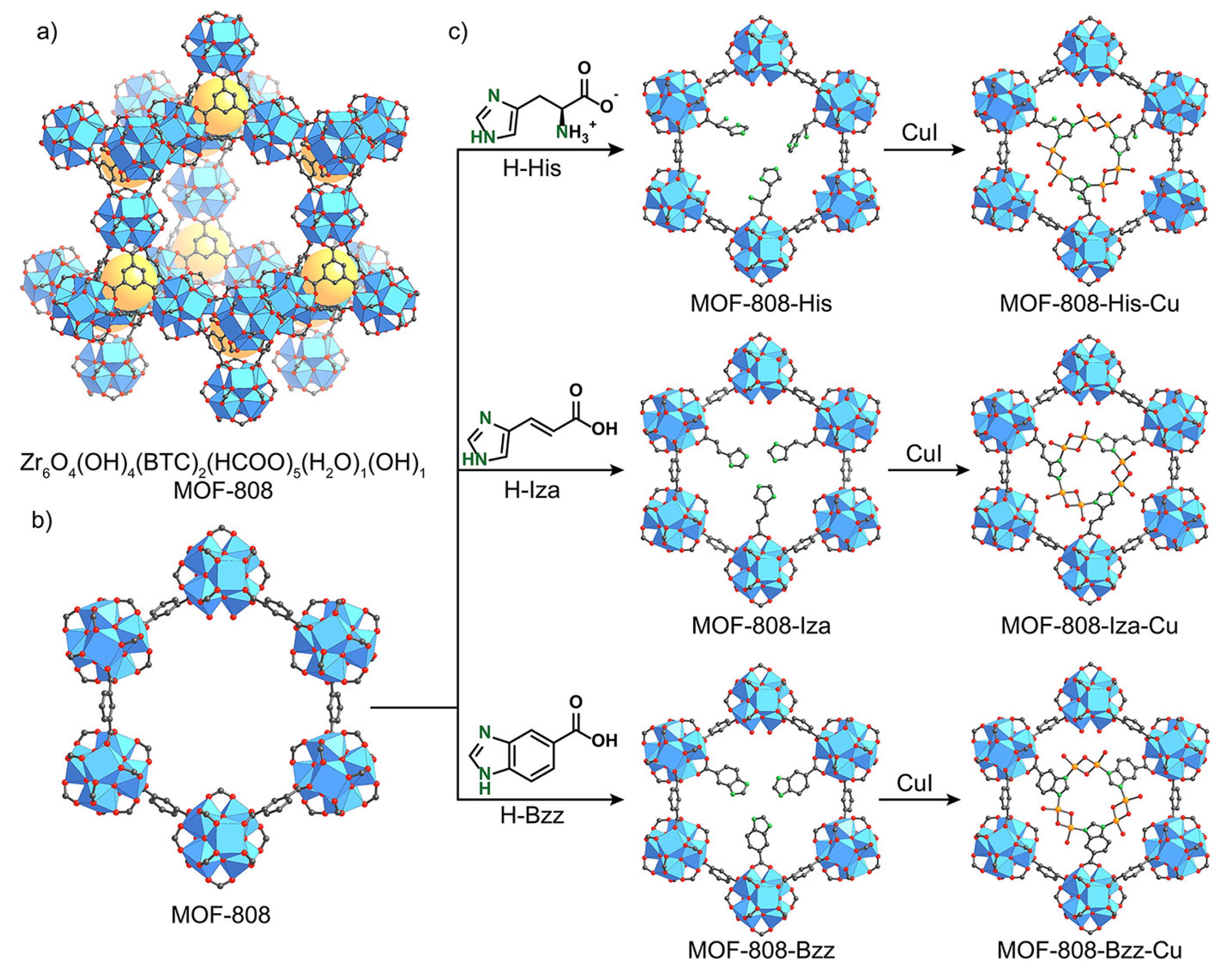
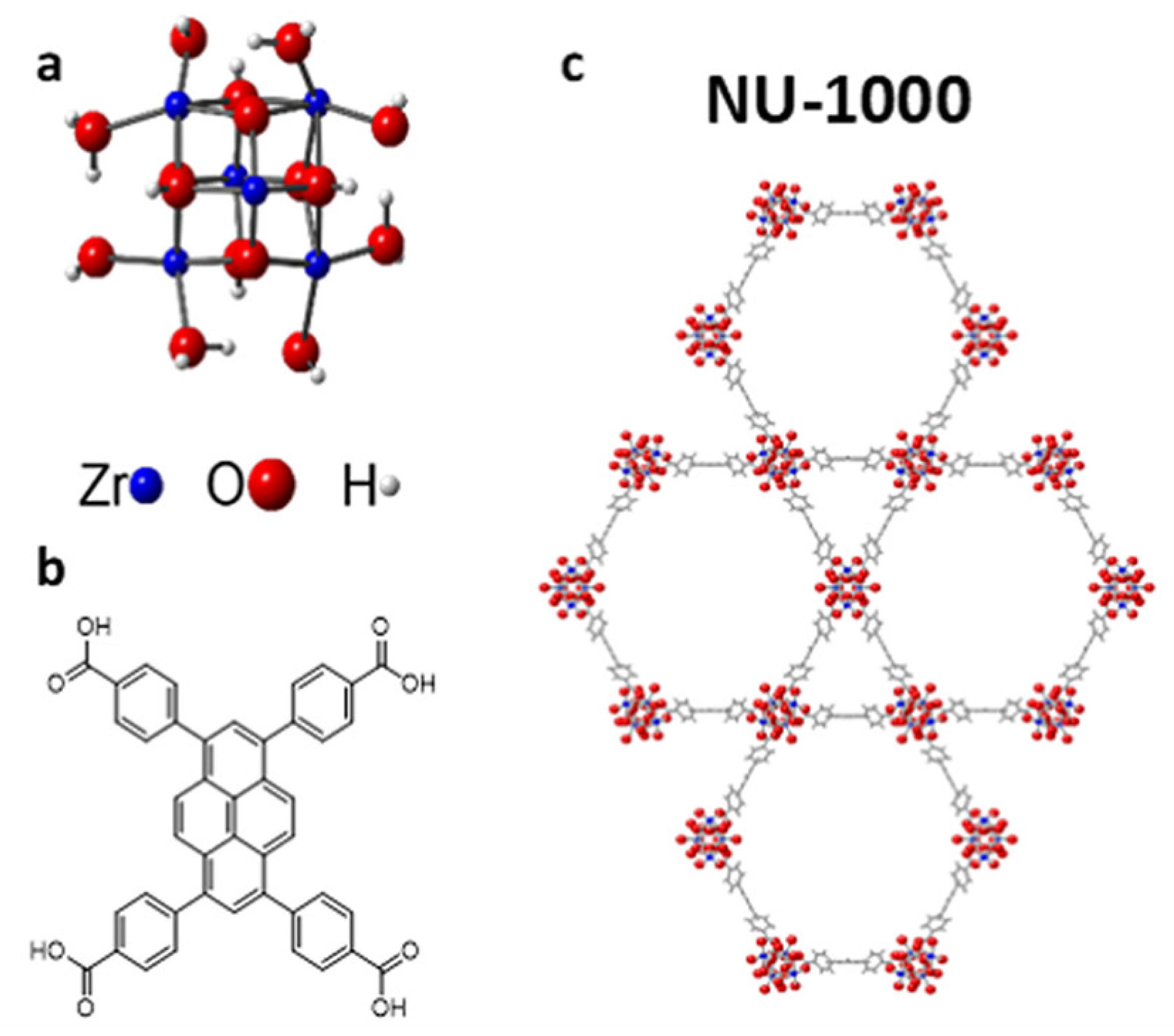
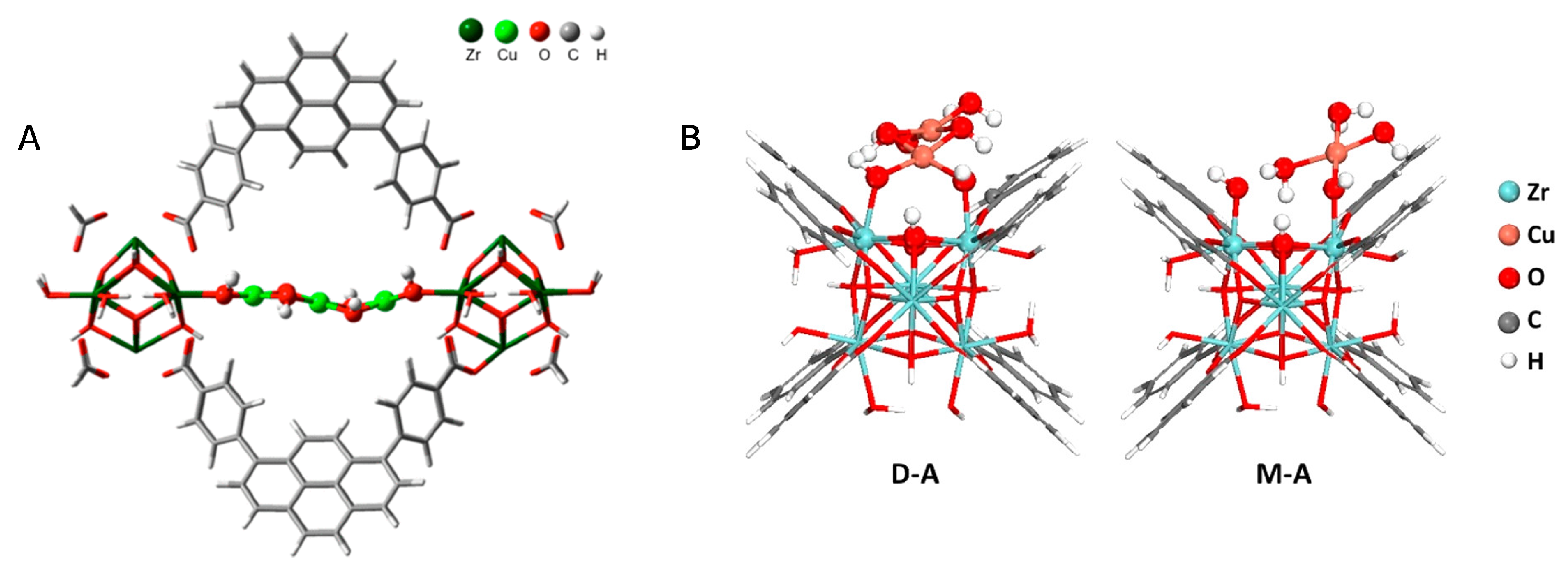
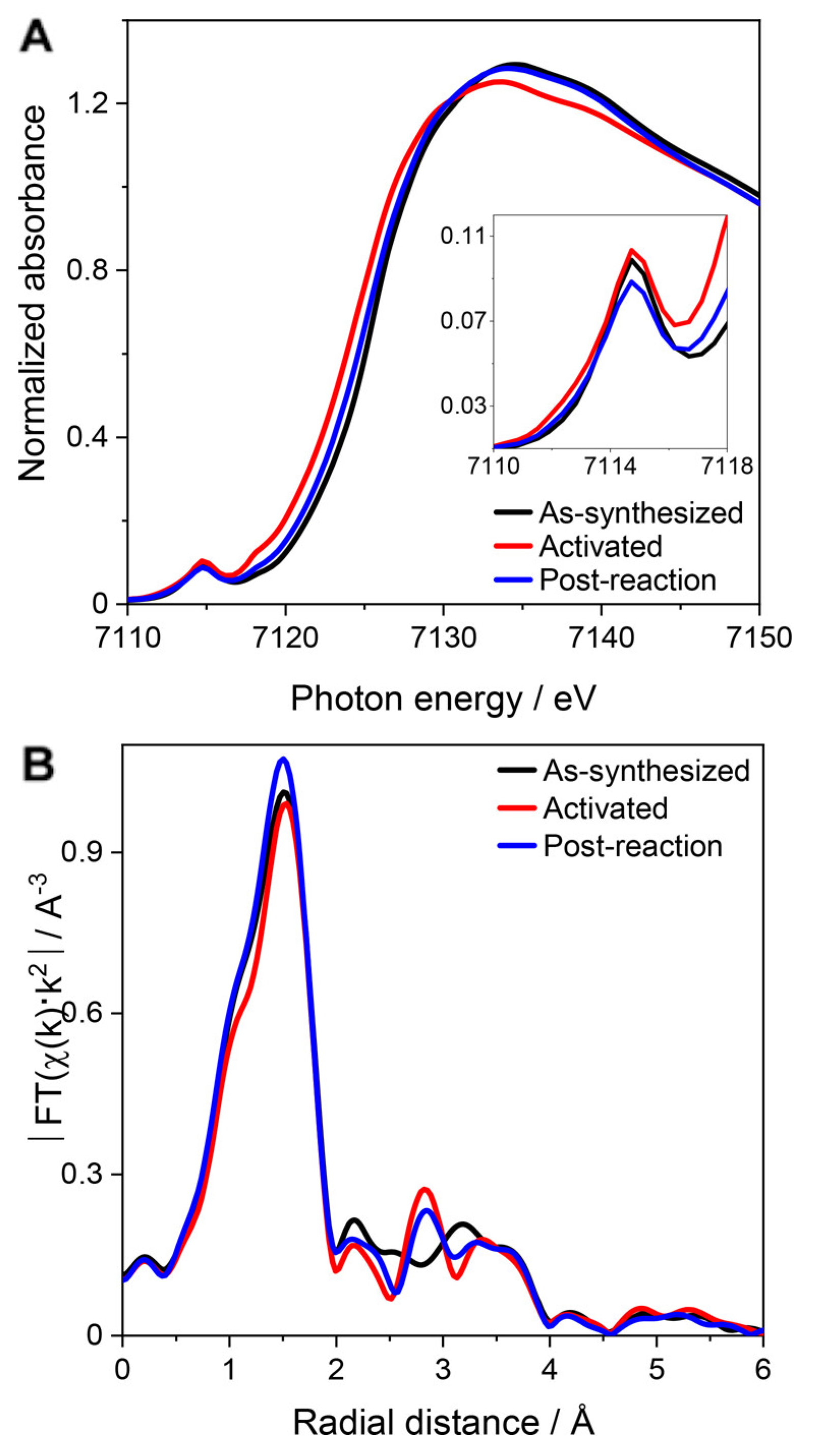
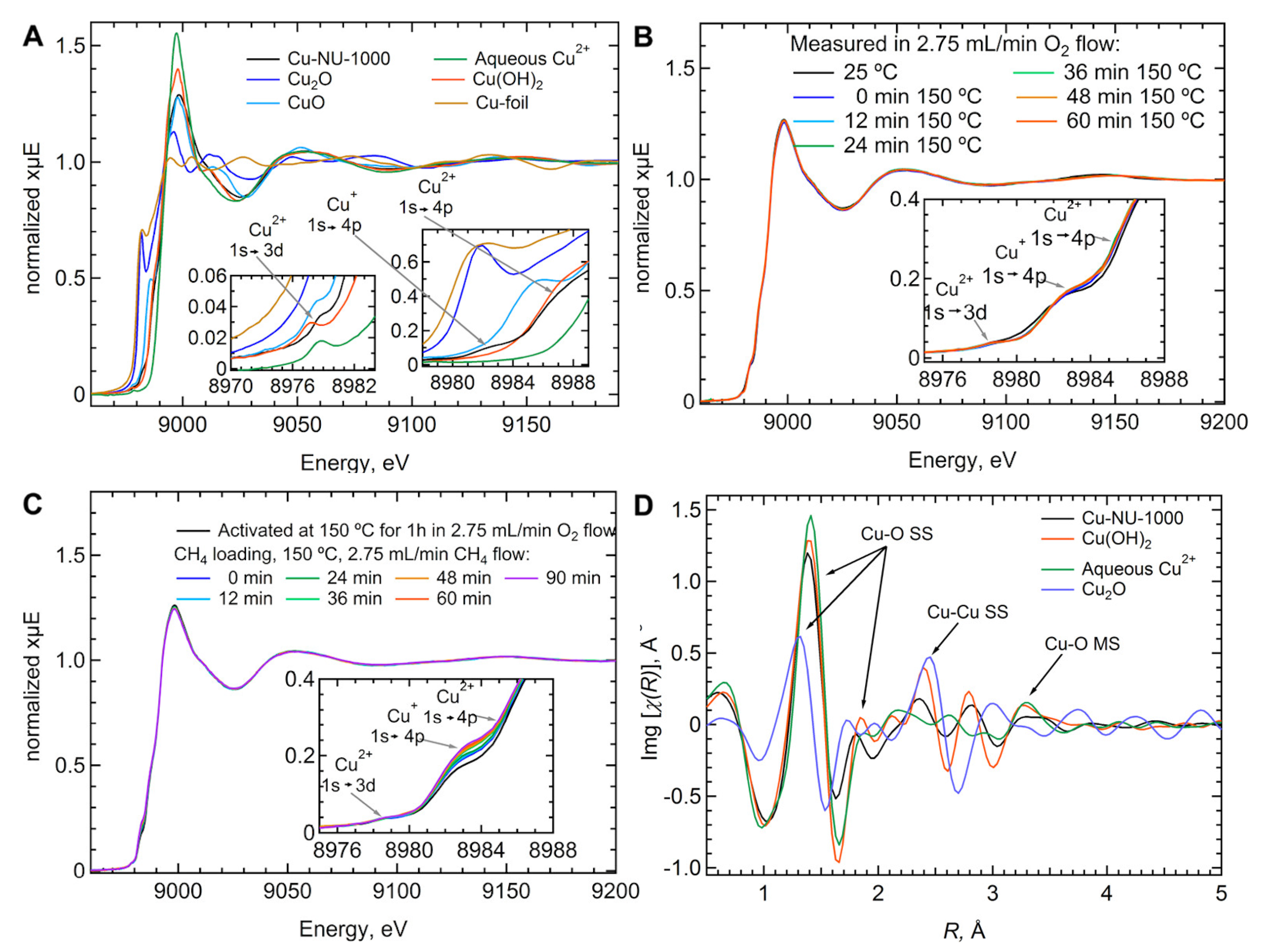
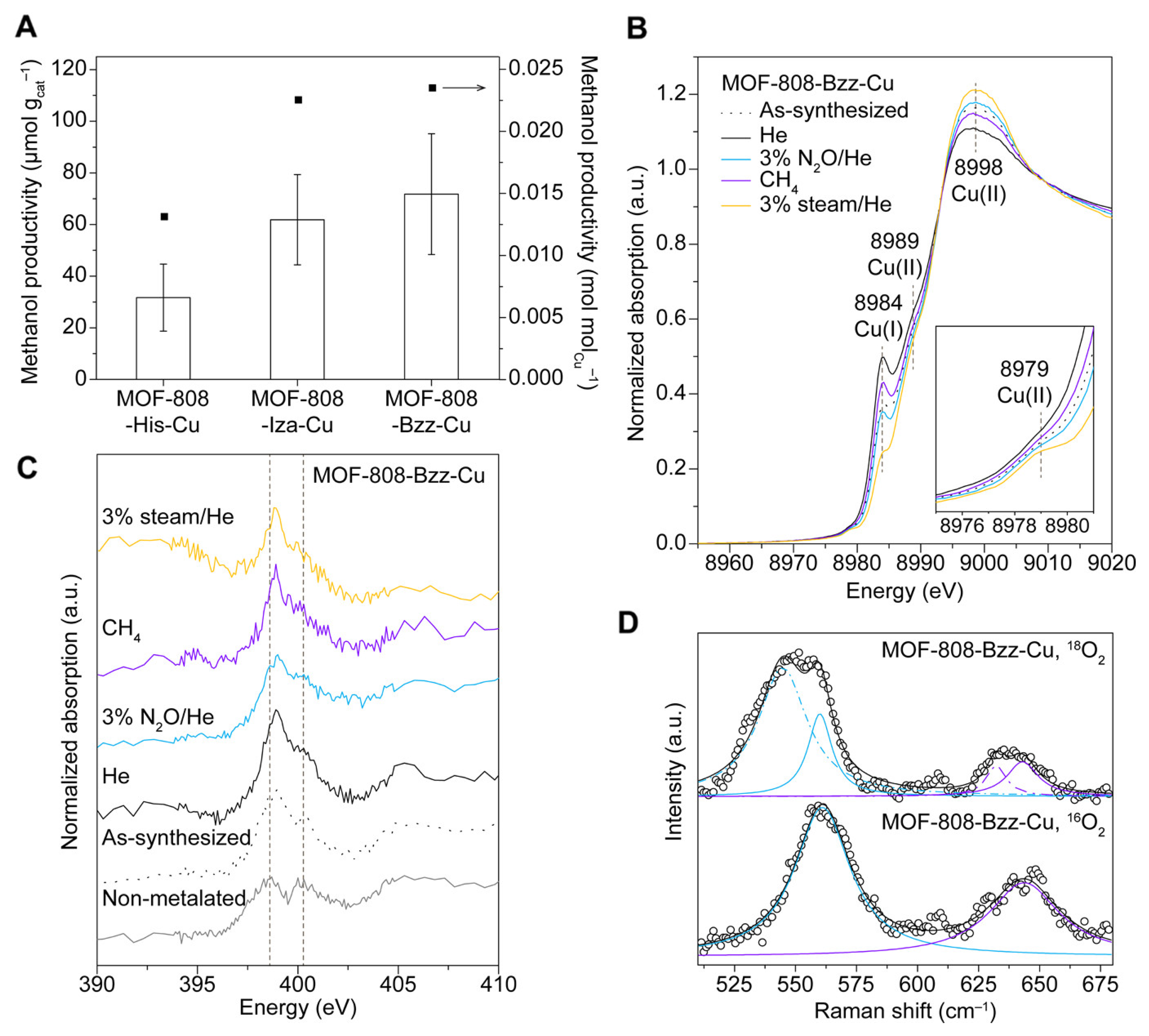
4. MOF-Supported Cu Clusters for the MTM Conversion
5. Spectroscopic Characterization of the MOF-Based Methane Oxidation
6. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| GWP | global warming potential |
| MTM | methane to methanol |
| BDE | bond dissociation energy |
| MOF | metal–organic framework |
| BDC | benzene-1,4-dicarboxylate |
| DOBDC | 2,5-2,5-dioxido-1,4-benzenedicarboxylate |
| BTC | benzene-1,3,5-tricarboxylate |
| ABTC | 3,3’,5,5’-azobenzene-tetracarboxylate |
| TBAPy | 1,3,6,8-tetrakis(p-benzoate)-pyrene |
| SBUs | secondary building units |
| DFT | density functional theory |
| sMMO | soluble methane monooxygenase |
| NADH | nicotinamide adenine dinucleotide |
| HTS | hydrothermal synthesis sample |
| ECS | electrochemical synthesis sample |
| LL | low loading |
| HL | high loading |
| TOF | turnover frequency |
| TON | turnover number |
| bpy | 2,2’-bipyridine |
| Hbpydc | 2,2’-bipyridine-5,5’-dicarboxylic acid |
| XPS | X-ray photoelectron spectroscopy |
| EPR | electron paramagnetic resonance |
| XAS | X-ray absorption spectroscopy |
| pMMO | particulate methane monooxygenase |
| QM/MM | quantum mechanics/molecular mechanics |
| EDS | energy-dispersive spectroscopy |
| XANES | X-ray absorption near edge structure |
| DRS | diffuse reflectance spectroscopy |
| PXRD | powder X-ray diffraction |
| HAADF-STEM | high-angle annular dark-field scanning transmission electron microscopy |
| pair distribution function | |
| EXAFS | extended X-ray absorption fine structure |
| FT | Fourier transform |
| CN | coordination number |
| SS | single scattering |
| MS | multiple scattering |
| XES | X-ray emission spectroscopy |
| MR | multi-reference |
| ML | machine learning |
References
- Caballero, A.; Pérez, P.J. Methane as Raw Material in Synthetic Chemistry: The Final Frontier. Chem. Soc. Rev. 2013, 42, 8809–8820. [Google Scholar] [CrossRef] [PubMed]
- Bakkaloglu, S.; Cooper, J.; Hawkes, A. Methane Emissions Along Biomethane and Biogas Supply Chains are Underestimated. One Earth 2022, 5, 724–736. [Google Scholar] [CrossRef]
- MacDonald, G. Role of Methane Clathrates in Past and Future Climates. Clim. Change 1990, 16, 247–281. [Google Scholar] [CrossRef]
- Dummer, N.F.; Willock, D.J.; He, Q.; Howard, M.J.; Lewis, R.J.; Qi, G.; Taylor, S.H.; Xu, J.; Bethell, D.; Kiely, C.J.; et al. Methane Oxidation to Methanol. Chem. Rev. 2022, 123, 6359–6411. [Google Scholar] [CrossRef] [PubMed]
- Saunois, M.; Stavert, A.R.; Poulter, B.; Bousquet, P.; Canadell, J.G.; Jackson, R.B.; Raymond, P.A.; Dlugokencky, E.J.; Houweling, S.; Patra, P.K.; et al. The Global Methane Budget 2000–2017. Earth Syst. Sci. Data 2020, 12, 1561–1623. [Google Scholar] [CrossRef]
- Andrade, L.S.; Lima, H.H.; Silva, C.T.; Amorim, W.L.; Poco, J.G.; Lopez-Castillo, A.; Kirillova, M.V.; Carvalho, W.A.; Kirillov, A.M.; Mandelli, D. Metal–Organic Frameworks as Catalysts and Biocatalysts for Methane Oxidation: The Current State of the Art. Coord. Chem. Rev. 2023, 481, 215042. [Google Scholar] [CrossRef]
- Mar, K.A.; Unger, C.; Walderdorff, L.; Butler, T. Beyond CO2 Equivalence: The Impacts of Methane on Climate, Ecosystems, and Health. Environ. Sci. Policy 2022, 134, 127–136. [Google Scholar] [CrossRef]
- Yuliati, L.; Yoshida, H. Photocatalytic Conversion of Methane. Chem. Soc. Rev. 2008, 37, 1592–1602. [Google Scholar] [CrossRef]
- Schwarz, H. Chemistry with Methane: Concepts Rather than Recipes. Angew. Chem. Int. Ed. 2011, 50, 10096–10115. [Google Scholar] [CrossRef]
- Sher Shah, M.S.A.; Oh, C.; Park, H.; Hwang, Y.J.; Ma, M.; Park, J.H. Catalytic Oxidation of Methane to Oxygenated Products: Recent Advancements and Prospects for Electrocatalytic and Photocatalytic Conversion at Low Temperatures. Adv. Sci. 2020, 7, 2001946. [Google Scholar] [CrossRef]
- Innovation Outlook: Renewable Methanol. IRENA. 2021. Available online: https://www.irena.org/publications/2021/Jan/Innovation-Outlook-Renewable-Methanol (accessed on 25 July 2023).
- Methanol Price and Supply/Demand. Methanol Institute. 2022. Available online: https://www.methanol.org/methanol-price-supply-demand/ (accessed on 25 July 2023).
