
Mechanistic Insights into Palladium(II)-Catalyzed Carboxylation of Thiophene and Carbon Dioxide

Abstract
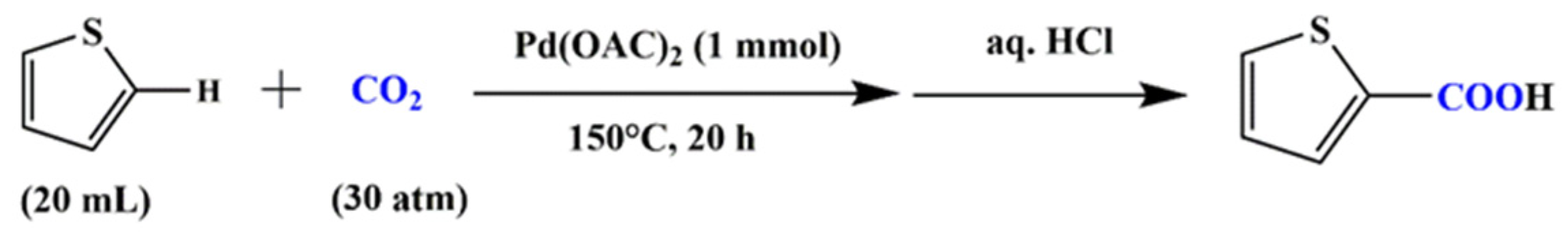
:1. Introduction
2. Results and Discussions
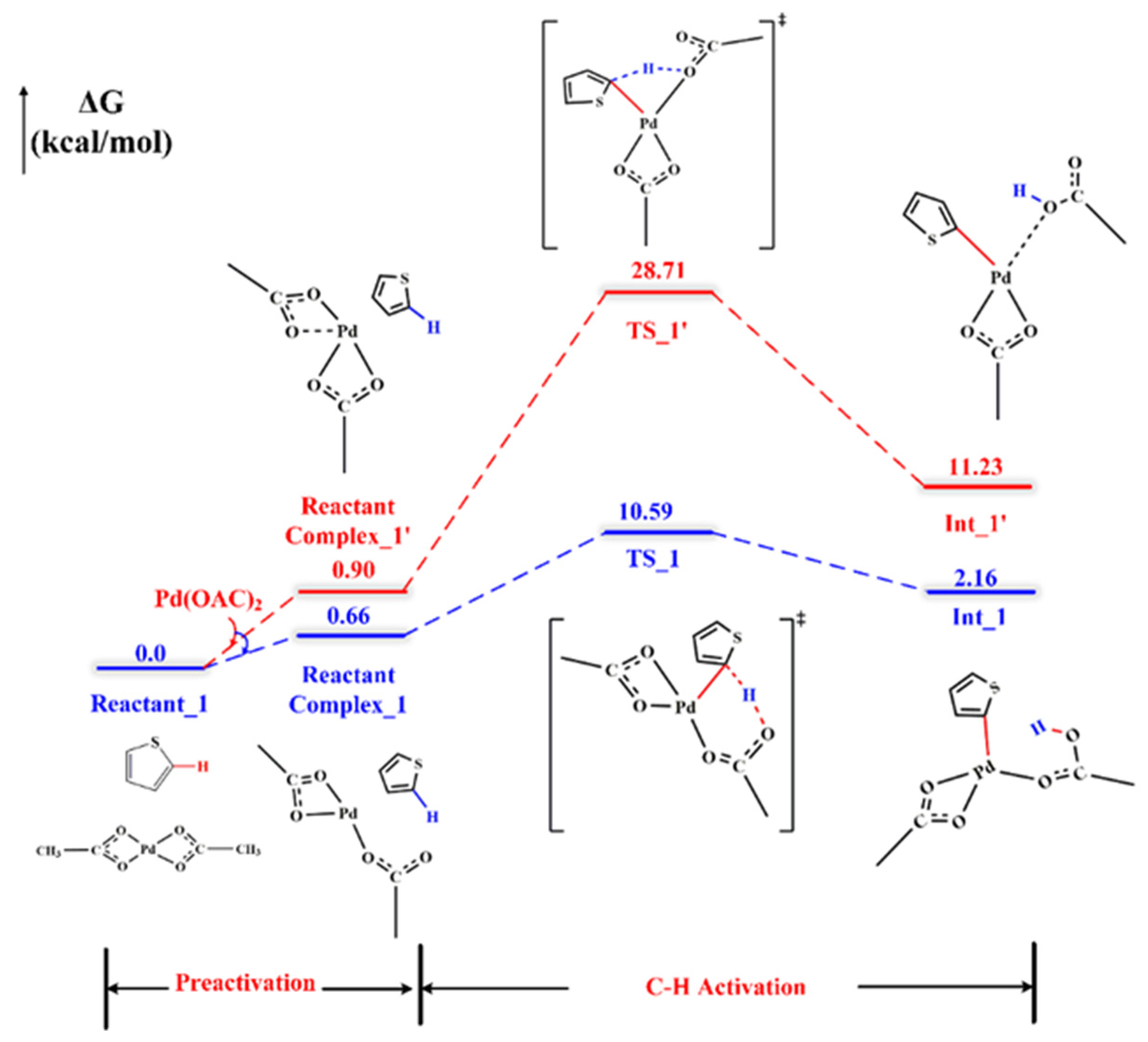
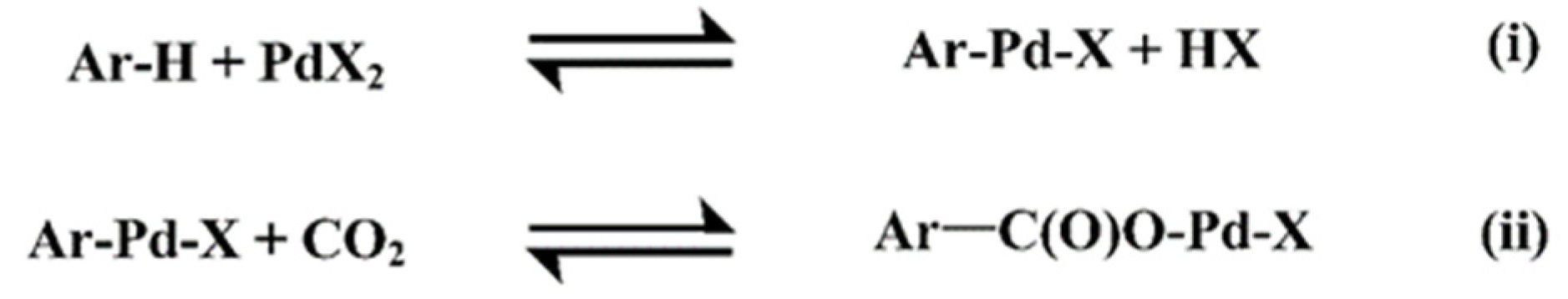
2.1. C–H Activation Step
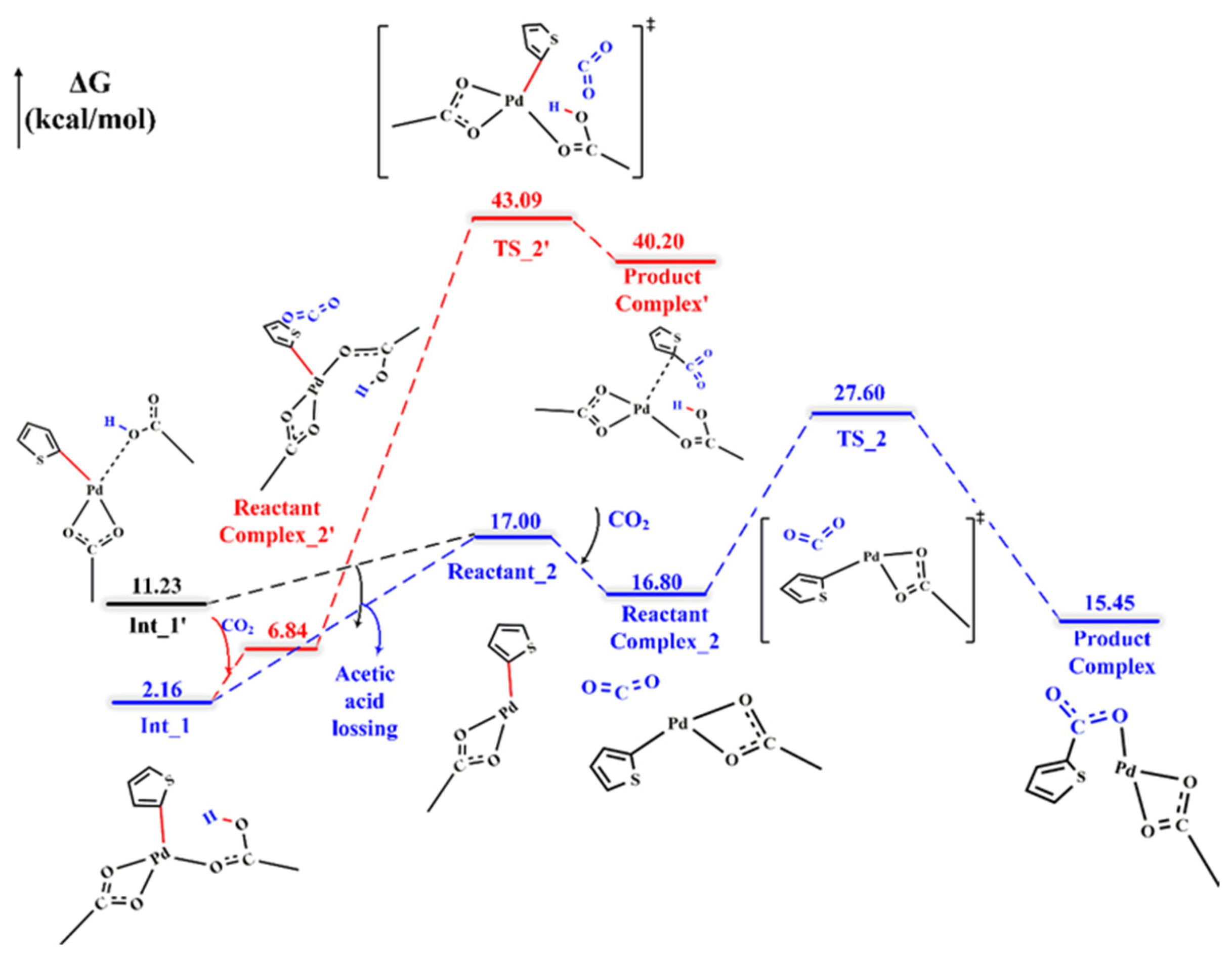
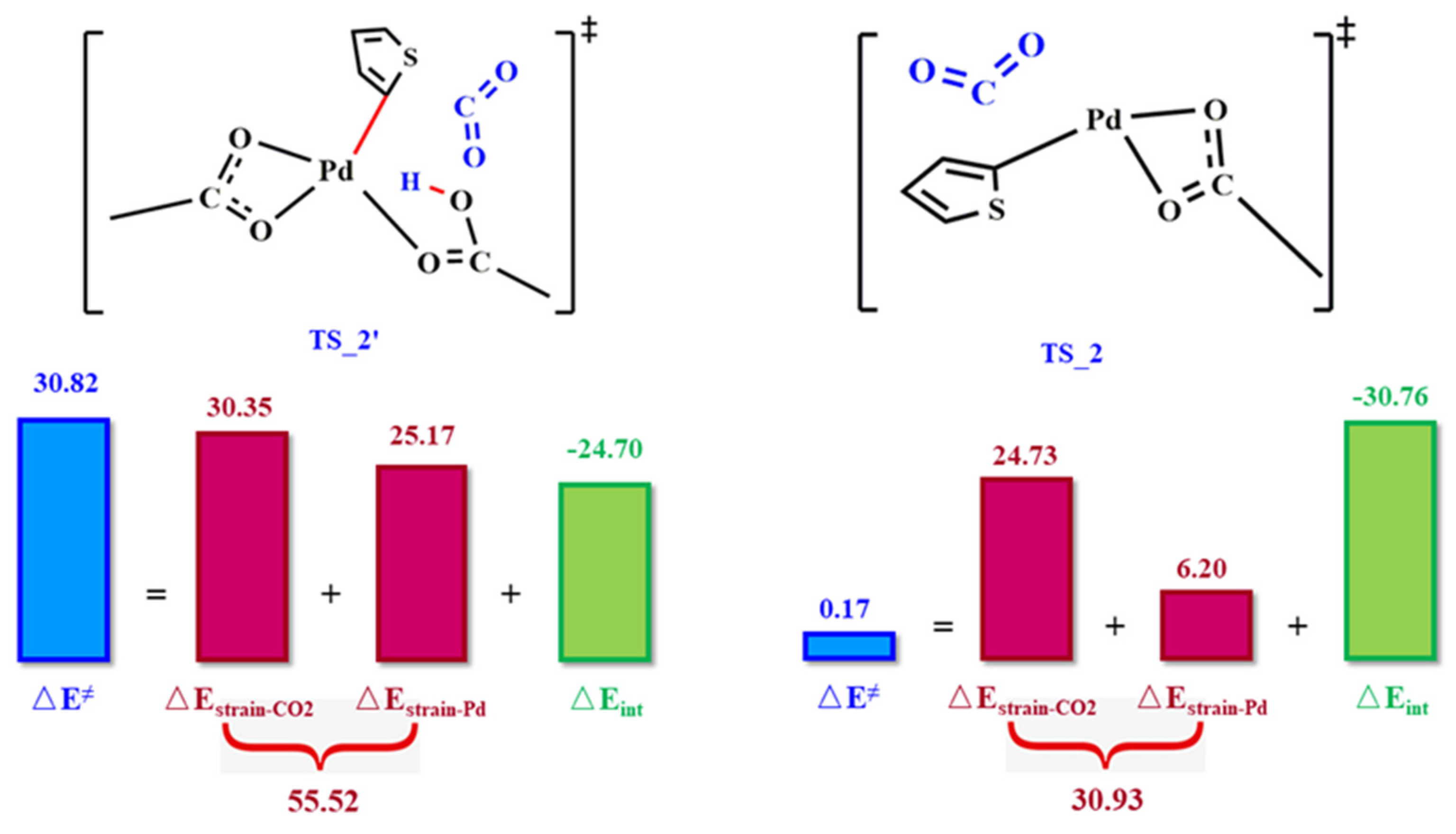
2.2. CO2 Insertion Step
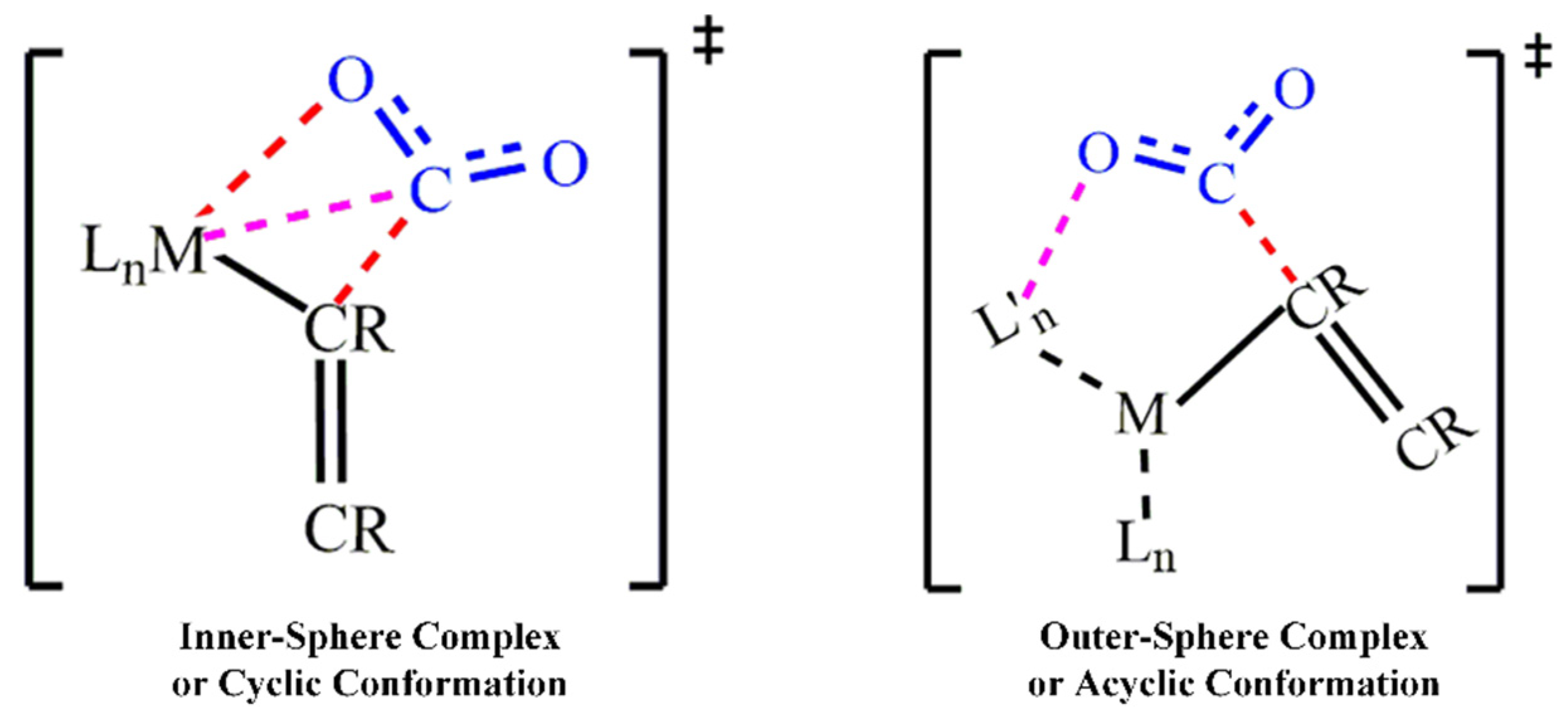
2.3. CO2 Interaction Modes
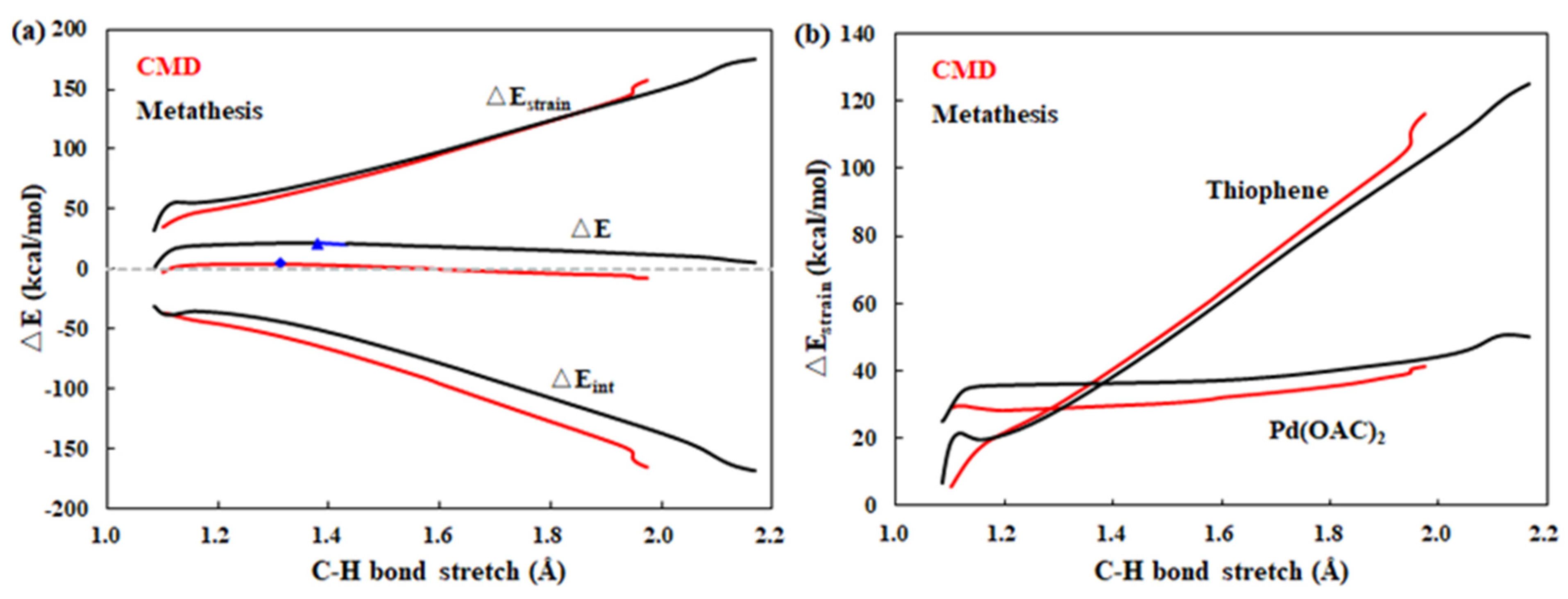
2.4. Clarification of the Origin of Difference in Energy Barriers for Different Modes
3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sakakura, T.; Choi, J.C.; Yasuda, H. Transformation of carbon dioxide. Chem. Rev. 2007, 107, 2365–2387. [Google Scholar] [CrossRef] [PubMed]
- Kula, K.; Kacka-Zych, A.; Lapczuk-Krygier, A.; Jasinski, R. Analysis of the possibility and molecular mechanism of carbon dioxide consumption in the Diets-Alder processes. Pure Appl. Chem. 2021, 93, 427–446. [Google Scholar] [CrossRef]
- Luo, J.; Larrosa, I. C–H carboxylation of aromatic compounds through CO2 fixation. ChemSusChem 2017, 10, 3317–3332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, J.; Li, M.; Zhang, J.; Sun, B.; Mo, F. C-H bond carboxylation with carbon dioxide. ChemSusChem 2019, 12, 6–39. [Google Scholar] [CrossRef] [PubMed]
