
Mechanistic Insights into the Effect of Sulfur on the Selectivity of Cobalt-Catalyzed Fischer–Tropsch Synthesis: A DFT Study


Abstract
:1. Introduction
2. Computational Methodology
2.1. Computational Model
2.2. Computational Details
3. Results and Discussion
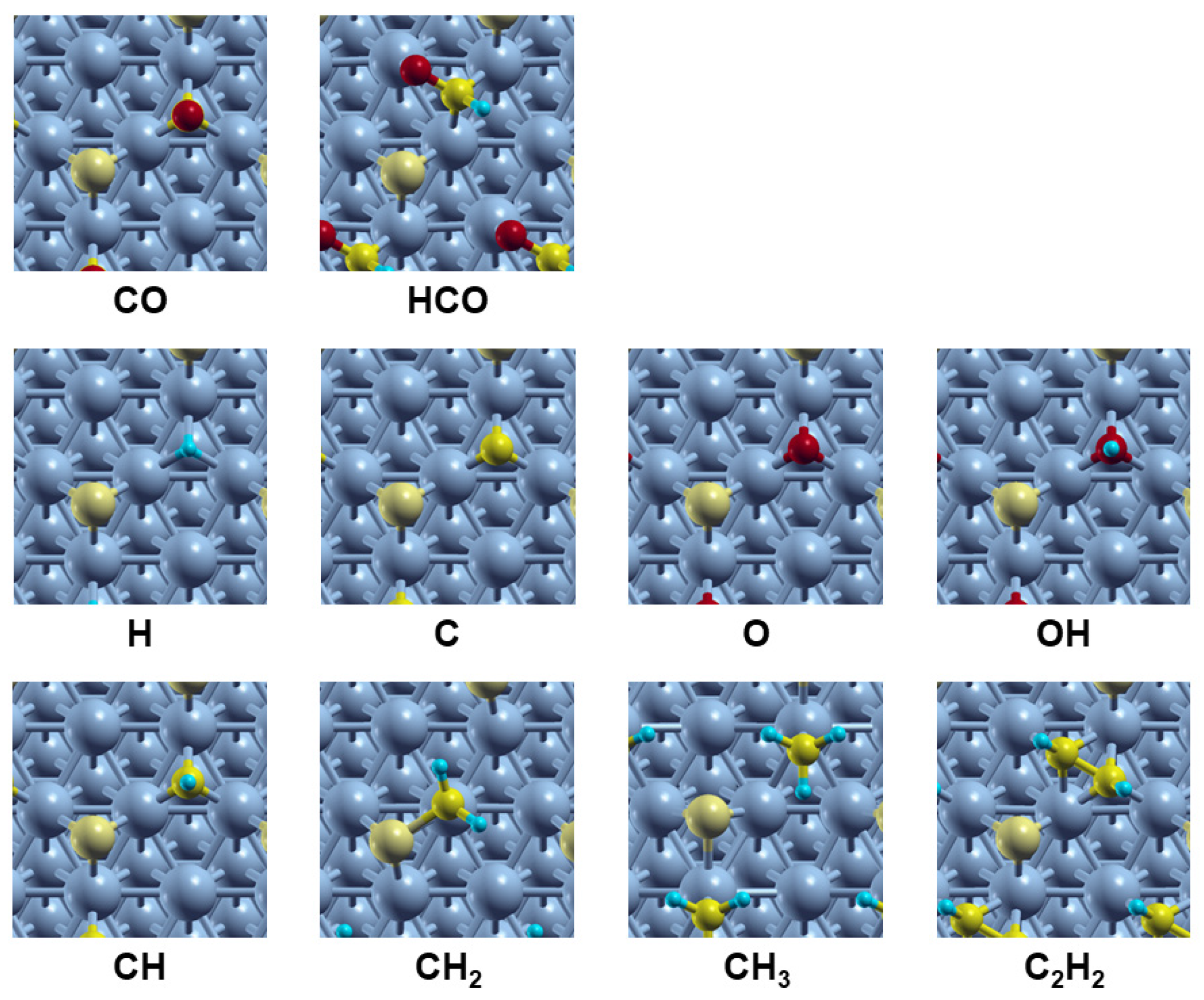
3.1. Effect of Sulfur on the Adsorption of the Reactants and Intermediates of FTS on Co(111)
3.2. Effect of Sulfur on the Elementary Reactions of FTS on Co(111)
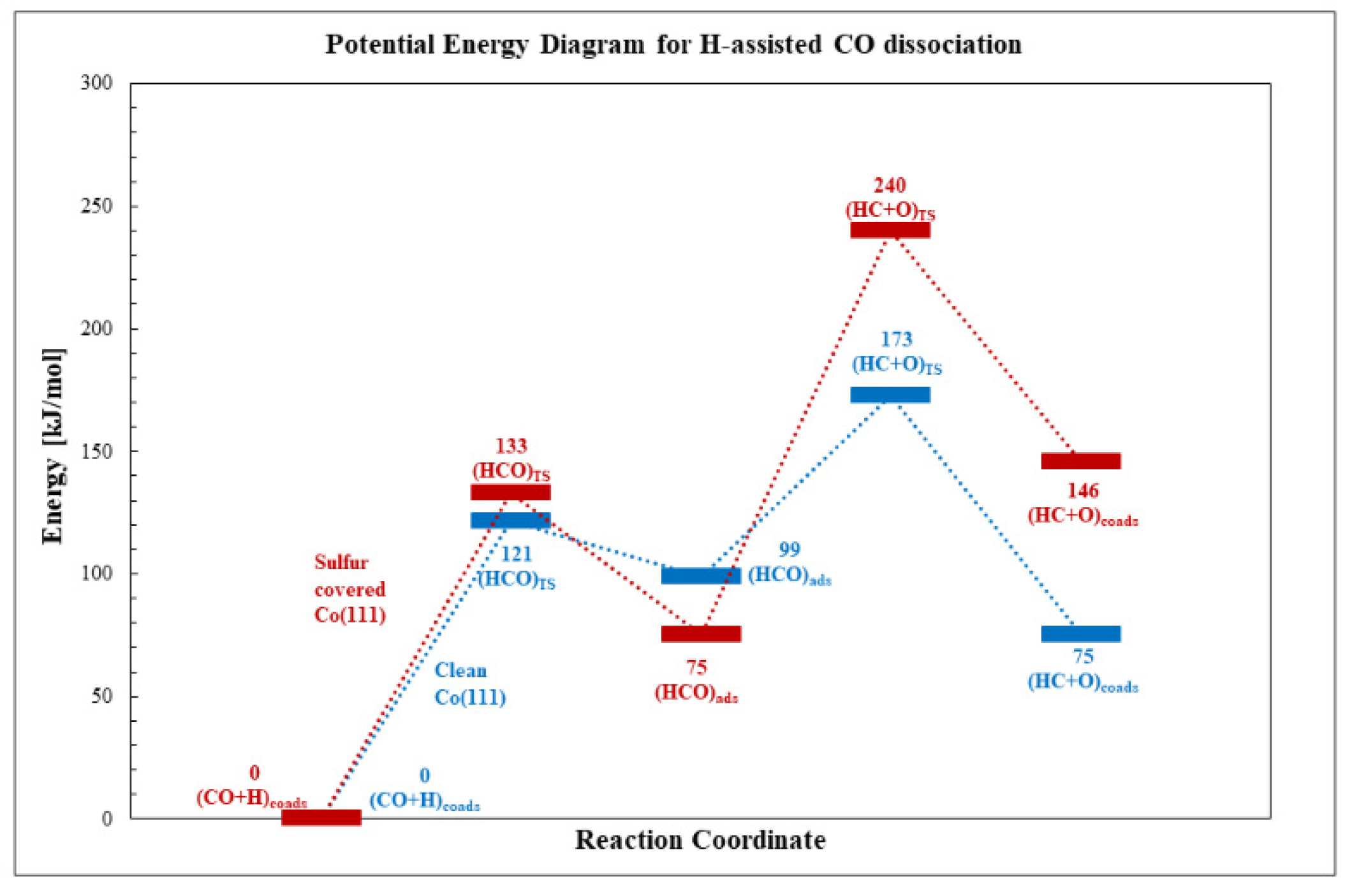
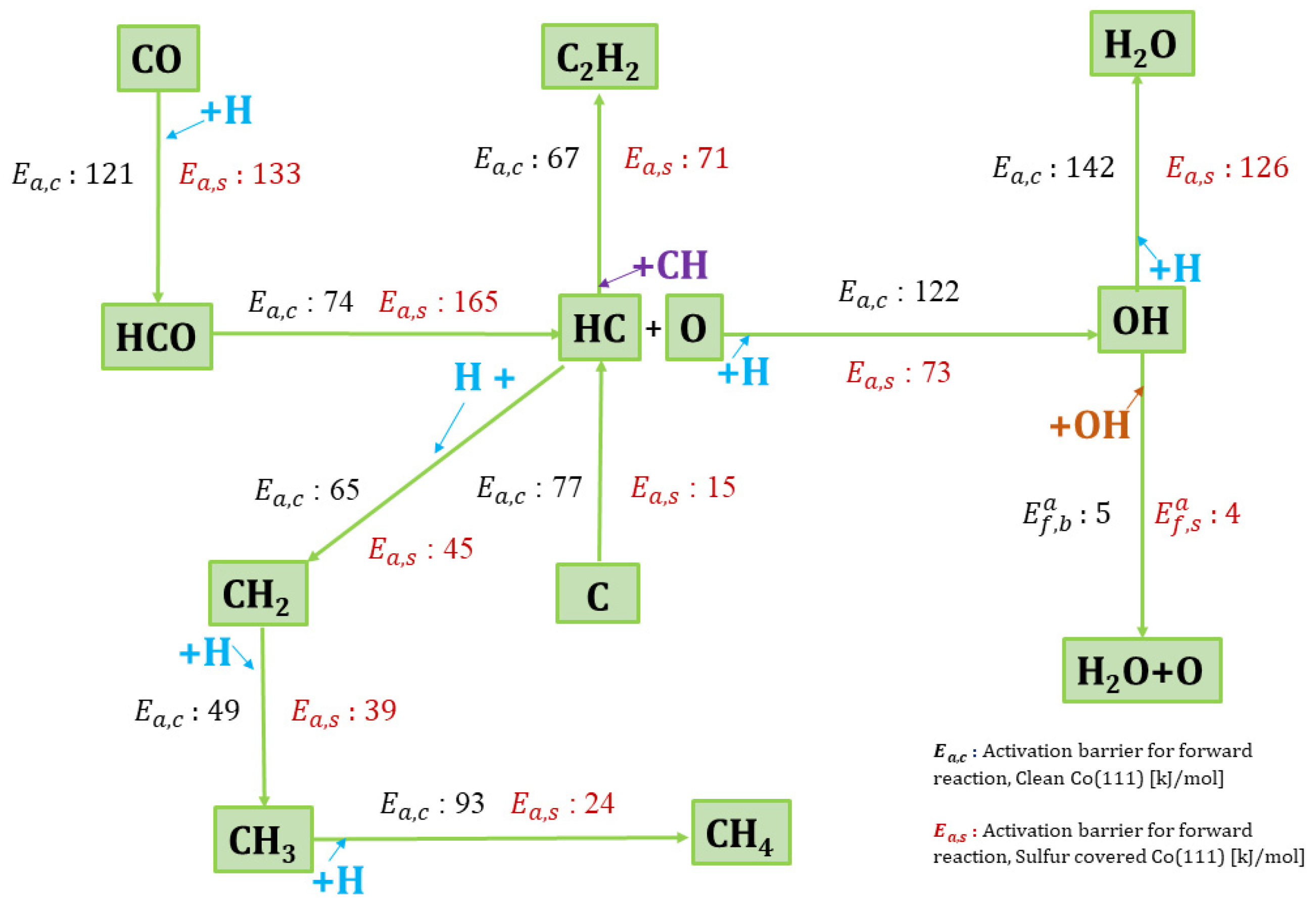
3.2.1. H-Assisted CO Dissociation
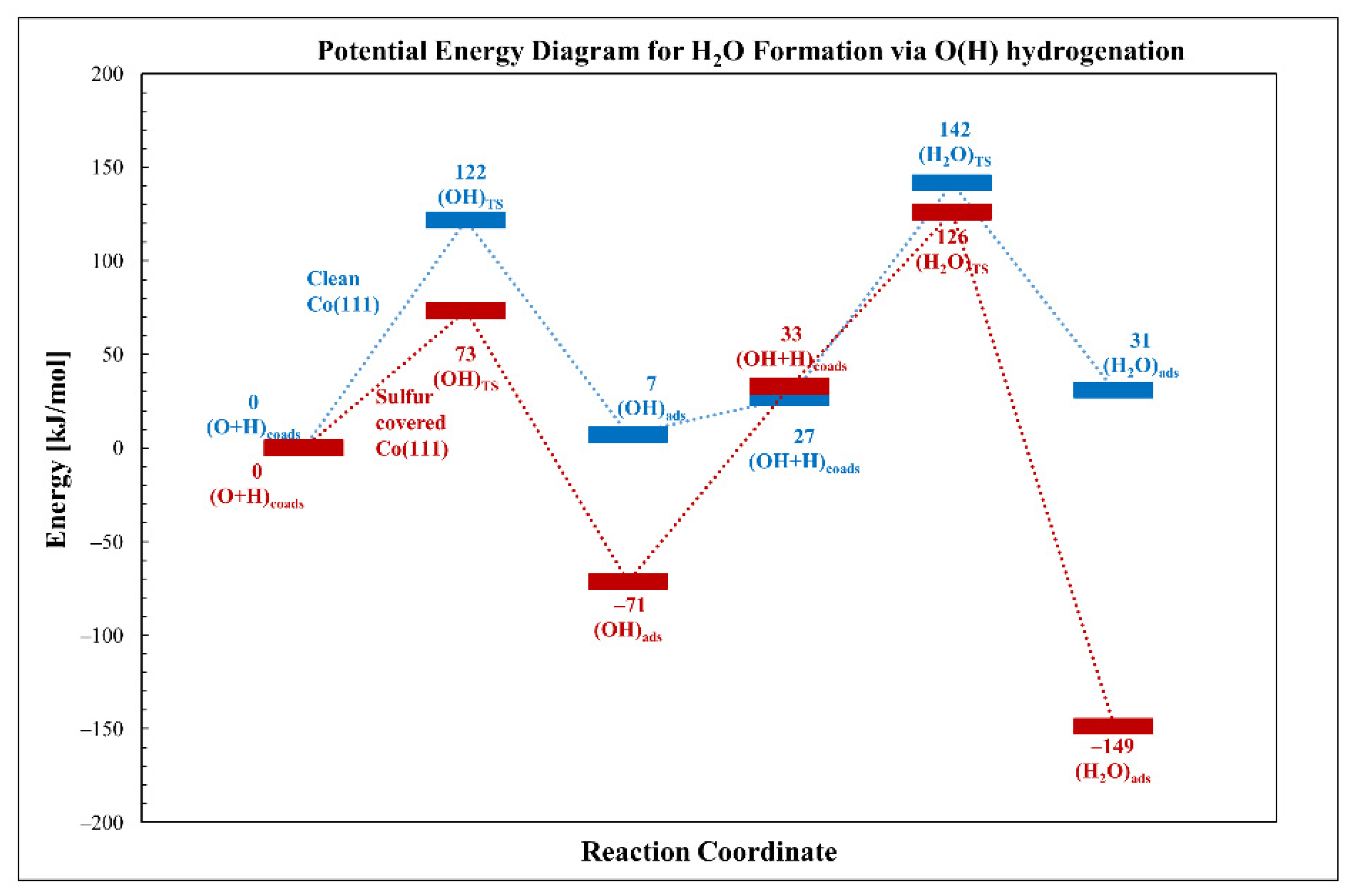
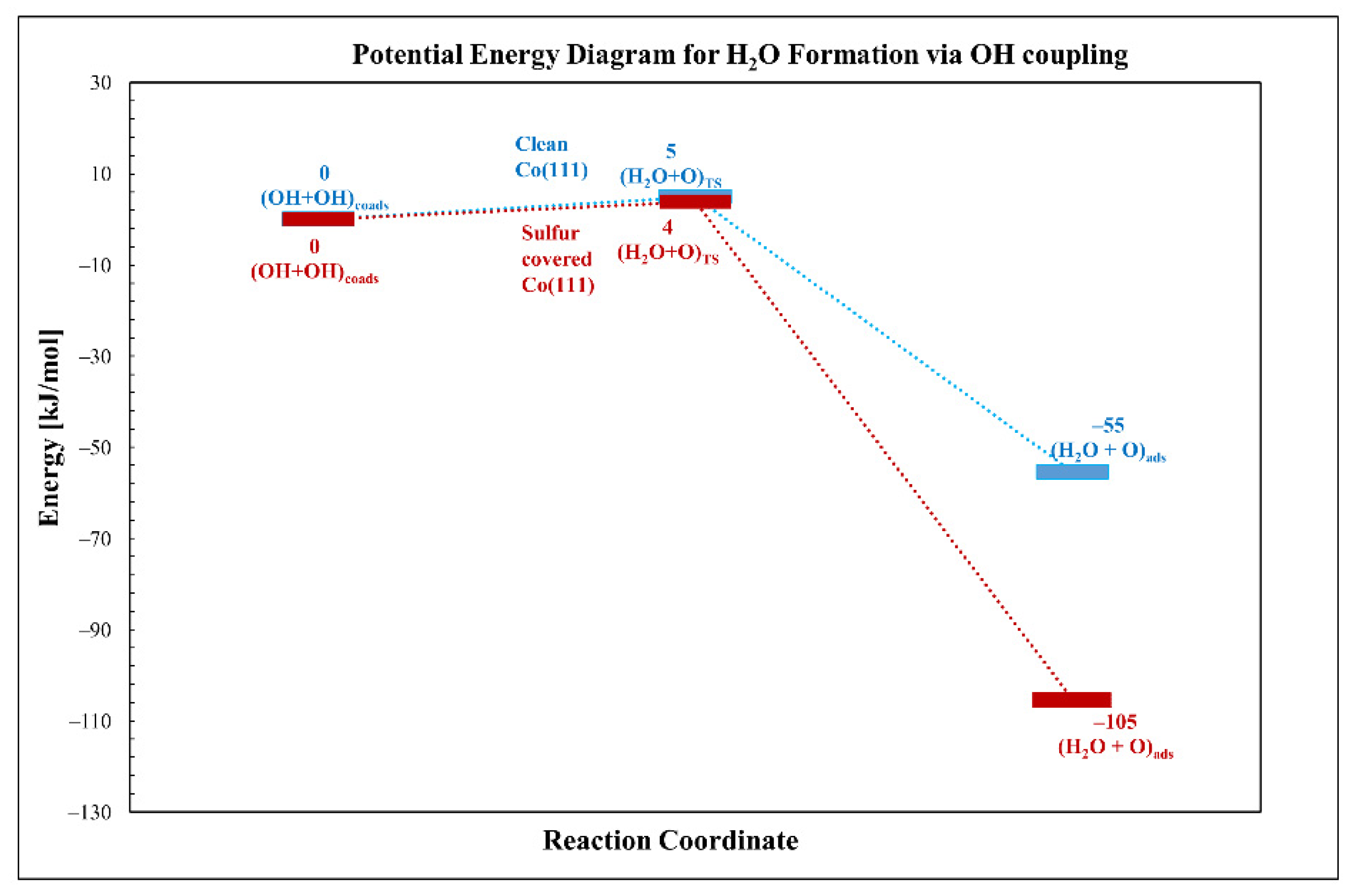
3.2.2. Oxygen Hydrogenation (Water Formation)
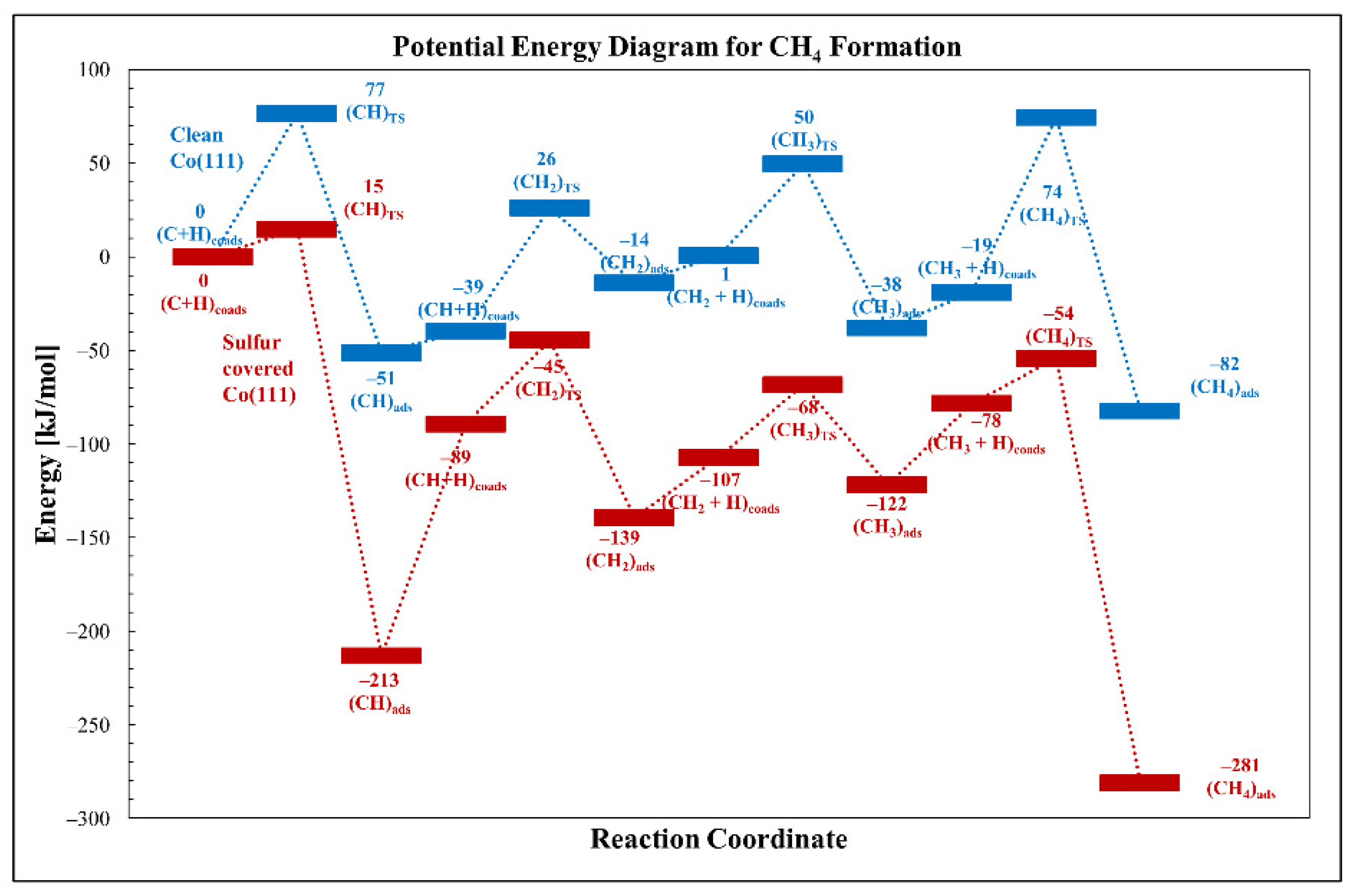
3.2.3. Carbon Hydrogenation (Methane Formation)
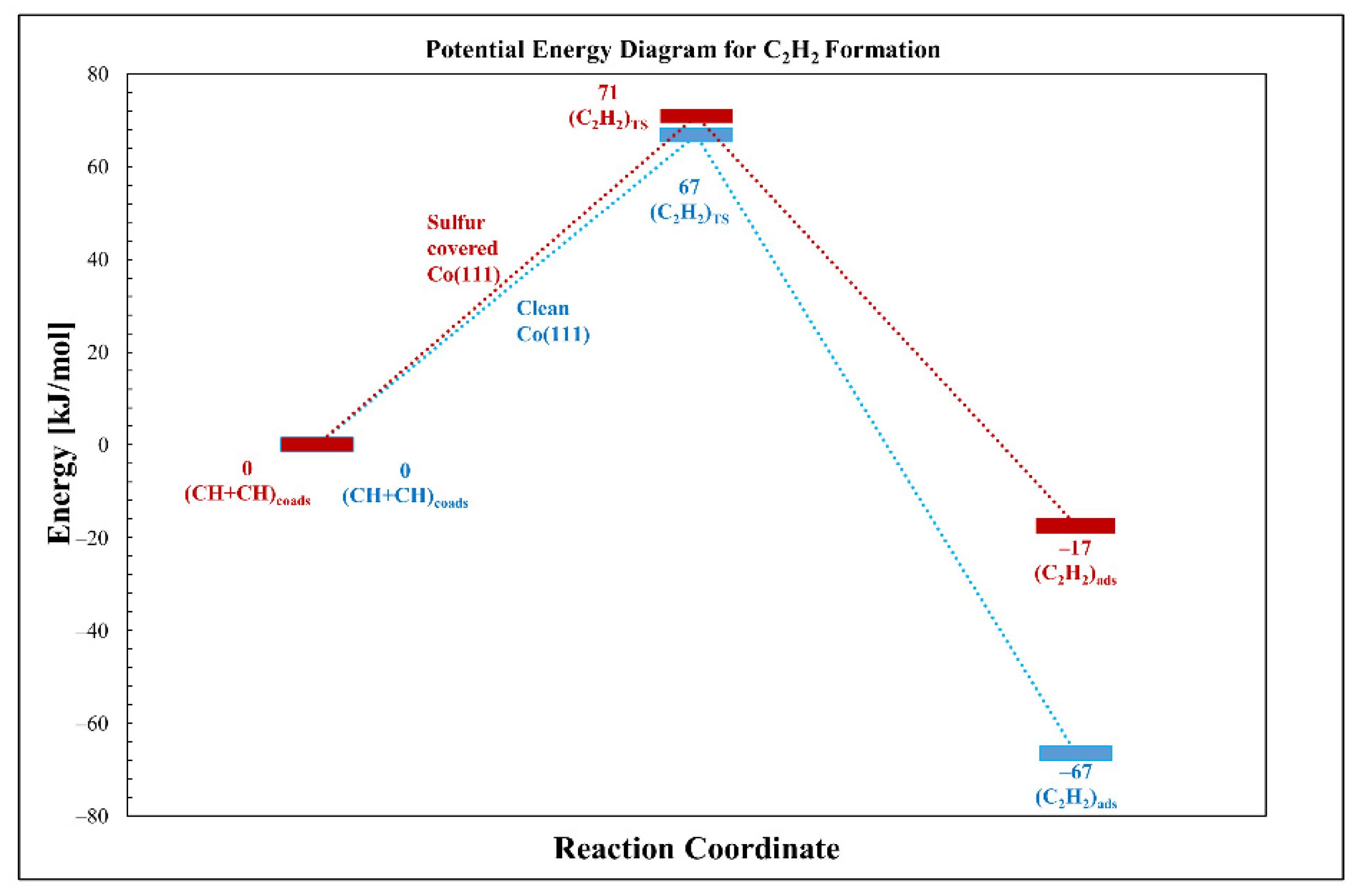
3.2.4. CH Coupling (Acetylene Formation)
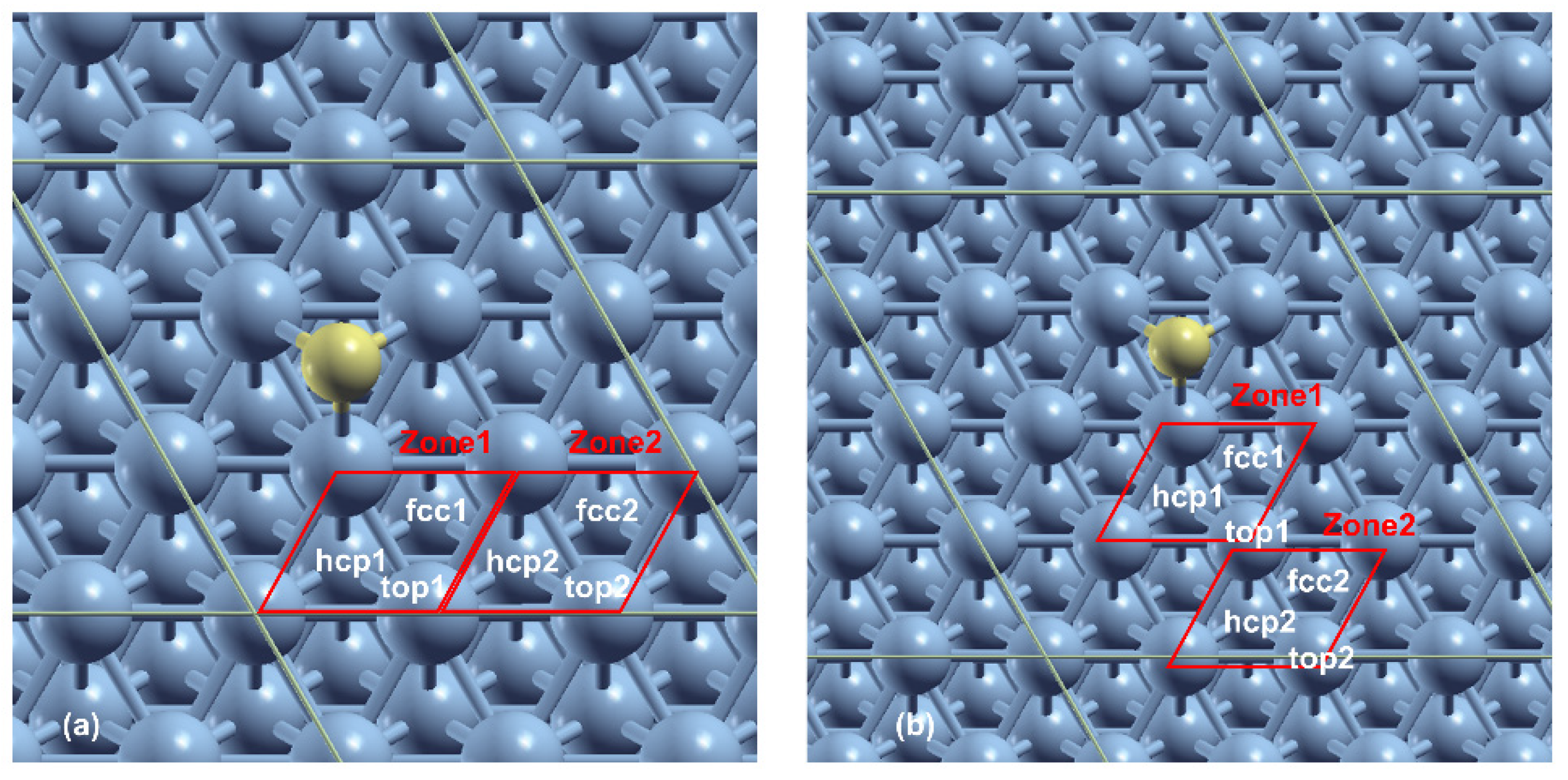
3.3. Effect of Sulfur Coverages Lower than 0.25 ML on FTS Adsorbates on Co(111)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bartholomew, C.H. Mechanisms of catalyst deactivation. Appl. Catal. A Gen. 2001, 212, 17–60. [Google Scholar] [CrossRef]
- Xie, J.; Paalanen, P.P.; van Deelen, T.W.; Weckhuysen, B.M.; Louwerse, M.J.; de Jong, K.P. Promoted cobalt metal catalysts suitable for the production of lower olefins from natural gas. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef]
- Tsakoumis, N.E.; Ronning, M.; Borg, O.; Rytter, E.; Holmen, A. Deactivation of Cobalt Based Fischer-Tropsch Catalysts: A Review. Catal. Today 2010, 154, 162–182. Available online: http://www.sciencedirect.com/science/article/pii/S0920586110002282 (accessed on 23 March 2022). [CrossRef]