- Acquarola, C.; Bhatelia, T.; Pareek, V.; Ao, M.; Shah, M.T. Optimized Process for Methanol Production via Bi-reforming Syngas. Ind. Eng. Chem. Res. 2022, 61, 5557–5567. [Google Scholar] [CrossRef]
- Bussche, K.; Froment, G. A Steady-State Kinetic Model for Methanol Synthesis and the Water Gas Shift Reaction on a Commercial Cu/ZnO/Al2O3Catalyst. J. Catal. 1996, 161, 1–10. [Google Scholar] [CrossRef]
- Latimer, A.A.; Kakekhani, A.; Kulkarni, A.R.; Nørskov, J.K. Direct Methane to Methanol: The Selectivity–Conversion Limit and Design Strategies. ACS Catal. 2018, 8, 6894–6907. [Google Scholar] [CrossRef]
- Liu, R.S.; Iwamoto, M.; Lunsford, J.H. Partial Oxidation of Methane by Nitrous Oxide over Molybdenum Oxide Supported on Silica. J. Chem. Soc. Chem. Commun. 1982, 78–79. [Google Scholar] [CrossRef]
- Khan, M.; Somorjai, G. A Kinetic Study of Partial Oxidation of Methane with Nitrous Oxide on a Molybdena-Silica Catalyst. J. Catal. 1985, 91, 263–271. [Google Scholar] [CrossRef]
- Suzuki, T.; Wada, K.; Shima, M.; Watanabe, Y. Photoinduced Partial Oxidation of Methane into Formaldehyde on Silica-Supported Molybdena. J. Chem. Soc. Chem. Commun. 1990, 1059–1060. [Google Scholar] [CrossRef]
- Zhang, X.; He, D.H.; Zhang, Q.J.; Xu, B.Q.; Zhu, Q.M. Methanol from Oxidation of Methane over MoOx/La-Co-Oxide Catalysts. In Natural Gas Conversion VII; Studies in Surface Science and Catalysis; Bao, X., Xu, Y., Eds.; Elsevier: Amsterdam, The Netherlands, 2004; Volume 147, pp. 541–546. [Google Scholar]
- Zhen, K.; Khan, M.; Mak, C.; Lewis, K.; Somorjai, G. Partial Oxidation of Methane with Nitrous Oxide over V2O5 SiO2 Catalyst. J. Catal. 1985, 94, 501–507. [Google Scholar] [CrossRef]
- Yu, T.; Li, Z.; Lin, L.; Chu, S.; Su, Y.; Song, W.; Wang, A.; Weckhuysen, B.M.; Luo, W. Highly Selective Oxidation of Methane into Methanol over Cu-Promoted Monomeric Fe/ZSM-5. ACS Catal. 2021, 11, 6684–6691. [Google Scholar] [CrossRef]
- Mahyuddin, M.H.; Staykov, A.; Shiota, Y.; Yoshizawa, K. Direct Conversion of Methane to Methanol by Metal-Exchanged ZSM-5 Zeolite (Metal = Fe, Co, Ni, Cu). ACS Catal. 2016, 6, 8321–8331. [Google Scholar] [CrossRef]
- Starokon, E.V.; Parfenov, M.V.; Arzumanov, S.S.; Pirutko, L.V.; Stepanov, A.G.; Panov, G.I. Oxidation of Methane to Methanol on the Surface of FeZSM-5 Zeolite. J. Catal. 2013, 300, 47–54. [Google Scholar] [CrossRef]
- Wood, B.R.; Reimer, J.A.; Bell, A.T. Studies of N2O Adsorption and Decomposition on Fe–ZSM-5. J. Catal. 2002, 209, 151–158. [Google Scholar] [CrossRef]
- Alayon, E.M.; Nachtegaal, M.; Ranocchiari, M.; van Bokhoven, J.A. Catalytic Conversion of Methane to Methanol over Cu-Mordenite. Chem. Commun. 2012, 48, 404–406. [Google Scholar] [CrossRef] [PubMed]
- Knorpp, A.J.; Newton, M.A.; Pinar, A.B.; van Bokhoven, J.A. Conversion of Methane to Methanol on Copper Mordenite: Redox Mechanism of Isothermal and High-Temperature-Activation Procedure. Ind. Eng. Chem. Res. 2018, 57, 12036–12039. [Google Scholar] [CrossRef]
- Yousefzadeh, H.; Bozbag, S.E.; Sushkevich, V.; van Bokhoven, J.A.; Erkey, C. Stepwise Conversion of Methane to Methanol over Cu-Mordenite Prepared by Supercritical and Aqueous Ion Exchange Routes and Quantification of Active Cu Species by H2-TPR. Catal. Commun. 2023, 174, 106574. [Google Scholar] [CrossRef]
- Bozbag, S.E.; Alayon, E.M.C.; Pecháĉek, J.; Nachtegaal, M.; Ranocchiari, M.; van Bokhoven, J.A. Methane to Methanol over Copper Mordenite: Yield Improvement through Multiple Cycles and Different Synthesis Techniques. Catal. Sci. Technol. 2016, 6, 5011–5022. [Google Scholar] [CrossRef]
- Pappas, D.K.; Martini, A.; Dyballa, M.; Kvande, K.; Teketel, S.; Lomachenko, K.A.; Baran, R.; Glatzel, P.; Arstad, B.; Berlier, G.; et al. The Nuclearity of the Active Site for Methane to Methanol Conversion in Cu-Mordenite: A Quantitative Assessment. J. Am. Chem. Soc. 2018, 140, 15270–15278. [Google Scholar] [CrossRef]
- Cai, X.; Fang, S.; Hu, Y.H. Unprecedentedly High Efficiency for Photocatalytic Conversion of Methane to Methanol over Au-Pd/TiO2 - what is the Role of each Component in the System? J. Mater. Chem. A 2021, 9, 10796–10802. [Google Scholar] [CrossRef]
- Zhou, W.; Qiu, X.; Jiang, Y.; Fan, Y.; Wei, S.; Han, D.; Niu, L.; Tang, Z. Highly Selective Aerobic Oxidation of Methane to Methanol over Gold Decorated Zinc Oxide via Photocatalysis. J. Mater. Chem. A 2020, 8, 13277–13284. [Google Scholar] [CrossRef]