- Olah, G.A.; Torok, B.; Joschek, J.P.; Bucsi, I.; Esteves, P.M.; Rasul, G.; Prakash GK, S. Efficient chemoselective carboxylation of aromatics to arylcarboxylic acids with a superelectrophilically activated carbon dioxide-Al2Cl6/Al system. J. Am. Chem. Soc. 2002, 124, 11379–11391. [Google Scholar] [CrossRef] [PubMed]
- Munshi, P.; Beckman, E.J. Effect of incubation of CO2 and Lewis acid on the generation of toluic acid from toluene and CO2. Ind. Eng. Chem. Res. 2009, 48, 1059–1062. [Google Scholar] [CrossRef]
- Munshi, P.; Beckman, E.J.; Padmanabhan, S. Combined influence of fluorinated solvent and base in Friedel-Crafts reaction of toluene and CO2. Ind. Eng. Chem. Res. 2010, 49, 6678–6682. [Google Scholar] [CrossRef]
- Nemoto, K.; Yoshida, H.; Egusa, N.; Morohashi, N.; Hattori, T. Direct carboxylation of arenes and halobenzenes with CO2 by the combined use of AlBr3 and R3SiCl. J. Org. Chem. 2010, 75, 7855–7862. [Google Scholar] [CrossRef]
- Nemoto, K.; Onozawa, S.; Egusa, N.; Morohashi, N.; Hattori, T. Carboxylation of indoles and pyrroles with CO2 in the presence of dialkylaluminum halides. Tetrahedron Lett. 2009, 50, 4512–4514. [Google Scholar] [CrossRef]
- Nemoto, K.; Onozawa, S.; Konno, M.; Morohashi, N.; Hattori, T. Direct carboxylation of thiophenes and benzothiophenes with the aid of EtAlCl2. Bull. Chem. Soc. Jpn. 2012, 85, 369–371. [Google Scholar] [CrossRef]
