
Modification of the Acidic and Textural Properties of HY Zeolite by AHFS Treatment and Its Coke Formation Performance in the Catalytic Cracking Reaction of N-Butene

Abstract
:1. Introduction
2. Results and Discussion
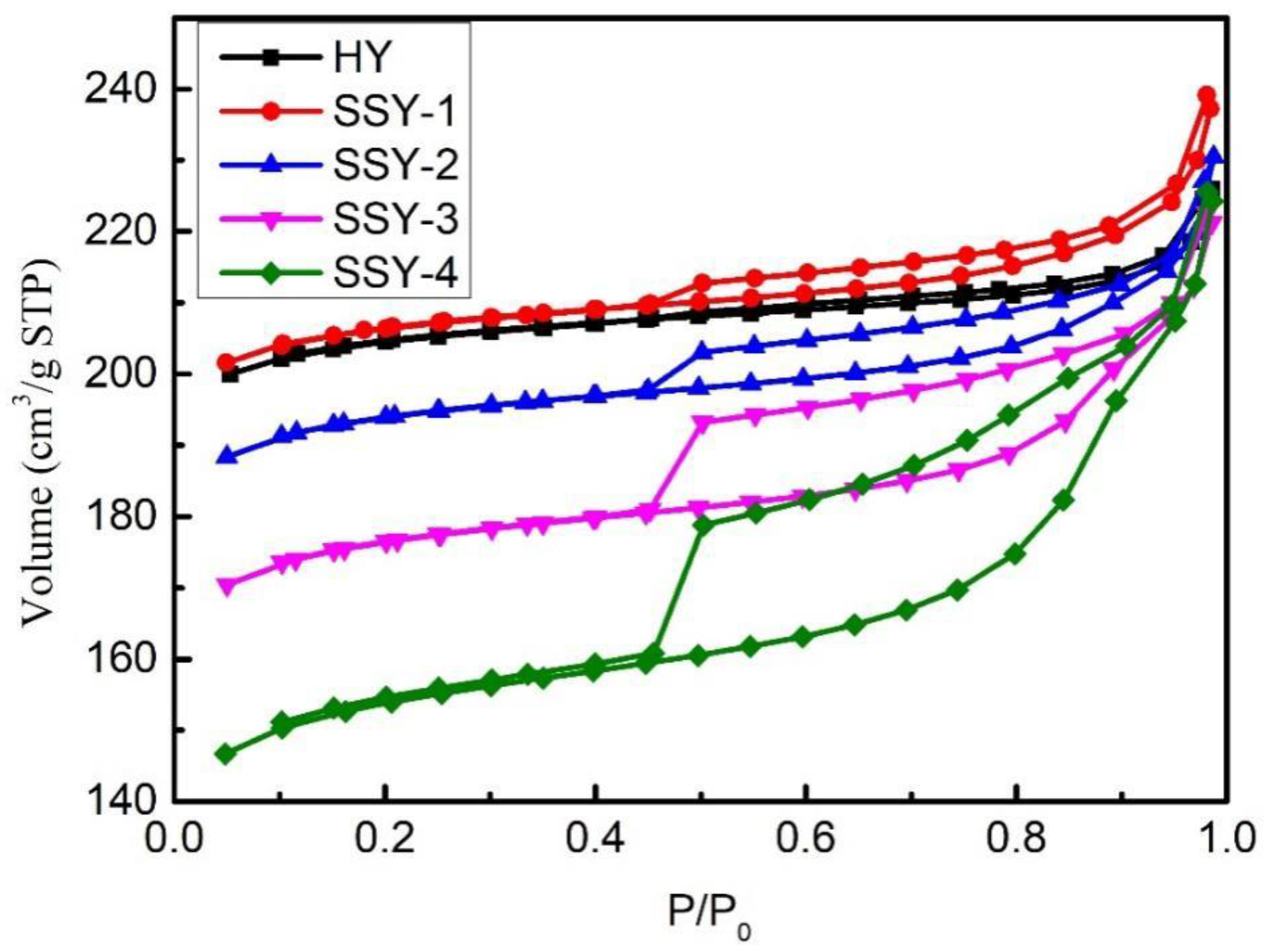