- Visconti, C.G.; Lietti, L.; Forzatti, P.; Zennaro, R. Fischer-Tropsch synthesis on sulphur poisoned Co/Al2O3 catalyst. Appl. Catal. A Gen. 2007, 330, 49–56. [Google Scholar] [CrossRef]
- Borg, Ø.; Hammer, N.; Enger, B.C.; Myrstad, R.; Lindvg, O.A.; Eri, S.; Skagseth, T.H.; Rytter, E. Effect of biomass-derived synthesis gas impurity elements on cobalt Fischer-Tropsch catalyst performance including in situ sulphur and nitrogen addition. J. Catal. 2011, 279, 163–173. [Google Scholar] [CrossRef]
- Pansare, S.S.; Allison, J.D. An investigation of the effect of ultra-low concentrations of sulfur on a Co/γ-Al2O3 Fischer-Tropsch synthesis catalyst. Appl. Catal. A Gen. 2010, 387, 224–230. [Google Scholar] [CrossRef]
- Barrientos, J.; Montes, V.; Boutonnet, M.; Järås, S. Further insights into the effect of sulfur on the activity and selectivity of cobalt-based Fischer–Tropsch catalysts. Catal. Today 2016, 275, 119–126. [Google Scholar] [CrossRef]
- Lahtinen, J.; Kantola, P.; Jaatinen, S.; Habermehl-Cwirzen, K.; Salo, P.; Vuorinen, J.; Lindroos, M.; Pussi, K.; Seitsonen, A.P. LEED and DFT investigation on the (2 × 2)-S overlayer on Co(0 0 0 1). Surf. Sci. 2005, 599, 113–121. [Google Scholar] [CrossRef]
- Habermehl-Cwirzen, K.M.E.; Kauraala, K.; Lahtinen, J. Hydrogen on cobalt: The effects of carbon monoxide and sulphur additives on the D-2/Co(0001) system. Phys. Scr. T 2004, 108, 28–32. [Google Scholar] [CrossRef] [Green Version]
- Habermehl-Cwirzen, K.; Lahtinen, J. Sulfur poisoning of the CO adsorption on Co(0001). Surf. Sci. 2004, 573, 183–190. [Google Scholar] [CrossRef]