- Tanaka, S.; Watanabe, K.; Tanaka, Y.; Hattori, T. EtAlCl2/2,6-disubstituted pyridine-mediated carboxylation of alkenes with carbon dioxide. Org. Lett. 2016, 18, 2576–2579. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Cheng, Z. Performance of combined use of chlorosilanes and AlCl3 in the carboxylation of toluene with CO2. AIChE J. 2017, 63, 185–191. [Google Scholar] [CrossRef]
- Boogaerts, F., II; Nolan, S.P. Carboxylation of C-H Bonds using N-heterocyclic carbene gold(I) complexes. J. Am. Chem. Soc. 2010, 132, 8858–8859. [Google Scholar] [CrossRef] [PubMed]
- Boogaerts, F., II; Fortman, G.C.; Furst, M.R.L.; Cazin, C.S.J.; Nolan, S.P. Carboxylation of N-H/C-H bonds using N-heterocyclic carbene copper(I) complexes. Angew. Chem. Int. Ed. 2010, 49, 8674–8677. [Google Scholar] [CrossRef]
- Yoo, W.J.; Capdevila, M.G.; Du, X.; Kobayashi, S. Base-mediated carboxylation of unprotected indole derivatives with carbon dioxide. Org. Lett. 2012, 14, 5326–5329. [Google Scholar] [CrossRef]
- Fenner, S.; Ackermann, L. C-H carboxylation of heteroarenes with ambient CO2. Green Chem. 2016, 18, 3804–3807. [Google Scholar] [CrossRef] [Green Version]
- Shigeno, M.; Hanasaka, K.; Sasaki, K.; Kumada-Nozawa, K.; Kondo, Y. Direct carboxylation of electron-rich heteroarenes promoted by LiO-tBu with CsF and [18]crown-6. Chem. -A Eur. J. 2019, 25, 3235–3239. [Google Scholar]