- Zecchina, A.; Rivallan, M.; Berlier, G.; Lamberti, C.; Ricchiardi, G. Structure and Nuclearity of Active Sites in Fe-Zeolites: Comparison with Iron Sites in Enzymes and Homogeneous Catalysts. Phys. Chem. Chem. Phys. 2007, 9, 3483–3499. [Google Scholar] [CrossRef]
- Zuo, H.; Xin, Q.; Meynen, V.; Klemm, E. Sensitivity of the Selective Oxidation of Methane Over Fe/ZSM-5 Zeolites in a Micro Fixed-Bed Reactor for the Catalyst Preparation Method. Appl. Catal. A Gen. 2018, 566, 96–103. [Google Scholar] [CrossRef]
- Hall, J.N.; Bollini, P. Low-Temperature, Ambient Pressure Oxidation of Methane to Methanol Over Every Tri-Iron Node in a Metal–Organic Framework Material. Chem. Eur. J. 2020, 26, 16639–16643. [Google Scholar] [CrossRef] [PubMed]
- Shen, K.; Kumari, S.; Huang, Y.C.; Jang, J.; Sautet, P.; Morales-Guio, C.G. Electrochemical Oxidation of Methane to Methanol on Electrodeposited Transition Metal Oxides. J. Am. Chem. Soc. 2023, 145, 6927–6943. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Li, S.; Wang, S.; Zhang, Y.; Long, C.; Xie, J.; Fan, X.; Zhao, W.; Xu, P.; Fan, Y.; et al. Enabling Specific Photocatalytic Methane Oxidation by Controlling Free Radical Type. J. Am. Chem. Soc. 2023, 145, 2698–2707. [Google Scholar] [CrossRef]
- Gan, Y.; Huang, M.; Yu, F.; Yu, X.; He, Z.; Liu, J.; Lin, T.; Dai, Y.; Niu, Q.; Zhong, L. Highly Selective Photocatalytic Methane Oxidation to Methanol Using CO2 as a Soft Oxidant. ACS Sustain. Chem. Eng. 2023, 11, 5537–5546. [Google Scholar] [CrossRef]
- Furukawa, H.; Cordova, K.E.; O’Keeffe, M.; Yaghi, O.M. The Chemistry and Applications of Metal-Organic Frameworks. Science 2013, 341, 1230444. [Google Scholar] [CrossRef]
- Stock, N.; Biswas, S. Synthesis of Metal-Organic Frameworks (MOFs): Routes to Various MOF Topologies, Morphologies, and Composites. Chem. Rev. 2012, 112, 933–969. [Google Scholar] [CrossRef]
- Rosen, A.S.; Notestein, J.M.; Snurr, R.Q. Structure–Activity Relationships That Identify Metal–Organic Framework Catalysts for Methane Activation. ACS Catal. 2019, 9, 3576–3587. [Google Scholar] [CrossRef]
- Rosen, A.S.; Notestein, J.M.; Snurr, R.Q. Identifying Promising Metal–Organic Frameworks for Heterogeneous Catalysis via High-Throughput Periodic Density Functional Theory. J. Comput. Chem. 2019, 40, 1305–1318. [Google Scholar] [CrossRef]
- Konnerth, H.; Matsagar, B.M.; Chen, S.S.; Prechtl, M.H.; Shieh, F.K.; Wu, K.C.W. Metal-Organic Framework (MOF)-Derived Catalysts for Fine Chemical Production. Coord. Chem. Rev. 2020, 416, 213319. [Google Scholar] [CrossRef]
- Maina, J.W.; Pozo-Gonzalo, C.; Kong, L.; Schütz, J.; Hill, M.; Dumée, L.F. Metal Organic Framework Based Catalysts for CO2 Conversion. Mater. Horiz. 2017, 4, 345–361. [Google Scholar] [CrossRef]
- Liu, M.; Wu, J.; Hou, H. Metal-Organic Framework (MOF)-Based Materials as Heterogeneous Catalysts for C-H Bond Activation. Chem. Eur. J. 2019, 25, 2935–2948. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.G.; Zhang, G.Y.; Hu, T.L.; Bu, X.H. Metal-Organic Framework-Based Heterogeneous Catalysts for the Conversion of C1 Chemistry: CO, CO2 and CH4. Coord. Chem. Rev. 2019, 387, 79–120. [Google Scholar] [CrossRef]
- Bavykina, A.; Kolobov, N.; Khan, I.S.; Bau, J.A.; Ramirez, A.; Gascon, J. Metal–Organic Frameworks in Heterogeneous Catalysis: Recent Progress, New Trends, and Future Perspectives. Chem. Rev. 2020, 120, 8468–8535. [Google Scholar] [CrossRef] [PubMed]
- Cavka, J.H.; Jakobsen, S.; Olsbye, U.; Guillou, N.; Lamberti, C.; Bordiga, S.; Lillerud, K.P. A New Zirconium Inorganic Building Brick Forming Metal Organic Frameworks with Exceptional Stability. J. Am. Chem. Soc. 2008, 130, 13850–13851. [Google Scholar] [CrossRef]
- Li, Y.; Fu, Y.; Ni, B.; Ding, K.; Chen, W.; Wu, K.; Huang, X.; Zhang, Y. Effects of Ligand Functionalization on the Photocatalytic Properties of Titanium-Based MOF: A Density Functional Theory Study. AIP Adv. 2018, 8, 035012. [Google Scholar] [CrossRef]
- Barona, M.; Snurr, R.Q. Exploring the Tunability of Trimetallic MOF Nodes for Partial Oxidation of Methane to Methanol. ACS Appl. Mater. Interfaces 2020, 12, 28217–28231. [Google Scholar] [CrossRef]
- Xiao, D.J.; Bloch, E.D.; Mason, J.A.; Queen, W.L.; Hudson, M.R.; Planas, N.; Borycz, J.; Dzubak, A.L.; Verma, P.; Lee, K.; et al. Oxidation of Ethane to Ethanol by N2O in a Metal-Organic Framework with Coordinatively Unsaturated Iron(II) Sites. Nat. Chem. 2014, 6, 590–595. [Google Scholar] [CrossRef]