- Ehrensperger, M.; Wintterlin, J. In situ scanning tunneling microscopy of the poisoning of a Co(0001) Fischer-Tropsch model catalyst by sulfur. J. Catal. 2015, 329, 49–56. [Google Scholar] [CrossRef]
- McAllister, B.; Hu, P. A density functional theory study of sulfur poisoning. J. Chem. Phys. 2005, 122, 084709. [Google Scholar] [CrossRef] [PubMed]
- Curulla-Ferré, D.; Govender, A.; Bromfield, T.C.; Niemantsverdriet, J.W. A DFT study of the adsorption and dissociation of CO on sulfur-precovered Fe(100). J. Phys. Chem. B 2006, 110, 13897–13904. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.H.; Jiao, Z.Y.; Yang, Z.X. Coverage effects on the adsorption of sulfur on Co(0 0 0 1): A DFT study. Surf. Sci. 2010, 604, 817–823. [Google Scholar] [CrossRef]
- Weststrate, C.J.; van Helden, P.; Niemantsverdriet, J.W. Reflections on the Fischer-Tropsch synthesis: Mechanistic issues from a surface science perspective. Catal. Today 2016, 275, 100–110. [Google Scholar] [CrossRef]
- den Breejen, J.P.; Radstake, P.B.; Bezemer, G.L.; Bitter, J.H.; Frøseth, V.; Holmen, A.; de Jong, K.P. On the origin of the cobalt particle size effects in Fischer-Tropsch catalysis. J. Am. Chem. Soc. 2009, 131, 7197–7203. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Zhang, Y.K.; Yang, W.T. Comment on “Generalized gradient approximation made simple”. Phys. Rev. Lett. 1998, 80, 890. [Google Scholar] [CrossRef]
- Dion, M.; Rydberg, H.; Schroder, E.; Langreth, D.C.; Lundqvist, B.I. Van der Waals density functional for general geometries. Phys. Rev. Lett. 2004, 92, 246401. [Google Scholar] [CrossRef] [Green Version]