- Shigeno, M.; Sasaki, K.; Kumada-Nozawa, K.; Kondo, Y. Double-carboxylation of two C-H bonds in 2-alkylheteroarenes using LiO-t-Bu/CsF. Org. Lett. 2019, 21, 4515–4519. [Google Scholar] [CrossRef]
- Shigeno, M.; Tohara, I.; Kumada-Nozawa, K.; Kondo, Y. Direct C-2 carboxylation of 3-substituted indoles using a combined Brønsted base consisting of LiO-tBu/CsF/18-crown-6. Eur. J. Org. Chem. 2020, 2020, 1987–1991. [Google Scholar] [CrossRef]
- Vechorkin, O.; Hirt, N.; Hu, X. Carbon dioxide as the C1 source for direct C-H functionalization of aromatic heterocycles. Org. Lett. 2010, 12, 3567–3569. [Google Scholar] [CrossRef]
- Banerjee, A.; Dick, G.R.; Yoshino, T.; Kanan, M.W. Carbon dioxide utilization via carbonate-promoted C-H carboxylation. Nature 2016, 531, 215–219. [Google Scholar] [CrossRef]
- Dick, G.R.; Frankhouser, A.D.; Banerjee, A.; Kanan, M.W. A scalable carboxylation route to furan-2,5-dicarboxylic acid. Green Chem. 2017, 19, 2966–2972. [Google Scholar] [CrossRef]
- Hiroshi, Y.; Tetsuhiko, O. Preparation of Thiophenecarboxylic Acid. JP58029783, 22 February 1983. [Google Scholar]
- George, C.; Woodbury, J.; John, W. Production of Thiophenecarboxylic Acid. U.S. Patent 2492645, 27 December 1949. [Google Scholar]
- Guo, H. The environmental-friendly synthesis of 2-thiophencarboxylic acid. Chem. Intermed. 2007, 12, 13–20. [Google Scholar]