- Mandal, S.; Natarajan, S.; Mani, P.; Pankajakshan, A. Post-Synthetic Modification of Metal-Organic Frameworks Toward Applications. Adv. Funct. Mater. 2021, 31, 2006291. [Google Scholar] [CrossRef]
- Asgari, M.; Semino, R.; Schouwink, P.A.; Kochetygov, I.; Tarver, J.; Trukhina, O.; Krishna, R.; Brown, C.M.; Ceriotti, M.; Queen, W.L. Understanding How Ligand Functionalization Influences CO2 and N2 Adsorption in a Sodalite Metal–Organic Framework. Chem. Mater. 2020, 32, 1526–1536. [Google Scholar] [CrossRef]
- Lalonde, M.; Bury, W.; Karagiaridi, O.; Brown, Z.; Hupp, J.T.; Farha, O.K. Transmetalation: Routes to Metal Exchange within Metal-Organic Frameworks. J. Mater. Chem. A 2013, 1, 5453–5468. [Google Scholar] [CrossRef]
- Zheng, L.L.; Zhang, L.S.; Chen, Y.; Tian, L.; Jiang, X.H.; Chen, L.S.; Xing, Q.J.; Liu, X.Z.; Wu, D.S.; Zou, J.P. A New Strategy for the Fabrication of Covalent Organic Framework-Metal-Organic Framework Hybrids via In-Situ Functionalization of Ligands for Improved Hydrogen Evolution Reaction Activity. Chin. J. Catal. 2022, 43, 811–819. [Google Scholar] [CrossRef]
- Li, B.; Zhang, Y.; Ma, D.; Li, L.; Li, G.; Li, G.; Shi, Z.; Feng, S. A Strategy Toward Constructing a Bifunctionalized MOF Catalyst: Post-Synthetic Modification of MOFs on Organic Ligands and Coordinatively Unsaturated Metal Sites. Chem. Comm. 2012, 48, 6151–6153. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, X.; Kanchanakungwankul, S.; Lu, Z.; Noh, H.; Syed, Z.H.; Farha, O.K.; Truhlar, D.G.; Hupp, J.T. Unexpected “Spontaneous” Evolution of Catalytic, MOF-Supported Single Cu(II) Cations to Catalytic, MOF-Supported Cu(0) Nanoparticles. J. Am. Chem. Soc. 2020, 142, 21169–21177. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, Y.; Zhao, Y.; Liu, C.J. Recent Progresses in the Size and Structure Control of MOF Supported Noble Metal Catalysts. Catal. Today 2016, 263, 61–68. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, G.; Chen, J.; Niu, H. Excellent Catalytic Performance of Ce–MOF with Abundant Oxygen Vacancies Supported Noble Metal Pt in the Oxidation of Toluene. Catalysts 2022, 12, 775. [Google Scholar] [CrossRef]
- Han, A.; Wang, B.; Kumar, A.; Qin, Y.; Jin, J.; Wang, X.; Yang, C.; Dong, B.; Jia, Y.; Liu, J.; et al. Recent Advances for MOF-Derived Carbon-Supported Single-Atom Catalysts. Small Methods 2019, 3, 1800471. [Google Scholar] [CrossRef]
- Huang, H.; Shen, K.; Chen, F.; Li, Y. Metal-Organic Frameworks as a Good Platform for the Fabrication of Single-Atom Catalysts. ACS Catal. 2020, 10, 6579–6586. [Google Scholar] [CrossRef]
- Song, Z.; Zhang, L.; Doyle-Davis, K.; Fu, X.; Luo, J.L.; Sun, X. Recent Advances in MOF-Derived Single Atom Catalysts for Electrochemical Applications. Adv. Energy Mater. 2020, 10, 2001561. [Google Scholar] [CrossRef]
- Li, Z.; Schweitzer, N.M.; League, A.B.; Bernales, V.; Peters, A.W.; Getsoian, A.B.; Wang, T.C.; Miller, J.T.; Vjunov, A.; Fulton, J.L.; et al. Sintering-Resistant Single-Site Nickel Catalyst Supported by Metal–Organic Framework. J. Am. Chem. Soc. 2016, 138, 1977–1982. [Google Scholar] [CrossRef]
- Verma, P.; Vogiatzis, K.D.; Planas, N.; Borycz, J.; Xiao, D.J.; Long, J.R.; Gagliardi, L.; Truhlar, D.G. Mechanism of Oxidation of Ethane to Ethanol at Iron(IV)–Oxo Sites in Magnesium-Diluted Fe2(dobdc). J. Am. Chem. Soc. 2015, 137, 5770–5781. [Google Scholar] [CrossRef]
- Simons, M.C.; Vitillo, J.G.; Babucci, M.; Hoffman, A.S.; Boubnov, A.; Beauvais, M.L.; Chen, Z.; Cramer, C.J.; Chapman, K.W.; Bare, S.R.; et al. Structure, Dynamics, and Reactivity for Light Alkane Oxidation of Fe(II) Sites Situated in the Nodes of a Metal–Organic Framework. J. Am. Chem. Soc. 2019, 141, 18142–18151. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.N.; Li, M.; Bollini, P. Light Alkane Oxidation over Well-Defined Active Sites in Metal-Organic Framework Materials. Catal. Sci. Technol. 2022, 12, 418–435. [Google Scholar] [CrossRef]
- Simons, M.C.; Prinslow, S.D.; Babucci, M.; Hoffman, A.S.; Hong, J.; Vitillo, J.G.; Bare, S.R.; Gates, B.C.; Lu, C.C.; Gagliardi, L.; et al. Beyond Radical Rebound: Methane Oxidation to Methanol Catalyzed by Iron Species in Metal–Organic Framework Nodes. J. Am. Chem. Soc. 2021, 143, 12165–12174. [Google Scholar] [CrossRef] [PubMed]
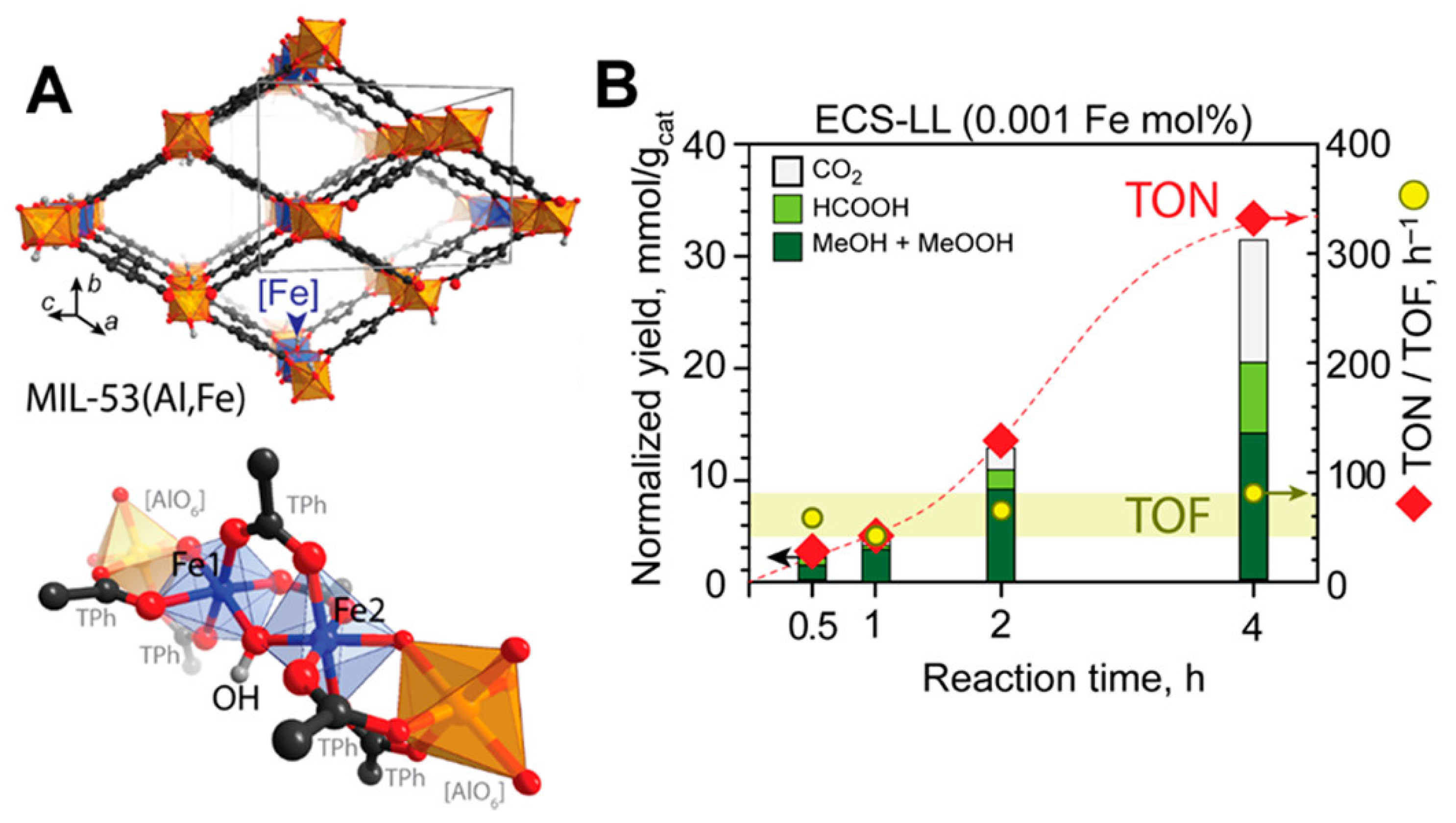
- Szécsényi, Á.; Li, G.; Gascon, J.; Pidko, E.A. Unraveling Reaction Networks Behind the Catalytic Oxidation of Methane with H2O2 Over a Mixed-Metal MIL-53(Al, Fe) MOF Catalyst. Chem. Sci. 2018, 9, 6765–6773. [Google Scholar] [CrossRef]