- Roman-Perez, G.; Soler, J.M. Efficient Implementation of a van der Waals Density Functional: Application to Double-Wall Carbon Nanotubes. Phys. Rev. Lett. 2009, 103, 096102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klimes, J.; Bowler, D.R.; Michaelides, A. Chemical accuracy for the van der Waals density functional. J. Phys. Condens. Matter 2010, 22, 022201. [Google Scholar] [CrossRef]
- Klimes, J.; Bowler, D.R.; Michaelides, A. Van der Waals density functionals applied to solids. Phys. Rev. B 2011, 83, 195131. [Google Scholar] [CrossRef] [Green Version]
- Gunasooriya, G.T.K.K.; van Bavel, A.P.; Kuipers, H.P.C.E.; Saeys, M. CO adsorption on cobalt: Prediction of stable surface phases. Surf. Sci. 2015, 642, L6–L10. [Google Scholar] [CrossRef]
- Chen, C.; Wang, Q.; Wang, G.; Hou, B.; Jia, L.; Li, D. Mechanistic insight into the C2 hydrocarbons formation from Syngas on fcc-Co(111) surface: A DFT study. J. Phys. Chem. C 2016, 120, 9132–9147. [Google Scholar] [CrossRef]
- Gunasooriya, G.T.K.K.; van Bavel, A.P.; Kuipers, H.P.C.E.; Saeys, M. Key Role of Surface Hydroxyl Groups in C-O Activation during Fischer-Tropsch Synthesis. ACS Catal. 2016, 6, 3660–3664. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Govender, A.; Curulla Ferré, D.; Niemantsverdriet, J.W. The surface chemistry of water on Fe(100): A density functional theory study. ChemPhysChem 2012, 13, 1583–1590. [Google Scholar] [CrossRef]
- Henkelman, G.; Arnaldsson, A.; Jónsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comput. Mater. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]