- Pan, T.; Deng, J.; Xu, Q.; Zuo, Y.; Guo, Q.-X.; Fu, Y. Catalytic conversion of furfural into a 2,5-furandicarboxylic acid-based polyester with total carbon utilization. ChemSusChem 2013, 6, 47–50. [Google Scholar] [CrossRef]
- Sadow, A.D.; Weinstein, Z.B.; Kraus, G.A. Green, Copper-Catalyzed Disproportionation of Aromatic and Heteroaromatic Carboxylates to Dicarboxylates. U.S. Patent 16685456, 15 November 2019. [Google Scholar]
- Mizuno, H.; Takaya, J.; Iwasawa, N. Rhodium(I)-catalyzed direct carboxylation of arenes with CO2 via chelation-assisted C-H bond activation. J. Am. Chem. Soc. 2011, 133, 1251–1253. [Google Scholar] [CrossRef]
- Lv, X.Y.; Zhang, L.H.; Sun, B.B.; Li, Z.; Wu, Y.-B.; Lu, G. Computational studies on the Rh-catalyzed carboxylation of a C(sp2)-H bond using CO2. Catal. Sci. Technol. 2017, 7, 3539–3545. [Google Scholar] [CrossRef]
- Suga, T.; Mizuno, H.; Takaya, J.; Iwasawa, N. Direct carboxylation of simple arenes with CO2 through a rhodium-catalyzed C-H bond activation. Chem. Commun. 2014, 50, 14360–14363. [Google Scholar] [CrossRef]
- Lee, M.J.; Hwang, Y.K.; Kwak, J. Ag(I)-catalyzed C-H carboxylation of thiophene derivatives. Organometallics 2021, 40, 3136–3144. [Google Scholar] [CrossRef]
- Sugimoto, H.; Kawata, I.; Taniguchi, H.; Fujiwara, Y. Palladium-catalyzed carboxylation of aromatic compounds with carbon dioxide. J. Organomet. Chem. 1984, 266, C44–C46. [Google Scholar] [CrossRef]