- An, B.; Li, Z.; Wang, Z.; Zeng, X.; Han, X.; Cheng, Y.; Sheveleva, A.M.; Zhang, Z.; Tuna, F.; McInnes, E.J.L.; et al. Direct Photo-Oxidation of Methane to Methanol over a Mono-Iron Hydroxyl Site. Nat. Mater. 2022, 21, 932–938. [Google Scholar] [CrossRef] [PubMed]
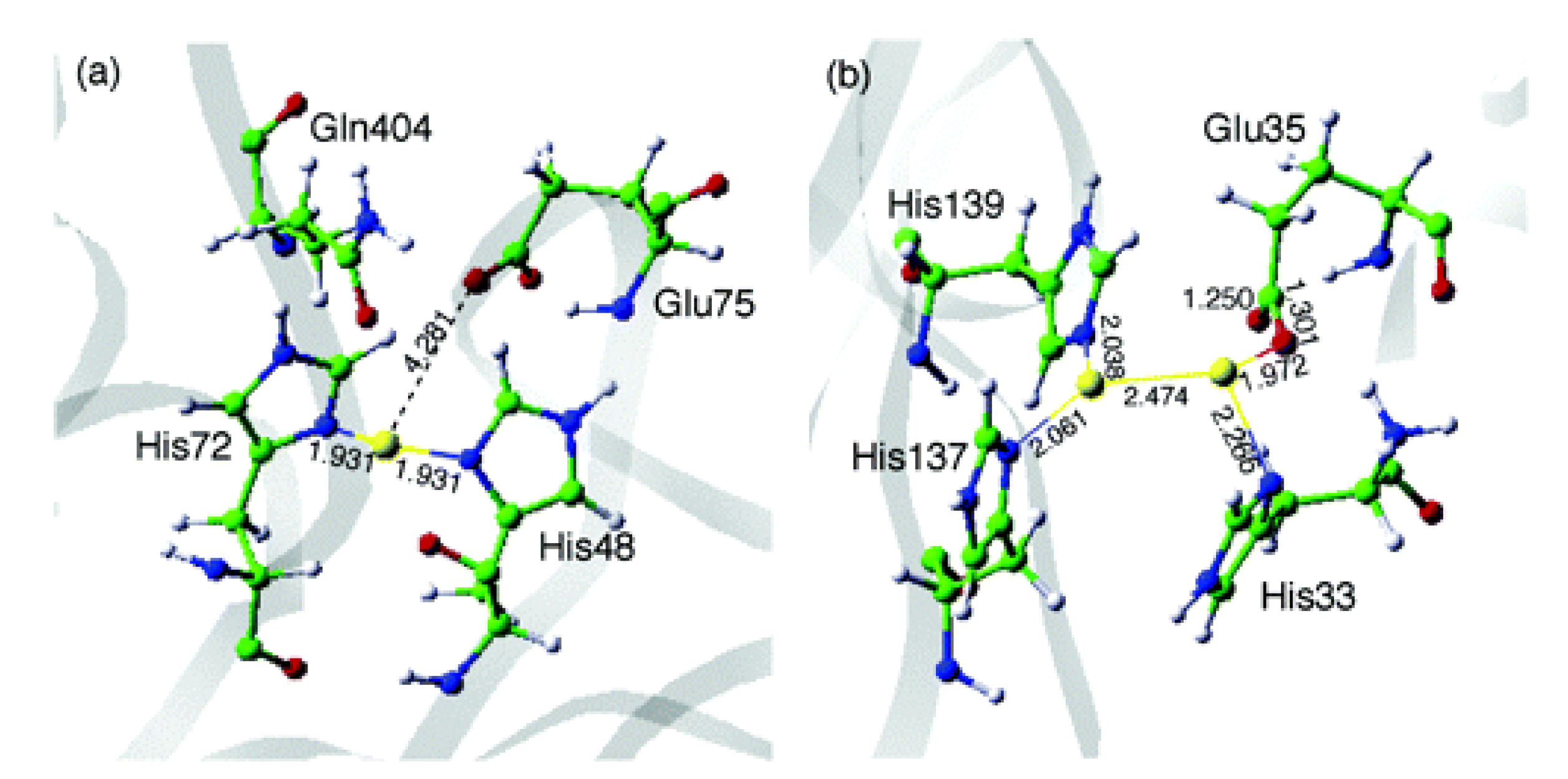
- Baek, J.; Rungtaweevoranit, B.; Pei, X.; Park, M.; Fakra, S.C.; Liu, Y.S.; Matheu, R.; Alshmimri, S.A.; Alshehri, S.; Trickett, C.A.; et al. Bioinspired Metal–Organic Framework Catalysts for Selective Methane Oxidation to Methanol. J. Am. Chem. Soc. 2018, 140, 18208–18216. [Google Scholar] [CrossRef]
- Zheng, J.; Ye, J.; Ortuño, M.A.; Fulton, J.L.; Gutiérrez, O.Y.; Camaioni, D.M.; Motkuri, R.K.; Li, Z.; Webber, T.E.; Mehdi, B.L.; et al. Selective Methane Oxidation to Methanol on Cu-oxo Dimers stabilized by Zirconia Nodes of an NU-1000 Metal–Organic Framework. J. Am. Chem. Soc. 2019, 141, 9292–9304. [Google Scholar] [CrossRef]
- Ikuno, T.; Zheng, J.; Vjunov, A.; Sanchez-Sanchez, M.; Ortuño, M.A.; Pahls, D.R.; Fulton, J.L.; Camaioni, D.M.; Li, Z.; Ray, D.; et al. Methane Oxidation to Methanol Catalyzed by Cu-oxo Clusters Stabilized in NU-1000 Metal–Organic Framework. J. Am. Chem. Soc. 2017, 139, 10294–10301. [Google Scholar] [CrossRef]
- Kaye, S.S.; Dailly, A.; Yaghi, O.M.; Long, J.R. Impact of Preparation and Handling on the Hydrogen Storage Properties of Zn4O(1,4-benzenedicarboxylate)3 (MOF-5). J. Am. Chem. Soc. 2007, 129, 14176–14177. [Google Scholar] [CrossRef]
- Bloch, E.D.; Murray, L.J.; Queen, W.L.; Chavan, S.; Maximoff, S.N.; Bigi, J.P.; Krishna, R.; Peterson, V.K.; Grandjean, F.; Long, G.J.; et al. Selective Binding of O2 over N2 in a Redox-Active Metal-Organic Framework with Open Iron(II) Coordination Sites. J. Am. Chem. Soc. 2011, 133, 14814–14822. [Google Scholar] [CrossRef]
- Bara, D.; Meekel, E.G.; Pakamorė, I.; Wilson, C.; Ling, S.; Forgan, R.S. Exploring and Expanding the Fe-Terephthalate Metal-Organic framework Phase space by Coordination and Oxidation Modulation. Mater. Horiz. 2021, 8, 3377–3386. [Google Scholar] [CrossRef]
- Hendon, C.H.; Walsh, A. Chemical Principles Underpinning the Performance of the Metal-Organic Framework HKUST-1. Chem. Sci. 2015, 6, 3674–3683. [Google Scholar] [CrossRef] [PubMed]
- Quijia, C.R.; Lima, C.; Silva, C.; Alves, R.C.; Frem, R.; Chorilli, M. Application of MIL-100 (Fe) in Drug Delivery and Biomedicine. J. Drug. Deliv. Sci. Technol. 2021, 61, 102217. [Google Scholar] [CrossRef]
- Yuan, S.; Sun, X.; Pang, J.; Lollar, C.; Qin, J.S.; Perry, Z.; Joseph, E.; Wang, X.; Fang, Y.; Bosch, M.; et al. PCN-250 Under Pressure: Sequential Phase Transformation and the Implications for MOF Densification. Joule 2017, 1, 806–815. [Google Scholar] [CrossRef]
- Chen, B.; Ockwig, N.W.; Millward, A.R.; Contreras, D.S.; Yaghi, O.M. High H2 Adsorption in a Microporous Metal–Organic Framework with Open Metal Sites. Angew. Chem. Int. Ed. 2005, 44, 4745–4749. [Google Scholar] [CrossRef] [PubMed]
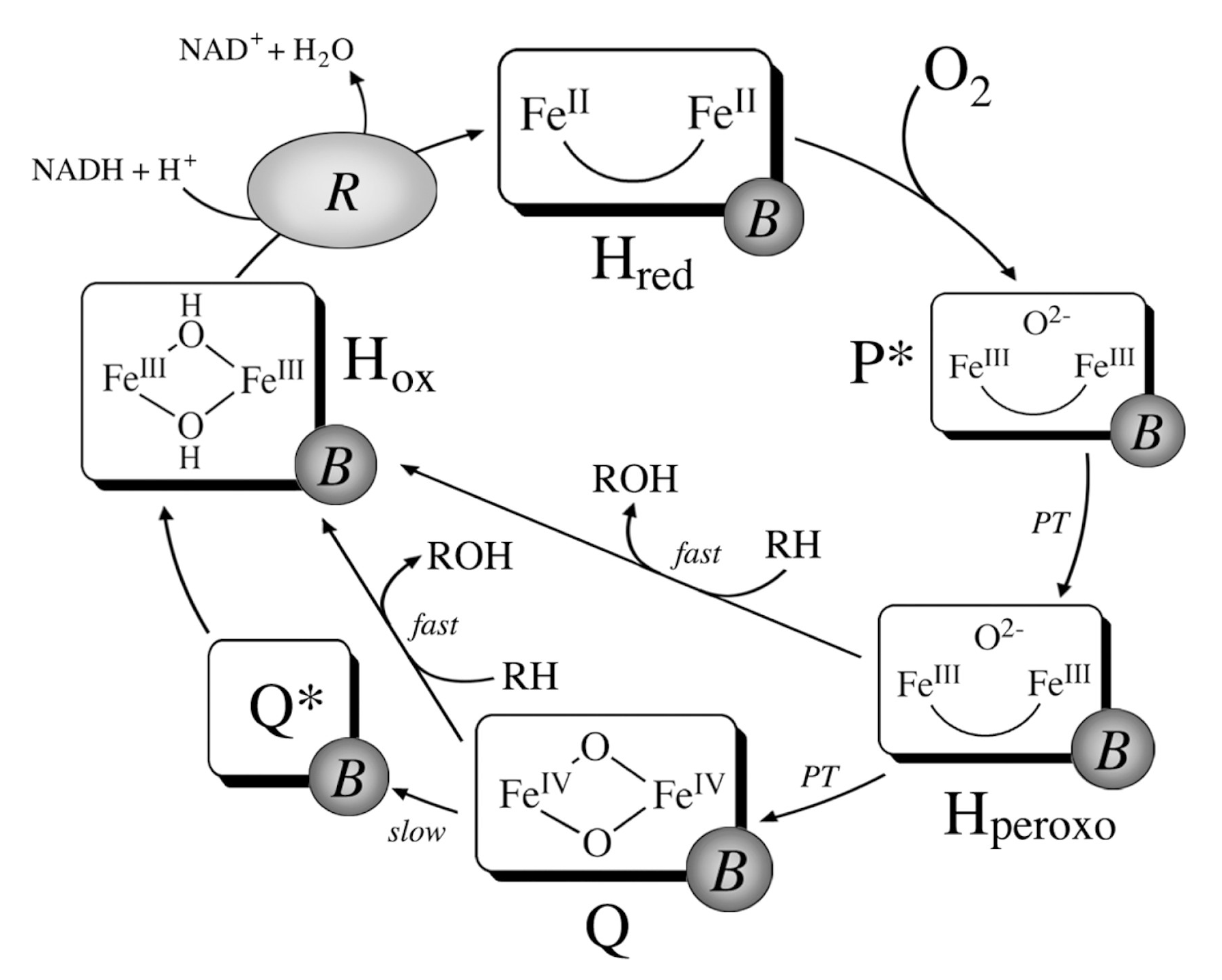
- Banerjee, R.; Proshlyakov, Y.; Lipscomb, J.D.; Proshlyakov, D.A. Structure of the Key Species in the Enzymatic Oxidation of Methane to Methanol. Nature 2015, 518, 431–434. [Google Scholar] [CrossRef]
- Wang, W.; Liang, A.D.; Lippard, S.J. Coupling Oxygen Consumption with Hydrocarbon Oxidation in Bacterial Multicomponent Monooxygenases. Acc. Chem. Res. 2015, 48, 2632–2639. [Google Scholar] [CrossRef]
- Wang, V.C.C.; Maji, S.; Chen, P.P.Y.; Lee, H.K.; Yu, S.S.F.; Chan, S.I. Alkane Oxidation: Methane Monooxygenases, Related Enzymes, and Their Biomimetics. Chem. Rev. 2017, 117, 8574–8621. [Google Scholar] [CrossRef]
- Baik, M.H.; Newcomb, M.; Friesner, R.A.; Lippard, S.J. Mechanistic Studies on the Hydroxylation of Methane by Methane Monooxygenase. Chem. Rev. 2003, 103, 2385–2420. [Google Scholar] [CrossRef]
- Liu, K.E.; Valentine, A.M.; Wang, D.; Huynh, B.H.; Edmondson, D.E.; Salifoglou, A.; Lippard, S.J. Kinetic and Spectroscopic Characterization of Intermediates and Component Interactions in Reactions of Methane Monooxygenase from Methylococcus Capsulatus (Bath). J. Am. Chem. Soc. 1995, 117, 10174–10185. [Google Scholar] [CrossRef]
- Jacobs, A.B.; Banerjee, R.; Deweese, D.E.; Braun, A.; Babicz, J.T.J.; Gee, L.B.; Sutherlin, K.D.; Böttger, L.H.; Yoda, Y.; Saito, M.; et al. Nuclear Resonance Vibrational Spectroscopic Definition of the Fe(IV)2 Intermediate Q in Methane Monooxygenase and Its Reactivity. J. Am. Chem. Soc. 2021, 143, 16007–16029. [Google Scholar] [CrossRef] [PubMed]