- Sanville, E.; Kenny, S.D.; Smith, R.; Henkelman, G. Improved grid-based algorithm for Bader charge allocation. J. Comput. Chem. 2007, 28, 899–908. [Google Scholar] [CrossRef]
- Tang, W.; Sanville, E.; Henkelman, G. A grid-based Bader analysis algorithm without lattice bias. J. Phys. Condens. Matter. 2009, 21, 084204. [Google Scholar] [CrossRef] [PubMed]
- Henkelman, G.; Uberuaga, B.P.; Jonsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, E.Ø.; Svenum, I.H.; Blekkan, E.A. Mn promoted Co catalysts for Fischer-Tropsch production of light olefins—An experimental and theoretical study. J. Catal. 2018, 361, 23–32. [Google Scholar] [CrossRef]
- Balakrishnan, N.; Joseph, B.; Bhethanabotla, V.R. Effect of platinum promoters on the removal of O from the surface of cobalt catalysts: A DFT study. Surf. Sci. 2012, 606, 634–643. [Google Scholar] [CrossRef]
- Jeske, K.; Kizilkaya, A.C.; López-Luque, I.; Pfänder, N.; Bartsch, M.; Concepción, P.; Prieto, G. Design of Cobalt Fischer-Tropsch Catalysts for the Combined Production of Liquid Fuels and Olefin Chemicals from Hydrogen-Rich Syngas. ACS Catal. 2021, 11, 4784–4798. [Google Scholar] [CrossRef] [PubMed]
- Weststrate, C.J.; van de Loosdrecht, J.; Niemantsverdriet, J.W. Spectroscopic insights into cobalt-catalyzed Fischer-Tropsch synthesis: A review of the carbon monoxide interaction with single crystalline surfaces of cobalt. J. Catal. 2016, 342, 1–16. [Google Scholar] [CrossRef]
- Zhuo, M.K.; Tan, K.F.; Borgna, A.; Saeys, M. Density Functional Theory Study of the CO Insertion Mechanism for Fischer-Tropsch Synthesis over Co Catalysts. J. Phys. Chem. C 2009, 113, 8357–8365. [Google Scholar] [CrossRef]