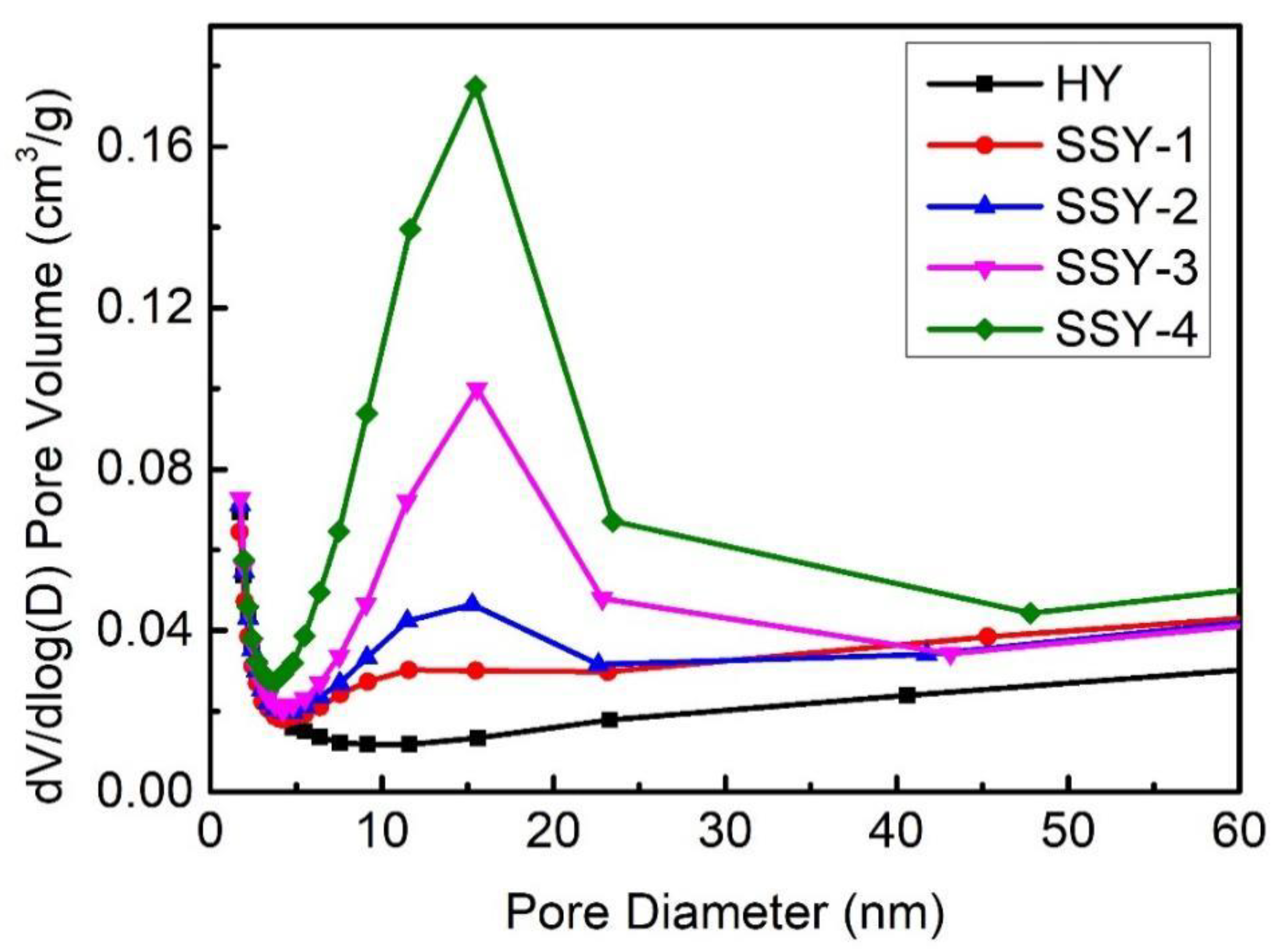
2.1. Influence of AHFS Treatment on Zeolite Structure
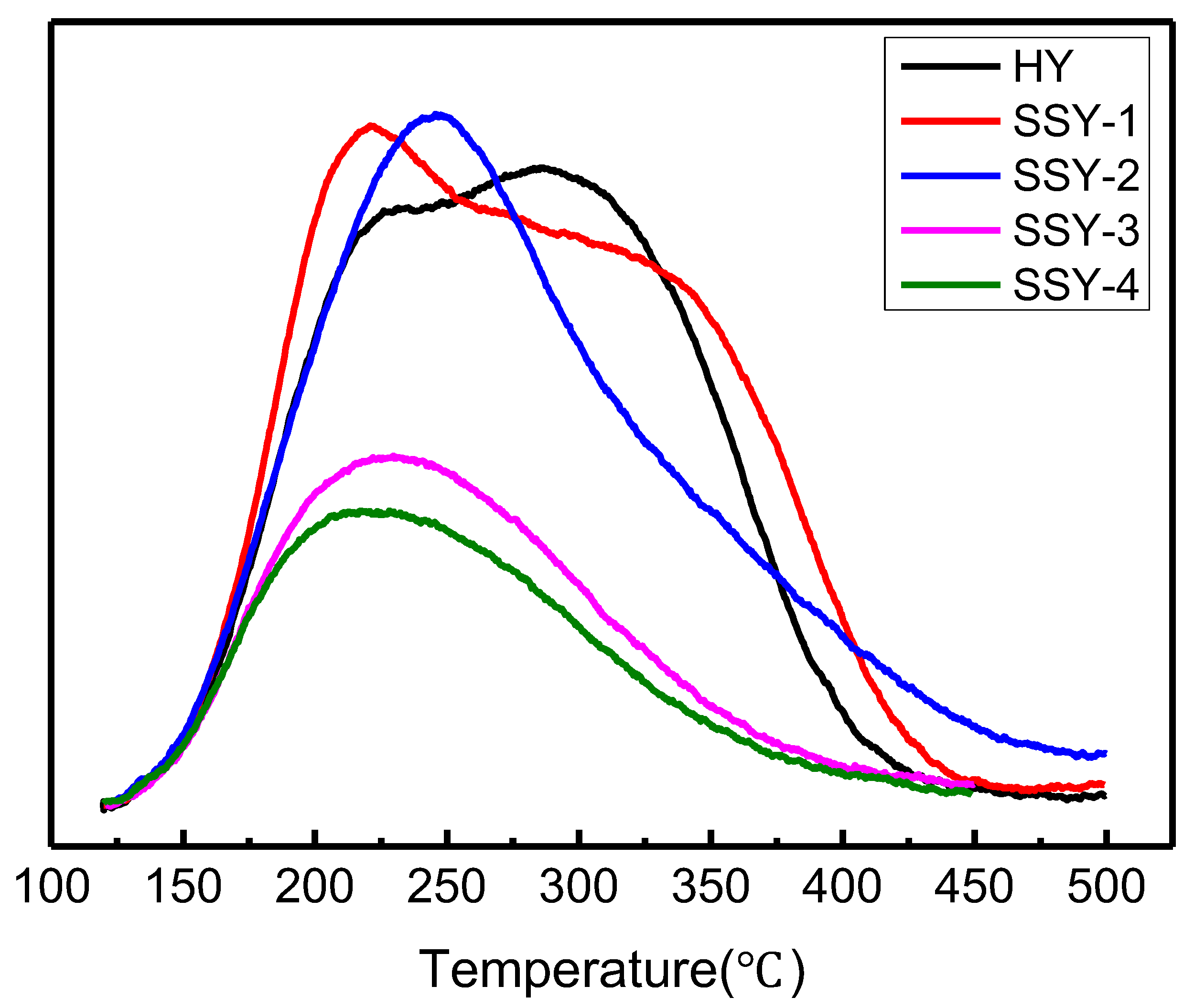
2.2. Acidic Properties of the Modified Zeolites
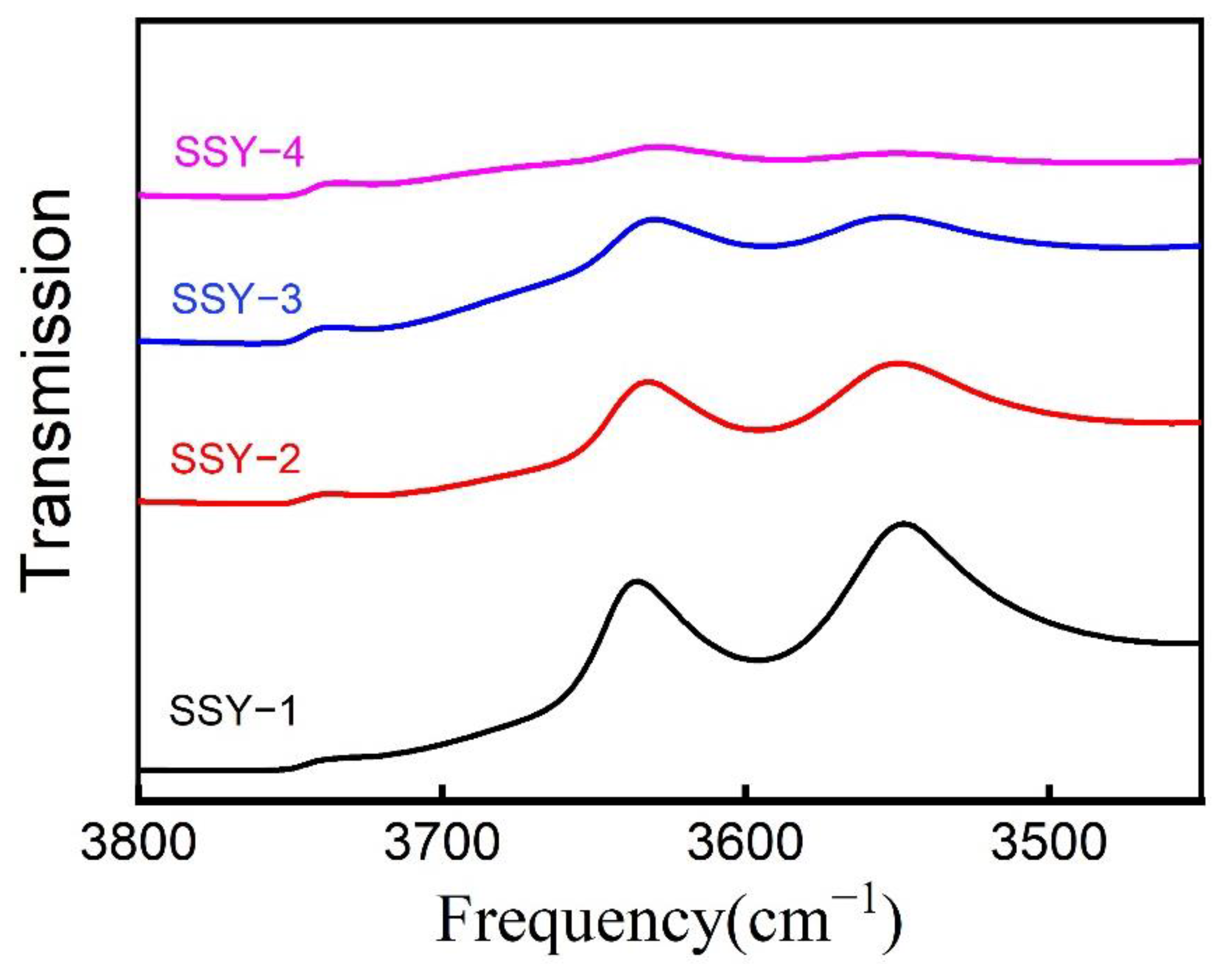
2.3. IR Spectra of the Hydroxy Groups