- Shan, C.; Bai, R.; Lan, Y. Theoretical advances of transition metals mediated C-H bonds cleavage. Acta Phys.-Chim. Sin. 2019, 35, 940–953. [Google Scholar] [CrossRef]
- Davies, D.; Macgregor, S.; McMullin, C. Computational studies of carboxylate-assisted C-H activation and functionalization at Group 8–10 transition metal centers. Chem. Rev. 2017, 117, 8649–9709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ackermann, L. Carboxylate-assisted transition-metal-catalyzed C-H bond functionalization: Mechanism and scope. Chem. Rev. 2011, 111, 1315–1345. [Google Scholar] [CrossRef] [PubMed]
- Wolters, L.P.; Bickelhaupt, F.M. The activation strain model and molecular orbital theory. WIREs Comput. Mol. Sci. 2015, 5, 324–343. [Google Scholar] [CrossRef] [PubMed]
- Bickelhaupt, F.M.; Houk, K.N. Analyzing reaction rates with the distortion/interaction-activation strain model. Angew. Chem. Int. Ed. 2017, 56, 10070–10086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, A.G.; Liu, P.; Merlic, C.A.; Houk, K.N. Distortion/interaction analysis reveals the origins of selectivities in Iridium-catalyzed C-H borylation of substituted arenes and 5-membered heterocycles. J. Am. Chem. Soc. 2014, 136, 4575–4583. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Xu, L.; Haines, B.; Ajitha, M.; Murakami, K.; Itami, K.; Musaev, D. Roles of base in the Pd-catalyzed annulative chlorophenylene dimerization. ACS Catal. 2020, 10, 3059–3073. [Google Scholar] [CrossRef]