- Kazaryan, A.; Baerends, E.J. Ligand Field Effects and the High Spin-High Reactivity Correlation in the H Abstraction by Non-Heme Iron(IV)-Oxo Complexes: A DFT Frontier Orbital Perspective. ACS Catal. 2015, 5, 1475–1488. [Google Scholar] [CrossRef]
- Isolated Fe Sites in Metal Organic Frameworks Catalyze the Direct Conversion of Methane to Methanol. ACS Catal. 2018, 8, 5542–5548. [CrossRef]
- Loiseau, T.; Serre, C.; Huguenard, C.; Fink, G.; Taulelle, F.; Henry, M.; Bataille, T.; Férey, G. A Rationale for the Large Breathing of the Porous Aluminum Terephthalate (MIL-53) Upon Hydration. Chem. Eur. J. 2004, 10, 1373–1382. [Google Scholar] [CrossRef]
- Férey, G.; Serre, C.; Mellot-Draznieks, C.; Millange, F.; Surblé, S.; Dutour, J.; Margiolaki, I. A Hybrid Solid with Giant Pores Prepared by a Combination of Targeted Chemistry, Simulation, and Powder Diffraction. Angew. Chem. Int. Ed. 2004, 43, 6296–6301. [Google Scholar] [CrossRef]
- Horcajada, P.; Surblé, S.; Serre, C.; Hong, D.Y.; Seo, Y.K.; Chang, J.S.; Grenèche, J.M.; Margiolaki, I.; Férey, G. Synthesis and Catalytic Properties of MIL-100(Fe), an Iron(iii) Carboxylate with Large Pores. Chem. Commun. 2007, 2820–2822. [Google Scholar] [CrossRef]
- Colby, J.; Stirling, D.I.; Dalton, H. The Soluble Methane Mono-Oxygenase of Methylococcus Capsulatus (Bath). Its Ability to Oxygenate n-Alkanes, n-Alkenes, Ethers, and Alicyclic, Aromatic and Heterocyclic Compounds. Biochem. J. 1977, 165, 395–402. [Google Scholar] [CrossRef]
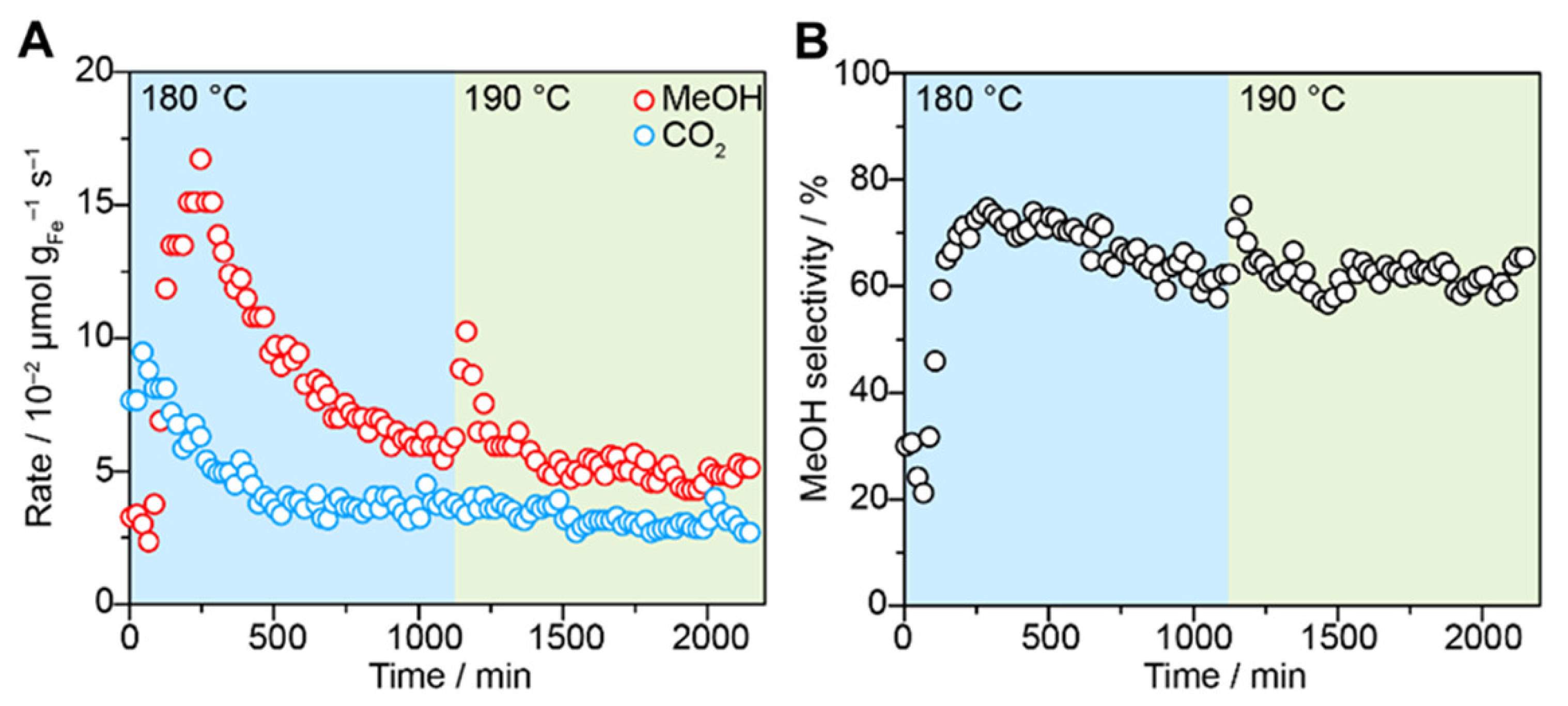
- Rungtaweevoranit, B.; Abdel-Mageed, A.M.; Khemthong, P.; Eaimsumang, S.; Chakarawet, K.; Butburee, T.; Kunkel, B.; Wohlrab, S.; Chainok, K.; Phanthasri, J.; et al. Structural Evolution of Iron-Loaded Metal-Organic Framework Catalysts for Continuous Gas-Phase Oxidation of Methane to Methanol. ACS Appl. Mater. Interfaces 2023, 15, 26700–26709. [Google Scholar] [CrossRef]
- Semrau, J.D.; DiSpirito, A.A.; Yoon, S. Methanotrophs and Copper. FEMS Microbiol. Rev. 2010, 34, 496–531. [Google Scholar] [CrossRef]
- Stanley, S.; Prior, S.; Leak, D.; Dalton, H. Copper Stress Underlies the Fundamental Change in Intracellular Location of Methane Mono-Oxygenase in Methane-Oxidizing Organisms: Studies in Batch and Continuous Cultures. Biotechnol. Lett. 1983, 5, 487–492. [Google Scholar] [CrossRef]
- Murrell, J.C.; McDonald, I.R.; Gilbert, B. Regulation of expression of methane monooxygenases by copper ions. Trends Microbiol. 2000, 8, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Lawton, T.J.; Rosenzweig, A.C. Methane-Oxidizing Enzymes: An Upstream Problem in Biological Gas-to-Liquids Conversion. J. Am. Chem. Soc. 2016, 138, 9327–9340. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.M.; Rawat, S.; Telser, J.; Hoffman, B.M.; Stemmler, T.L.; Rosenzweig, A.C. Crystal Structure and Characterization of Particulate Methane Monooxygenase from Methylocystis Species Strain M. Biochemistry 2011, 50, 10231–10240. [Google Scholar] [CrossRef] [PubMed]
- Ross, M.O.; Rosenzweig, A.C. A Tale of Two Methane Monooxygenases. J. Biol. Inorg. Chem. 2017, 22, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Cutsail, G.E.; Ross, M.O.; Rosenzweig, A.C.; DeBeer, S. Towards a Unified Understanding of the Copper Sites in Particulate Methane Monooxygenase: An X-ray Absorption Spectroscopic Investigation. Chem. Sci. 2021, 12, 6194–6209. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa, K.; Shiota, Y. Conversion of Methane to Methanol at the Mononuclear and Dinuclear Copper Sites of Particulate Methane Monooxygenase (pMMO): A DFT and QM/MM Study. J. Am. Chem. Soc. 2006, 128, 9873–9881. [Google Scholar] [CrossRef]
- Citek, C.; Herres-Pawlis, S.; Stack, T.D.P. Low Temperature Syntheses and Reactivity of Cu2O2 Active-Site Models. Acc. Chem. Res. 2015, 48, 2424–2433. [Google Scholar] [CrossRef]
- Woertink, J.S.; Smeets, P.J.; Groothaert, M.H.; Vance, M.A.; Sels, B.F.; Schoonheydt, R.A.; Solomon, E.I. A [Cu2O]2+ Core in Cu-ZSM-5, the Active Site in the Oxidation of Methane to Methanol. Proc. Nat. Ac. Sci. USA 2009, 106, 18908–18913. [Google Scholar] [CrossRef]