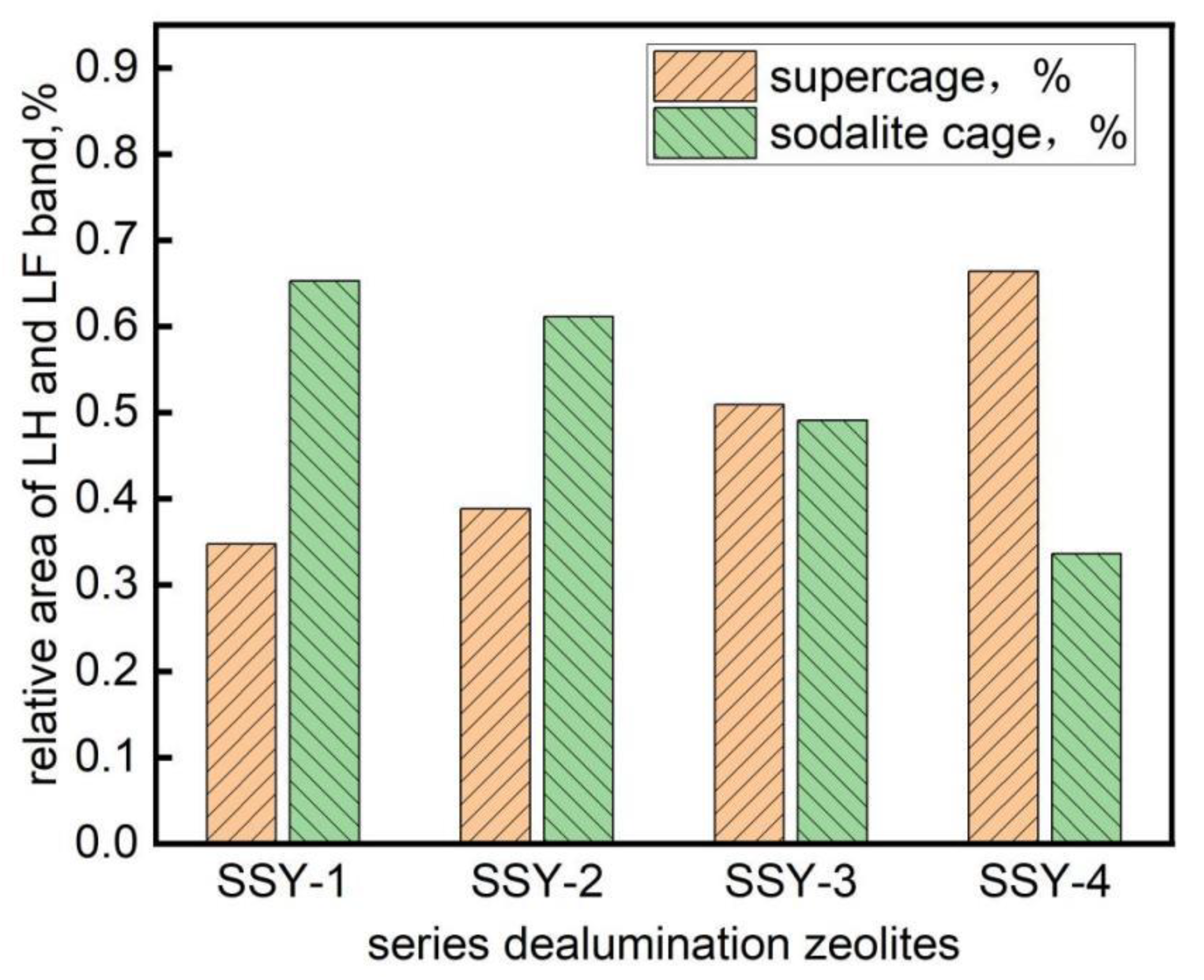
2.4. Pyridine-IR Study of the Acid Properties
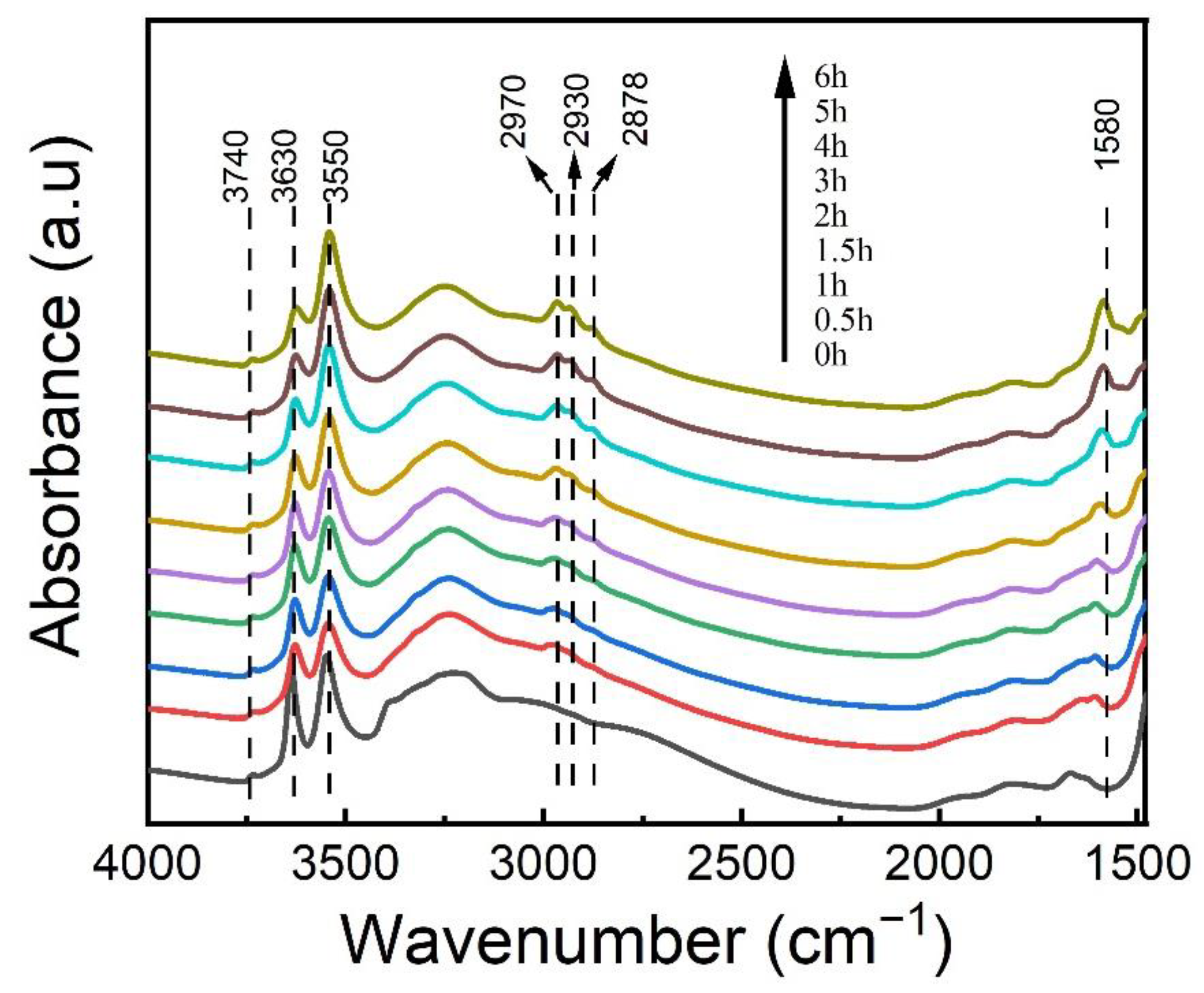
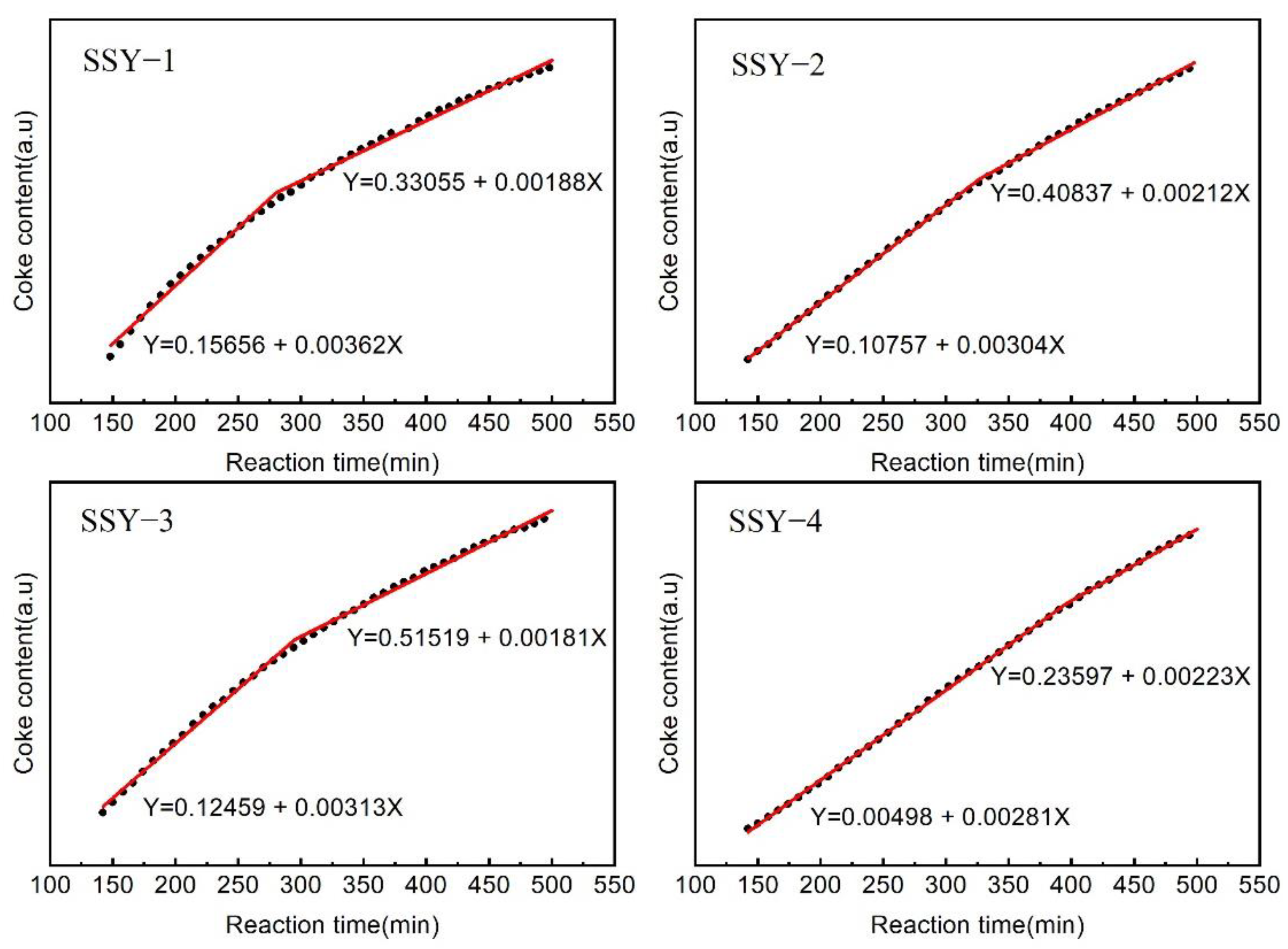
2.5. FT-IR Study of the Coke Formation
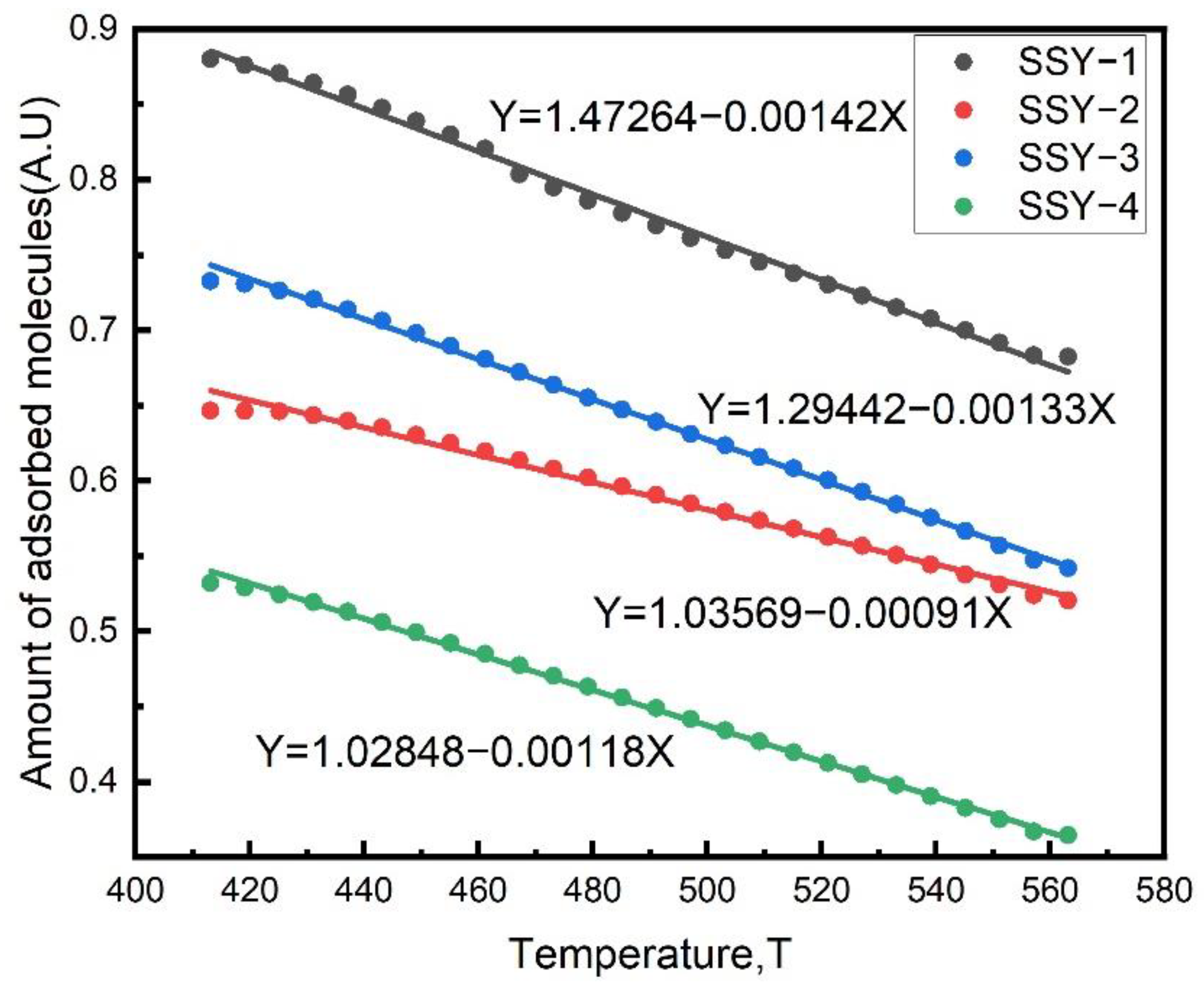
2.6. Effect of Acidity on the Nature of Coke
3. Experimental
3.1. Materials
3.2. Dealumination
3.3. Physicochemical Characterization
3.4. Acidity Measurements
3.5. In Situ FTIR Reaction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fukudome, K.; Suzuki, T. Highly Selective Oxidative Dehydrogenation of Propane to Propylene over VOx–SiO2 Catalysts. Catal. Surv. Asia 2015, 19, 172–187. [Google Scholar] [CrossRef]
- Agency, I.E. The future of petrochemicals. 2018. Available online: https://www.iea.org/reports/the-future-of-petrochemicals (accessed on 29 April 2022).