- Ojeda, M.; Nabar, R.; Nilekar, A.U.; Ishikawa, A.; Mavrikakis, M.; Iglesia, E. CO activation pathways and the mechanism of Fischer-Tropsch synthesis. J. Catal. 2010, 272, 287–297. [Google Scholar] [CrossRef]
- Qi, Y.; Yang, J.; Chen, D.; Holmen, A. Recent Progresses in Understanding of Co-Based Fischer–Tropsch Catalysis by Means of Transient Kinetic Studies and Theoretical Analysis. Catal. Lett. 2015, 145, 145–161. [Google Scholar] [CrossRef] [Green Version]
- Govender, S.; Gambu, T.G.; van Heerden, T.; van Steen, E. Mechanistic pathways for oxygen removal on Pt-doped Co(111) in the Fischer-Tropsch reaction. Catal. Today 2020, 342, 142–151. [Google Scholar] [CrossRef]
- Kizilkaya, A.C.; Niemantsverdriet, J.W.; Weststrate, C.J. Oxygen Adsorption and Water Formation on Co(0001). J. Phys. Chem. C 2016, 120, 4833–4842. [Google Scholar] [CrossRef]
- Chen, W.; Filot, I.A.W.; Pestman, R.; Hensen, E.J.M. Mechanism of Cobalt-Catalyzed CO Hydrogenation: 2. Fischer-Tropsch Synthesis. ACS Catal. 2017, 7, 8061–8071. [Google Scholar] [CrossRef]
- Qi, Y.; Yang, J.; Holmen, A.; Chen, D. Investigation of C1 + C1 coupling reactions in cobalt-catalyzed fischer-tropsch synthesis by a combined dft and kinetic isotope study. Catalysts 2019, 9, 551. [Google Scholar] [CrossRef] [Green Version]
- Weststrate, C.J.; Niemantsverdriet, J.W. CO as a Promoting Spectator Species of CxHy Conversions Relevant for Fischer-Tropsch Chain Growth on Cobalt: Evidence from Temperature-Programmed Reaction and Reflection Absorption Infrared Spectroscopy. ACS Catal. 2018, 8, 10826–10835. [Google Scholar] [CrossRef]