- Scalmani, G.; Frisch, M. Continuous surface charge polarizable continuum models of solvation. I. General formalism. J. Chem. Phys. 2010, 132, 114110–114115. [Google Scholar] [CrossRef]
- Dolg, M.; Wedig, U.; Stoll, H.; Preuss, H. Energy-adjusted ab inito pseudopotentials for the first row transition elements. J. Chem. Phys. 1987, 86, 866–872. [Google Scholar] [CrossRef]
- Andrae, D.; Haubermann, U.; Dolg, M.; Stoll, H.; Preub, H. Energy-adjusted ab inito pseudopotentials for the second and third row transition elements. Theor. Chim. Acta. 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Marenich, A.; Cramer, C.; Truhlar, D. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Sobereva. Available online: http://sobereva.com/327 (accessed on 10 October 2021).
- Lu, T.; Chen, Q. Interaction region indicator: A simple real space function clearly revealing both chemical bonds and weak interactions. Chem. -Methods 2021, 1, 231–239. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| M | M-Complex | C–OA (Å) | C–OB (Å) | ∠O–C–O° | Cthio–CO2 (Å) | M–Cthio (Å) | M–CCO2 (Å) | M–OCO2 (Å) |
|---|---|---|---|---|---|---|---|---|
| Pd(Ⅱ) | Complex with AA | 1.22 | 1.22 | 140.78 | 1.82 | 2.11 | 3.02 | 3.32 |