- Sheng, J.; Yan, B.; Lu, W.-D.; Qiu, B.; Gao, X.-Q.; Wang, D.; Lu, A.-H. Oxidative dehydrogenation of light alkanes to olefins on metal-free catalysts. Chem. Soc. Rev. 2021, 50, 1438–1468. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Xu, W.; Li, X.; Cen, J.; Xu, Y.; Lin, L.; Yao, S. Non-oxidative dehydrogenation of propane to propene over Pt-Sn/Al2O3 catalysts: Identification of the nature of active site. Chem. Eng. J. 2022, 443, 136393. [Google Scholar] [CrossRef]
- Kogan, S.B.; Herskowitz, M. Selective propane dehydrogenation to propylene on novel bimetallic catalysts. Catal. Commun. 2001, 2, 179–185. [Google Scholar] [CrossRef]
- Del Campo, P.; Martínez, C.; Corma, A. Activation and conversion of alkanes in the confined space of zeolite-type materials. Chem. Soc. Rev. 2021, 50, 8511–8595. [Google Scholar] [CrossRef]
- Li, Z.; Shi, W.; Wang, X.; Jiang, F. Deep Catalytic Cracking Process for Light-Olefins Production. In Fluid Catalytic Cracking III; American Chemical Society: Washington, DC, USA, 1994; Volume 571, pp. 33–42. [Google Scholar]
- Eschenbacher, A.; Varghese, R.J.; Delikonstantis, E.; Mynko, O.; Goodarzi, F.; Enemark-Rasmussen, K.; Oenema, J.; Abbas-Abadi, M.S.; Stefanidis, G.D.; Van Geem, K.M. Highly selective conversion of mixed polyolefins to valuable base chemicals using phosphorus-modified and steam-treated mesoporous HZSM-5 zeolite with minimal carbon footprint. Appl. Catal. B Environ. 2022, 309, 121251. [Google Scholar]
- Zhu, X.; Liu, S.; Song, Y.; Xu, L. Catalytic cracking of C4 alkenes to propene and ethene: Influences of zeolites pore structures and Si/Al2 ratios. Appl. Catal. A Gen. 2005, 288, 134–142. [Google Scholar] [CrossRef]
- Chen, S.; Manos, G. Study of Coke and Coke Precursors During Catalytic Cracking of n-Hexane and 1-Hexene over Ultrastable Y Zeolite. Catal. Lett. 2004, 96, 195–200. [Google Scholar] [CrossRef]
- Groten, W.A.; Wojciechowski, B.W.; Hunter, B.K. On the relationship between coke formation chemistry and catalyst deactivation. J. Catal. 1992, 138, 343–350. [Google Scholar] [CrossRef]
- Quintana-Solórzano, R.; Thybaut, J.W.; Marin, G.B. Catalytic cracking and coking of (cyclo)alkane/1-octene mixtures on an equilibrium catalyst. Appl. Catal. A Gen. 2006, 314, 184–199. [Google Scholar] [CrossRef]
- Shi, Z.C.; Arora, S.S.; Trahan, D.W.; Hickman, D.; Bhan, A. Methanol to hydrocarbons conversion: Why dienes and monoenes contribute differently to catalyst deactivation? Chem. Eng. J. 2022, 437, 12. [Google Scholar] [CrossRef]
- Wang, B.; Gong, X.; Zhang, Z.; Zhu, Q.; He, W. Modelling and understanding deposit formation of hydrocarbon fuels from the coke characteristics. Fuel 2022, 319, 123745. [Google Scholar] [CrossRef]
- Zhao, G.; Teng, J.; Xie, Z.; Jin, W.; Yang, W.; Chen, Q.; Tang, Y. Effect of phosphorus on HZSM-5 catalyst for C4-olefin cracking reactions to produce propylene. J. Catal. 2007, 248, 29–37. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, Z. Catalytic cracking of 1-butene to propylene by Ag modified HZSM-5. Chin. J. Chem. Eng. 2015, 23, 1131–1137. [Google Scholar] [CrossRef]
- Lin, L.; Qiu, C.; Zhuo, Z.; Zhang, D.; Zhao, S.; Wu, H.; Liu, Y.; He, M. Acid strength controlled reaction pathways for the catalytic cracking of 1-butene to propene over ZSM-5. J. Catal. 2014, 309, 136–145. [Google Scholar] [CrossRef]
- Zhu, X.; Liu, S.; Song, Y.; Xie, S.; Xu, L. Catalytic cracking of 1-butene to propene and ethene on MCM-22 zeolite. Appl. Catal. A Gen. 2005, 290, 191–199. [Google Scholar] [CrossRef]
- Liu, B.; Xie, K.; Oh, S.C.; Sun, D.; Fang, Y.; Xi, H. Direct synthesis of hierarchical USY zeolite for retardation of catalyst deactivation. Chem. Eng. Sci. 2016, 153, 374–381. [Google Scholar] [CrossRef]
- Cañizares, P.; Carrero, A. Dealumination of ferrierite by ammonium hexafluorosilicate treatment: Characterization and testing in the skeletal isomerization of n-butene. Appl. Catal. A Gen. 2003, 248, 227–237. [Google Scholar] [CrossRef]
- Wang, Q.L.; Torrealba, M.; Giannetto, G.; Guisnet, M.; Perot, G.; Cahoreau, M.; Caisso, J. Dealumination of Y zeolite with ammonium hexafluorosilicate: A SIMS-XPS study of the aluminum distribution. Zeolites 1990, 10, 703–706. [Google Scholar] [CrossRef]
- Shertukde, P.V.; Hall, W.K.; Marcelin, G. Effect of dealumination of the structure and acidity of H-Y zeolites. Catal. Today 1992, 15, 491–502. [Google Scholar] [CrossRef]
