

Catalytic Efficiency of Primary α-Amino Amides as Multifunctional Organocatalysts in Recent Asymmetric Organic Transformations

Abstract
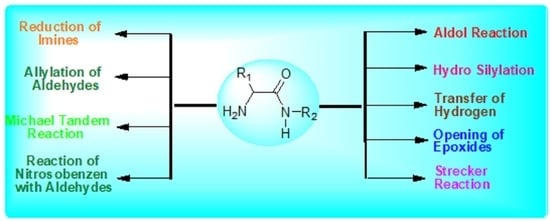
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
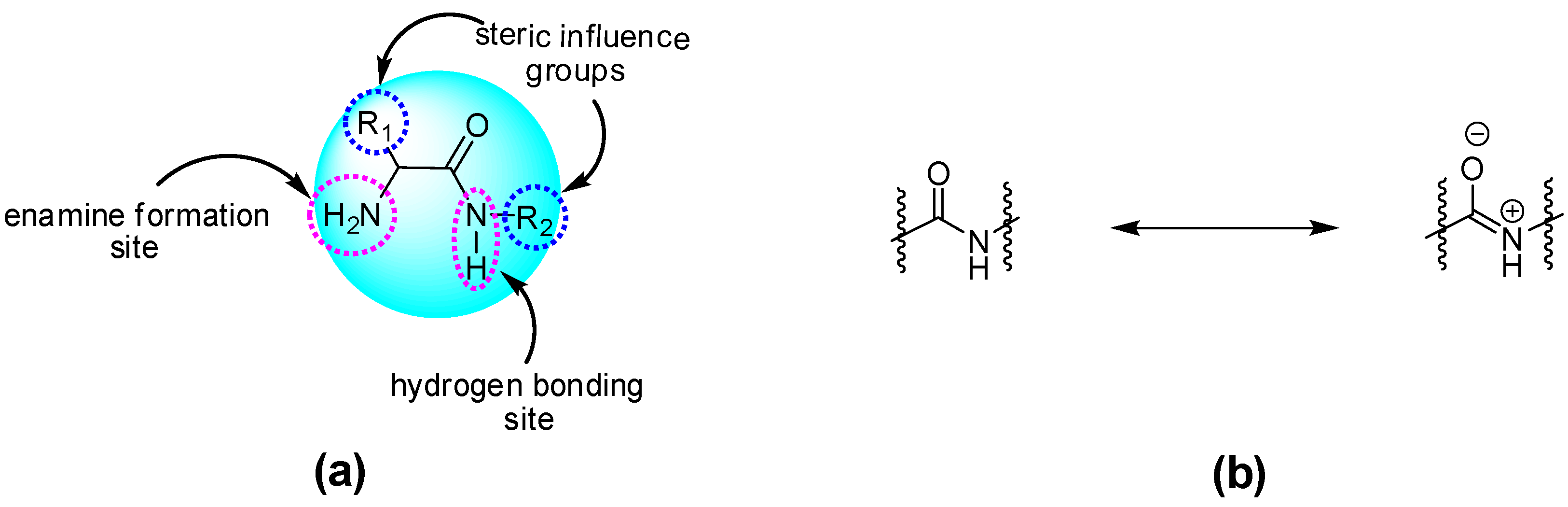{kind=link}
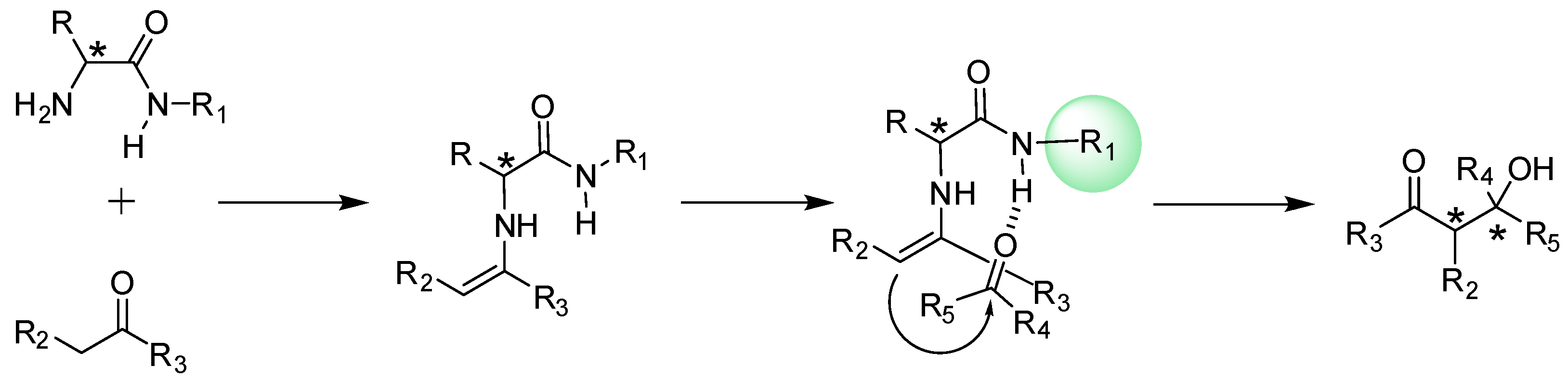{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