| Complex without AA | 1.24 | 1.18 | 145.39 | 2.01 | 2.01 | 2.65 | 2.10 | |
| Cu(Ⅱ) | Complex with AA | 1.22 | 1.20 | 144.89 | 1.95 | 2.05 | 2.66 | 2.60 |
| Complex without AA | 1.23 | 1.18 | 149.50 | 2.11 | 2.00 | 2.58 | 2.07 | |
| Ni(Ⅱ) | Complex with AA | 1.21 | 1.22 | 141.85 | 1.86 | 2.01 | 2.88 | 3.14 |
| Complex without AA | 1.23 | 1.18 | 151.03 | 2.14 | 1.92 | 2.50 | 1.92 | |
| Rh(Ⅱ) | Complex with AA | 1.22 | 1.22 | 140.01 | 1.81 | 2.13 | 2.92 | 3.08 |
| Complex without AA | 1.24 | 1.18 | 145.70 | 2.05 | 2.03 | 2.67 | 2.09 | |
| Cu(Ⅰ) | Complex with AA | 1.22 | 1.21 | 142.99 | 1.95 | 1.96 | 2.45 | 2.80 |
| Complex without AA | 1.23 | 1.19 | 145.10 | 2.02 | 2.00 | 2.50 | 2.19 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Q.; Ma, Y.; Zeng, A. Mechanistic Insights into Palladium(II)-Catalyzed Carboxylation of Thiophene and Carbon Dioxide. Catalysts 2022, 12, 654. https://doi.org/10.3390/catal12060654
Zhang Q, Ma Y, Zeng A. Mechanistic Insights into Palladium(II)-Catalyzed Carboxylation of Thiophene and Carbon Dioxide. Catalysts. 2022; 12(6):654. https://doi.org/10.3390/catal12060654
Chicago/Turabian StyleZhang, Qingjun, Youguang Ma, and Aiwu Zeng. 2022. "Mechanistic Insights into Palladium(II)-Catalyzed Carboxylation of Thiophene and Carbon Dioxide" Catalysts 12, no. 6: 654. https://doi.org/10.3390/catal12060654