- Rouquerol, F.; Rouquerol, J.; Sing, K. Adsorption by Powders and Porous Solids; Academic Press: San Diego, CA, USA, 1999. [Google Scholar]
- Gautier, M.; Muller, F.; Le Forestier, L.; Beny, J.M.; Guegan, R. NH4-smectite: Characterization, hydration properties and hydro mechanical behaviour. Appl. Clay Sci. 2010, 49, 247–254. [Google Scholar] [CrossRef] [Green Version]
- Qin, Z.; Shen, B.; Yu, Z.; Deng, F.; Zhao, L.; Zhou, S.; Yuan, D.; Gao, X.; Wang, B.; Zhao, H.; et al. A defect-based strategy for the preparation of mesoporous zeolite Y for high-performance catalytic cracking. J. Catal. 2013, 298, 102–111. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, Q.; Chen, H.; Zhang, X. Hydroconversion of waste cooking oil into green biofuel over hierarchical USY-supported NiMo catalyst: A comparative study of desilication and dealumination. Catalysts 2017, 7, 281. [Google Scholar] [CrossRef] [Green Version]
- Matharu, A.P.; Gladden, L.F.; Carr, S.W. Characterization and catalytic properties of dealuminated zeolite-Y: A comparison of ammonium hexafluorosilicate and hydrothermal treatments. Stud. Surf. Sci. Catal. 1995, 94, 147–154. [Google Scholar]
- Gola, A.; Rebours, B.; Milazzo, E.; Lynch, J.; Benazzi, E.; Lacombe, S.; Delevoye, L.; Fernandez, C. Effect of leaching agent in the dealumination of stabilized Y zeolites. Microporous Mesoporous Mater. 2000, 40, 73–83. [Google Scholar] [CrossRef]
- Pu, X.; Liu, N.-w.; Shi, L. Acid properties and catalysis of USY zeolite with different extra-framework aluminum concentration. Microporous Mesoporous Mater. 2015, 201, 17–23. [Google Scholar] [CrossRef]
- Katada, N.; Miyamoto, T.; Begum, H.A.; Naito, N.; Niwa, M.; Matsumoto, A.; Tsutsumi, K. Strong Acidity of MFI-Type Ferrisilicate Determined by Temperature-Programmed Desorption of Ammonia. J. Phys. Chem. B 2000, 104, 5511–5518. [Google Scholar] [CrossRef]
- Montanari, T.; Finocchio, E.; Busca, G. Infrared Spectroscopy of Heterogeneous Catalysts: Acidity and Accessibility of Acid Sites of Faujasite-Type Solid Acids. J. Phys. Chem. C 2011, 115, 937–943. [Google Scholar] [CrossRef]
- Corma, A.; Fornes, V.; Rey, F. Quinoline as a probe molecule for determination of external Broensted and Lewis acidity in zeolites. Zeolites 1993, 13, 56–59. [Google Scholar] [CrossRef]
- Corma, A.; Melo, F.V.; Rawlence, D.J. Effect of the nonuniform dealumination on the acidity and catalytic activity of faujasite. Zeolites 1990, 10, 690–694. [Google Scholar] [CrossRef]
- King, D.A. Thermal desorption from metal surfaces. Review. Surf. Sci. 1975, 47, 384–402. [Google Scholar] [CrossRef]
- Quanzhi, L.; Ruiming, Z.; Zhiyuan, X. Studies on the Surface Acidity of HY Zeolite by Combined IR and TPD. Stud. Surf. Sci. Catal. 1986, 28, 487–494. [Google Scholar]
- Karge, H.G.; Nießen, W.; Bludau, H. In-situ FTIR measurements of diffusion in coking zeolite catalysts. Appl. Catal. A Gen. 1996, 146, 339–349. [Google Scholar] [CrossRef]
- Caeiro, G.; Lopes, J.M.; Magnoux, P.; Ayrault, P.; Ribeiro, F.R. A FT-IR study of deactivation phenomena during methylcyclohexane transformation on H-USY zeolites: Nitrogen poisoning, coke formation, and acidity-activity correlations. J. Catal. 2007, 249, 234–243. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, Y.; Guo, G.S. In situ FTIR study of methanol to hydrocarbons conversion process on HZSM-5 zeolite. Adv. Mater. Res. 2011, 236–238, 958–963. [Google Scholar] [CrossRef]
- Mosqueda-Jimenez, B.I.; Jentys, A.; Seshan, K.; Lercher, J.A. On the surface reactions during NO reduction with propene and propane on Ni-exchanged mordenite. Appl. Catal. B-Environ. 2003, 46, 189–202. [Google Scholar] [CrossRef]
- Delara, E.C.; Kahn, R.; Seloudoux, R. Effect of an electric-field on a methane molecule. 1. infrared-analysis of methane (CH4-CD4) adsorbed in NaA zeolite in the temperature-range 150-20-K. J. Chem. Phys. 1985, 83, 2646–2652. [Google Scholar] [CrossRef]
- Li, L.; Gao, J.; Xu, C.; Meng, X. Reaction behaviors and mechanisms of catalytic pyrolysis of C4 hydrocarbons. Chem. Eng. J. 2006, 116, 155–161. [Google Scholar] [CrossRef]
- Kim, Y.H.; Lee, K.H.; Lee, J.S. The effect of pre-coking and regeneration on the activity and stability of Zn/ZSM-5 in aromatization of 2-methyl-2-butene. Catal. Today 2011, 178, 72–78. [Google Scholar] [CrossRef]