- Grundner, S.; Markovits, M.A.; Li, G.; Tromp, M.; Pidko, E.A.; Hensen, E.J.; Jentys, A.; Sanchez-Sanchez, M.; Lercher, J.A. Single-Site Trinuclear Copper Oxygen Clusters in Mordenite for Selective Conversion of Methane to Methanol. Nat. Commun. 2015, 6, 7546. [Google Scholar] [CrossRef]
- Pappas, D.K.; Borfecchia, E.; Dyballa, M.; Pankin, I.A.; Lomachenko, K.A.; Martini, A.; Signorile, M.; Teketel, S.; Arstad, B.; Berlier, G.; et al. Methane to Methanol: Structure-Activity Relationships for Cu-CHA. J. Am. Chem. Soc. 2017, 139, 14961–14975. [Google Scholar] [CrossRef]
- Platero-Prats, A.E.; Li, Z.; Gallington, L.C.; Peters, A.W.; Hupp, J.T.; Farha, O.K.; Chapman, K.W. Addressing the Characterisation Challenge to Understand Catalysis in MOFs: The Case of Nanoscale Cu Supported in NU-1000. Faraday Discuss. 2017, 201, 337–350. [Google Scholar] [CrossRef]
- Webber, T.E.; Desai, S.P.; Combs, R.L.; Bingham, S.; Lu, C.C.; Penn, R.L. Size Control of the MOF NU-1000 through Manipulation of the Modulator/Linker Competition. Cryst. Growth Des. 2020, 20, 2965–2972. [Google Scholar] [CrossRef]
- Tomkins, P.; Mansouri, A.; Bozbag, S.E.; Krumeich, F.; Park, M.B.; Alayon, E.M.C.; Ranocchiari, M.; van Bokhoven, J.A. Isothermal Cyclic Conversion of Methane into Methanol over Copper-Exchanged Zeolite at Low Temperature. Angew. Chem. Int. Ed. 2016, 128, 5557–5561. [Google Scholar] [CrossRef]
- Sheppard, T.; Hamill, C.; Goguet, A.; Rooney, D.; Thompson, J. A Low temperature, Isothermal Gas-Phase System for Conversion of Methane to Methanol Over Cu–ZSM-5. Chem. Commun. 2014, 50, 11053–11055. [Google Scholar] [CrossRef] [PubMed]
- Ren, M.; Shi, Q.; Mi, L.; Liang, W.; Yuan, M.; Wang, L.; Gao, Z.; Huang, W.; Huang, J.; Zuo, Z. Isothermal Conversion of Methane to Methanol over CuxOy@UiO-bpy. Mater. Today Sustain. 2021, 11–12, 100061. [Google Scholar] [CrossRef]
- Beznis, N.V.; Weckhuysen, B.M.; Bitter, J.H. Cu-ZSM-5 Zeolites for the Formation of Methanol from Methane and Oxygen: Probing the Active Sites and Spectator Species. Catal. Lett. 2010, 138, 14–22. [Google Scholar] [CrossRef]
- Lee, H.; Kwon, C.; Keum, C.; Kim, H.E.; Lee, H.; Han, B.; Lee, S.Y. Methane Partial Oxidation by Monomeric Cu Active Center Confined on ZIF-7. Chem. Eng. J. 2022, 450, 138472. [Google Scholar] [CrossRef]
- Delgass, W. Spectroscopy in Heterogeneous Catalysis; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Braglia, L.; Tavani, F.; Mauri, S.; Edla, R.; Krizmancic, D.; Tofoni, A.; Colombo, V.; D’Angelo, P.; Torelli, P. Catching the Reversible Formation and Reactivity of Surface Defective Sites in Metal-Organic Frameworks: An Operando Ambient Pressure-NEXAFS Investigation. J. Phys. Chem. Lett. 2021, 12, 9182–9187. [Google Scholar] [CrossRef]
- Tavani, F.; Fracchia, M.; Tofoni, A.; Braglia, L.; Jouve, A.; Morandi, S.; Manzoli, M.; Torelli, P.; Ghigna, P.; D’Angelo, P. Structural and Mechanistic Insights into Low-Temperature CO Oxidation over a Prototypical High Entropy Oxide by Cu L-edge Operando Soft X-ray Absorption Spectroscopy. Phys. Chem. Chem. Phys. 2021, 23, 26575–26584. [Google Scholar] [CrossRef]
- Tavani, F.; Busato, M.; Braglia, L.; Mauri, S.; Torelli, P.; D’Angelo, P. Caught while Dissolving: Revealing the Interfacial Solvation of the Mg2+ Ions on the MgO Surface. ACS Appl. Mater. Interfaces 2022, 14, 38370–38378. [Google Scholar] [CrossRef]
- Tavani, F.; Busato, M.; Veclani, D.; Braglia, L.; Mauri, S.; Torelli, P.; D’Angelo, P. Investigating the High-Temperature Water/MgCl2 Interface through Ambient Pressure Soft X-ray Absorption Spectroscopy. ACS Appl. Mater. Interfaces 2023, 15, 26166–26174. [Google Scholar] [CrossRef] [PubMed]
- Capocasa, G.; Sessa, F.; Tavani, F.; Monte, M.; Olivo, G.; Pascarelli, S.; Lanzalunga, O.; Di Stefano, S.; D’Angelo, P. Coupled X-ray Absorption/UV-vis Monitoring of Fast Oxidation Reactions Involving a Nonheme Iron-Oxo Complex. J. Am. Chem. Soc. 2019, 141, 2299–2304. [Google Scholar] [CrossRef] [PubMed]
- Tavani, F.; Martini, A.; Capocasa, G.; Di Stefano, S.; Lanzalunga, O.; D’Angelo, P. Direct Mechanistic Evidence for a Non-Heme Complex Reaction Through a Multivariate XAS Analysis. Inorg. Chem. 2020, 59, 9979–9989. [Google Scholar] [CrossRef]
- Tavani, F.; Fracchia, M.; Pianta, N.; Ghigna, P.; Quartarone, E.; D’Angelo, P. Multivariate Curve Resolution Analysis of Operando XAS Data for the Investigation of the Lithiation Mechanisms in High Entropy Oxides. Chem. Phys. Lett. 2020, 760, 137968. [Google Scholar] [CrossRef]