- Weststrate, C.J.; Sharma, D.; Rodriguez, D.G.; Gleeson, M.A.; Fredriksson, H.O.A.; Niemantsverdriet, J.W. Mechanistic insight into carbon-carbon bond formation on cobalt under simulated Fischer-Tropsch synthesis conditions. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Rytter, E.; Holmen, A. Perspectives on the Effect of Water in Cobalt Fischer-Tropsch Synthesis. ACS Catal. 2017, 7, 5321–5328. [Google Scholar] [CrossRef]
- Rytter, E.; Borg, Ø.; Tsakoumis, N.E.; Holmen, A. Water as key to activity and selectivity in Co Fischer-Tropsch synthesis: Γ-alumina based structure-performance relationships. J. Catal. 2018, 365, 334–343. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Co(111) | S-Co(111) | ΔEad (%) | |||
|---|---|---|---|---|---|
| Energy | Adsorption Site | Energy | Adsorption Site | ||
| CO | −140 | top | −70 | hcp | −50 |
| HCO | −194 | hcp | −87 | fcc | −55 |
| H | −288 | fcc | −262 | hcp | −9 |
| C | −647 | hcp | −562 | hcp | −13 |
| CH | −635 | hcp | −600 | hcp | −6 |
| CH2 | −414 | fcc | −342 | top | −17 |
| CH3 | −229 | fcc | −117 | top | −49 |
| CH4 | −22 | top | −24 | top | 9 |
| C2H2 | −206 | hcp and fcc | −6 | hcp and fcc | −97 |
| O | −545 | hcp | −451 | hcp | −7 |
| OH | −385 | hcp | −288 | hcp | −25 |
| H2O | −28 | top | −25 | top | −11 |
| Co(111) | S-Co(111) | ΔCharge (%) | ΔEad (%) | |
|---|---|---|---|---|
| S | NA | −0.68 | NA | NA |
| CO | −0.37 (top) | −0.54 (hcp)/−0.28 (top) | −25 | −50 |
| HCO | −0.73 | −0.53 | −27 | −55 |
| H | −0.41 | −0.34 | −16 | −9 |
| C | −0.88 | −0.78 | −11 | −13 |
| CH | −0.74 | −0.62 | −17 | −6 |
| CH2 | −0.64 | −0.25 | −62 | −17 |
| CH3 | −0.45 | −0.20 | −56 | −49 |
| C2H2 | −0.95 | −0.82 | −14 | −97 |