- Sahoo, S.K.; Viswanadham, N.; Ray, N.; Gupta, J.K.; Singh, I.D. Studies on acidity, activity and coke deactivation of ZSM-5 during n-heptane aromatization. Appl. Catal. A: Gen. 2001, 205, 1–10. [Google Scholar] [CrossRef]
- Boveri, M.; Márquez-Álvarez, C.; Laborde, M.Á.; Sastre, E. Steam and acid dealumination of mordenite: Characterization and influence on the catalytic performance in linear alkylbenzene synthesis. Catal. Today 2006, 114, 217–225. [Google Scholar] [CrossRef]
- Agudelo, J.L.; Mezari, B.; Hensen, E.J.M.; Giraldo, S.A.; Hoyos, L.J. On the effect of EDTA treatment on the acidic properties of USY zeolite and its performance in vacuum gas oil hydrocracking. Appl. Catal. A Gen. 2014, 488, 219–230. [Google Scholar] [CrossRef]
- Garralon, G.; Fornes, V.; Corma, A. Faujasites dealuminated with ammonium hexafluorosilicate: Variables affecting the method of preparation. Zeolites 1988, 8, 268–272. [Google Scholar] [CrossRef]
- Kuehne, M.A.; Babitz, S.M.; Kung, H.H.; Miller, J.T. Effect of framework Al content on HY acidity and cracking activity. Appl. Catal. A 1998, 166, 293–299. [Google Scholar] [CrossRef]
- Lonyi, F.; Lunsford, J.H. The development of strong acidity in hexafluorosilicate-modified Y-type zeolites. J. Catal. 1992, 136, 566–577. [Google Scholar] [CrossRef]
- Le Bail, A.; Duroy, H.; Fourquet, J.L. The ab-initio structure determination of lithium antimony tungstate (LiSbWO6) by x-ray powder diffraction. Mater. Res. Bull. 1988, 23, 447–452. [Google Scholar] [CrossRef]
- Sohn, J.R.; DeCanio, S.J.; Lunsford, J.H.; O’Donnell, D.J. Determination of framework aluminum content in dealuminated Y-type zeolites: A comparison based on unit cell size and wavenumber of IR bands. Zeolites 1986, 6, 225–227. [Google Scholar] [CrossRef]
- Breck, D.W. Zeolite Molecular Sieves: Structure, Chemistry and Use; Interscience-Wiley: New York, NY, USA, 1974; 771p. [Google Scholar]
- Cherkasov, N.; Vazhnova, T.; Lukyanov, D.B. Quantitative infra-red studies of Brønsted acid sites in zeolites: Case study of the zeolite mordenite. Vib. Spectrosc. 2016, 83, 170–179. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Smicro a | Sext b | Vmicro c | Vmeso d |
|---|---|---|---|---|
| m2/g | m2/g | cm3/g | cm3/g | |
| HY | 605 | 46 | 0.30 | 0.04 |
| SSY-1 | 608 | 49 | 0.30 | 0.06 |
| SSY-2 | 566 | 52 | 0.28 | 0.07 |
| SSY-3 | 504 | 60 | 0.25 | 0.10 |
| SSY-4 | 420 | 74 | 0.21 | 0.14 |
| Sample | a0 (Å) | Crystallinity% | Framework Si/Al Ratio | Global Si/Al Ratio | Total Al | Framework Al a | Extra-Framework Al b | B Acid Sites (×10−4 mol/g) |
|---|---|---|---|---|---|---|---|---|
| HY | 24.641 | 100 | 3.021 | 2.550 | 54 | 48 | 6 | 43 |
| SSY-1 | 24.582 | 151.8 | 3.660 | 3.720 | 41 | 41 | 0 | 37 |
| SSY-2 | 24.536 | 162.5 | 4.325 | 3.905 | 39 | 36 | 3 | 32 |
| SSY-3 | 24.492 | 147.3 | 5.145 | 4.535 | 35 | 31 | 3 | 28 |
| SSY-4 | 24.467 | 126.2 | 5.755 | 5.495 | 30 | 28 | 1 | 25 |
| Sample | Acid Type | Total Acid Sites (10−4 mol/g) | B/L Ratio | Strong Acid Sites (10−4 mol/g) | Weak Acid Sites (10−4 mol/g) |
|---|---|---|---|---|---|
| SSY-1 | Brønsted acid | 8.71 | 62.214 | 6.76 | 1.95 |
| Lewis acid | 0.14 | 0.03 | 0.11 | ||
| SSY-2 | Brønsted acid | 7.25 | 18.13 | 5.37 | 1.88 |
| Lewis acid | 0.40 | 0.15 | 0.25 | ||
| SSY-3 | Brønsted acid | 6.40 | 20.64 | 5.15 | 1.25 |
| Lewis acid | 0.31 | 0.05 | 0.25 | ||
| SSY-4 | Brønsted acid | 5.27 | 17 | 3.61 | 1.66 |
| Lewis acid | 0.31 | 0.14 | 0.17 |
| Zeolite | Ed Strong (KJ/mol) | Ed Weak (KJ/mol) |
|---|---|---|
| SSY-1 | 11.5 | 4.9 |
| SSY-2 | 11.6 | 5.7 |
| SSY-3 | 15 | 8.2 |
| SSY-4 | 20.4 | 10.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, X.; Wei, C.; Zhao, L.; Gao, J.; Xu, C. Modification of the Acidic and Textural Properties of HY Zeolite by AHFS Treatment and Its Coke Formation Performance in the Catalytic Cracking Reaction of N-Butene. Catalysts 2022, 12, 640. https://doi.org/10.3390/catal12060640
Lu X, Wei C, Zhao L, Gao J, Xu C. Modification of the Acidic and Textural Properties of HY Zeolite by AHFS Treatment and Its Coke Formation Performance in the Catalytic Cracking Reaction of N-Butene. Catalysts. 2022; 12(6):640. https://doi.org/10.3390/catal12060640
Chicago/Turabian StyleLu, Xu, Chenhao Wei, Liang Zhao, Jinsen Gao, and Chunming Xu. 2022. "Modification of the Acidic and Textural Properties of HY Zeolite by AHFS Treatment and Its Coke Formation Performance in the Catalytic Cracking Reaction of N-Butene" Catalysts 12, no. 6: 640. https://doi.org/10.3390/catal12060640