- Tavani, F.; Capocasa, G.; Martini, A.; Sessa, F.; Di Stefano, S.; Lanzalunga, O.; D’Angelo, P. Direct Structural and Mechanistic Insights into Fast Bimolecular Chemical Reactions in Solution through a Coupled XAS/UV-Vis Multivariate Statistical Analysis. Dalton Trans. 2021, 50, 131–142. [Google Scholar] [CrossRef]
- Tavani, F.; Capocasa, G.; Martini, A.; Sessa, F.; Di Stefano, S.; Lanzalunga, O.; D’Angelo, P. Activation of C-H Bonds by a Nonheme Iron(iv)-Oxo Complex: Mechanistic Evidence through a Coupled EDXAS/UV-Vis Multivariate Analysis. Phys. Chem. Chem. Phys. 2021, 23, 1188–1196. [Google Scholar] [CrossRef]
- Del Giudice, D.; Tavani, F.; Di Berto Mancini, M.; Frateloreto, F.; Busato, M.; Oliveira De Souza, D.; Cenesi, F.; Lanzalunga, O.; Di Stefano, S.; D’Angelo, P. Two Faces of the Same Coin: Coupling X-Ray Absorption and NMR Spectroscopies to Investigate the Exchange Reaction Between Prototypical Cu Coordination Complexes. Chem. Eur. J. 2022, 28, e202103825. [Google Scholar] [CrossRef] [PubMed]
- Frateloreto, F.; Tavani, F.; Di Berto Mancini, M.; Del Giudice, D.; Isabelle, K.; Lanzalunga, O.; Di Stefano, S.; D’Angelo, P. Following a Silent Metal Ion: A Combined X-ray Absorption and Nuclear Magnetic Resonance Spectroscopic Study of the Zn2+ Cation Dissipative Translocation between Two Different Ligands. J. Phys. Chem. Lett. 2022, 13, 5522–5529. [Google Scholar] [CrossRef]
- Feng, D.; Wang, K.; Wei, Z.; Chen, Y.P.; Simon, C.M.; Arvapally, R.K.; Martin, R.L.; Bosch, M.; Liu, T.F.; Fordham, S.; et al. Kinetically Tuned Dimensional Augmentation as a Versatile Synthetic Route Towards Robust Metal-Organic Frameworks. Nat. Commun. 2014, 5, 5723. [Google Scholar] [CrossRef]
- Snyder, B.E.R.; Bols, M.L.; Rhoda, H.M.; Plessers, D.; Schoonheydt, R.A.; Sels, B.F.; Solomon, E.I. Cage effects control the mechanism of methane hydroxylation in zeolites. Science 2021, 373, 327–331. [Google Scholar] [CrossRef]
- Kirillov, A.M.; Shul’pin, G.B. Pyrazinecarboxylic acid and analogs: Highly efficient co-catalysts in the metal-complex-catalyzed oxidation of organic compounds. Coord. Chem. Rev. 2013, 257, 732–754. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MOF | Active Site Type | Oxidant | Methanol Yield | Methanol Selectivity | Reference |
|---|---|---|---|---|---|
| MIL-53(Al,Fe) | FeO SBU | HO | ∼10–30 molg * | ∼10–40 * | [86] |
| MIL-100(Fe) | Fe--oxo trimer | NO | 0.34 mol(mol Fe) | ∼99% at 453 K | [34] |
| PCN-250 | Fe--oxo trimer | NO | 200 molg | 70% | [66] |
| PMOF-RuFe(OH) | mono-iron hydroxyl | O | 8.81 ± 0.34 mmolgh | 100% | [68] |
| Fe-UiO-66 | mono-iron hydroxyl | O | 0.167 molgs | 62% | [91] |
| MOF | Active Site Type | Oxidant | Methanol Yield (molg) | Methanol Selectivity | Reference |
|---|---|---|---|---|---|
| MOF-808-Bzz-Cu | histidine-supported Cu-oxo dimer | NO | 71.8 ± 23.4 | 100% | [69] |
| Cu-NU-1000 | Cu-oxo cluster | O | 17.7 | 45% (including DME) | [71] |
| Cu-2.9-NU-1000 | Cu-oxo dimer | O | 4.4 | 70% | [70] |
| Cu-UiO-bpy | CuO clusters | O | 24.3 | 88% | [108] |
| Cu-ZIF-7 | ligand-supported tetrahedral Cu | HO | 48 | 8% | [110] |
| Information Mainly Provided on: | |||||
|---|---|---|---|---|---|
| Spectroscopy | Energy (eV) | Kind of Transition | Solid Surface | Bond between Adsorbed Molecule and Surface | Adsorbed Molecule Structure |
| Infrared | (50–2.5) × 10 | Vibrational | N | Y | Y |
| Raman | (50–0.6) × 10 | Vibrational | N | Y | Y |
| Visible/near infrared | 0.5–6.5 | Electronic, vibrational | Y | Y | Y |
| Mössbauer | 10–10 | Nuclear | Y | N | N |
| EPR | (14–3.8) × 10 | Nuclear spin | Y | N | Y |
| NMR | (2.5–8.3) × 10 | Nuclear spin | Y | Y | Y |
| XPS | (0.1–1500) | Bound electron to the continuum | Y | Y | Y |
| XAS | (0.1–100) × 10 | Bound electron to the continuum | Y | Y | Y |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tavani, F.; Tofoni, A.; D’Angelo, P. Exploring the Methane to Methanol Oxidation over Iron and Copper Sites in Metal–Organic Frameworks. Catalysts 2023, 13, 1338. https://doi.org/10.3390/catal13101338
Tavani F, Tofoni A, D’Angelo P. Exploring the Methane to Methanol Oxidation over Iron and Copper Sites in Metal–Organic Frameworks. Catalysts. 2023; 13(10):1338. https://doi.org/10.3390/catal13101338
Chicago/Turabian StyleTavani, Francesco, Alessandro Tofoni, and Paola D’Angelo. 2023. "Exploring the Methane to Methanol Oxidation over Iron and Copper Sites in Metal–Organic Frameworks" Catalysts 13, no. 10: 1338. https://doi.org/10.3390/catal13101338