| O | −1.03 | −0.95 | −8 | −17 |
| OH | −0.67 | −0.60 | −10 | −25 |
| Ea | ΔEa | RE | Δ(RE) | |||
|---|---|---|---|---|---|---|
| Co(111) | S-Co(111) | Co(111) | S-Co(111) | |||
| H + CO→HCO | 121 | 133 | 12 | 99 | 75 | −24 |
| HCO→HC + O | 74 | 165 | 91 | −24 | 71 | 95 |
| C + H→CH | 77 | 15 | −62 | −51 | −214 | −163 |
| CH + H→CH2 | 65 | 45 | −20 | 25 | −50 | −75 |
| CH2 + H→CH3 | 49 | 39 | −10 | −38 | −15 | 23 |
| CH3 + H→CH4 | 93 | 24 | −69 | −64 | −203 | −139 |
| CH + CH→C2H2 | 67 | 71 | 4 | −67 | −17 | 50 |
| O + H→OH | 122 | 73 | −49 | 7 | −71 | −78 |
| O + H→H2O | 142 | 126 | −16 | 31 | −149 | −180 |
| OH + OH→H2O + O | 5 | 4 | −1 | −55 | −105 | −50 |
| ΔEad (%) | |||
|---|---|---|---|
| θS = 0.25 ML | θS = 0.11 ML | θS = 0.06 ML | |
| CO | −50 | −9 | −9 |
| H | −9 | −4 | −5 |
| C | −13 | −6 | −4 |
| CH | −6 | −4 | −4 |
| C2H2 | −97 | −19 | −10 |
| O | −17 | −8 | −9 |
| OH | −25 | −8 | −9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daga, Y.; Kizilkaya, A.C. Mechanistic Insights into the Effect of Sulfur on the Selectivity of Cobalt-Catalyzed Fischer–Tropsch Synthesis: A DFT Study. Catalysts 2022, 12, 425. https://doi.org/10.3390/catal12040425
Daga Y, Kizilkaya AC. Mechanistic Insights into the Effect of Sulfur on the Selectivity of Cobalt-Catalyzed Fischer–Tropsch Synthesis: A DFT Study. Catalysts. 2022; 12(4):425. https://doi.org/10.3390/catal12040425
Chicago/Turabian StyleDaga, Yagmur, and Ali Can Kizilkaya. 2022. "Mechanistic Insights into the Effect of Sulfur on the Selectivity of Cobalt-Catalyzed Fischer–Tropsch Synthesis: A DFT Study" Catalysts 12, no. 4: 425. https://doi.org/10.3390/catal12040425