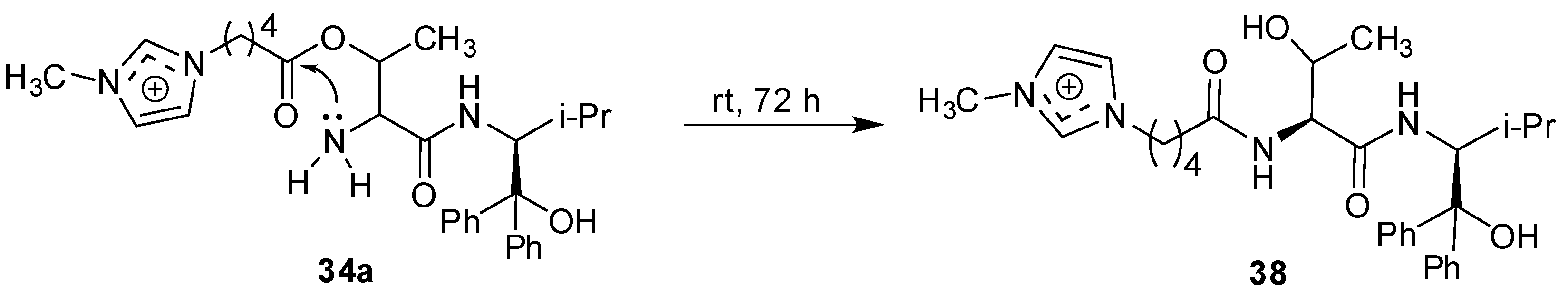{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
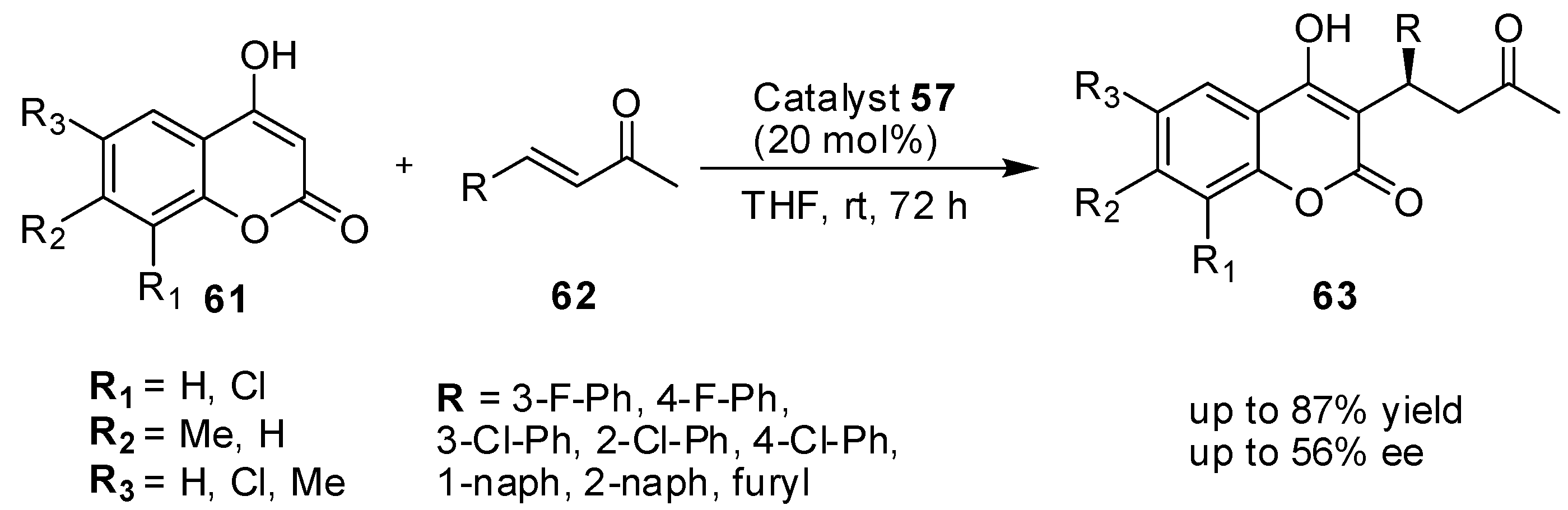{kind=link}
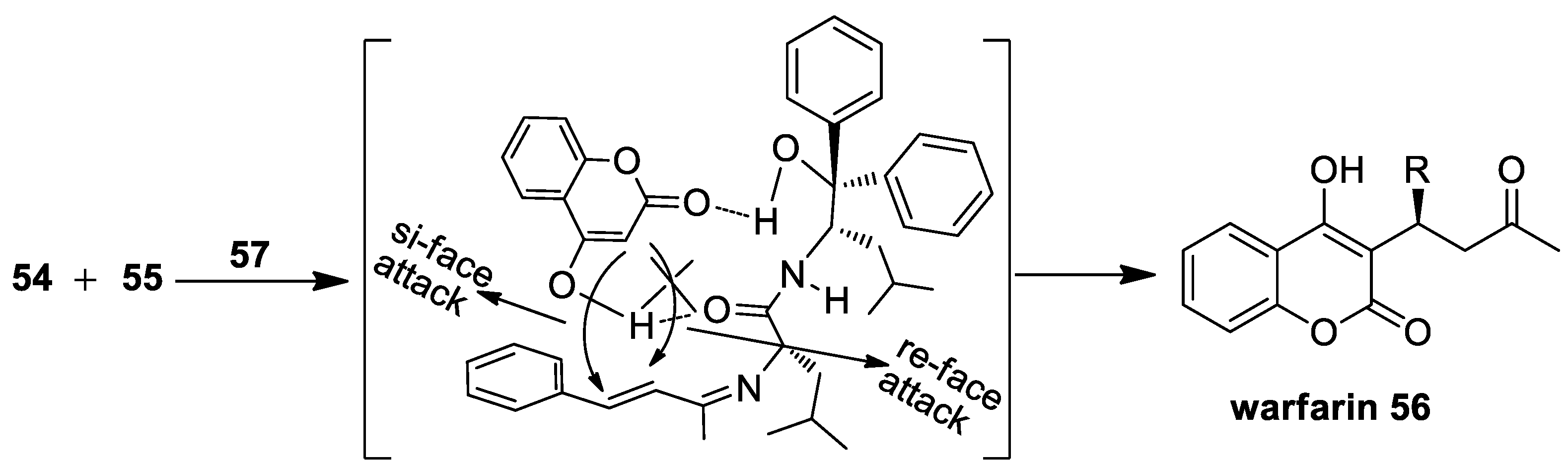{kind=link}
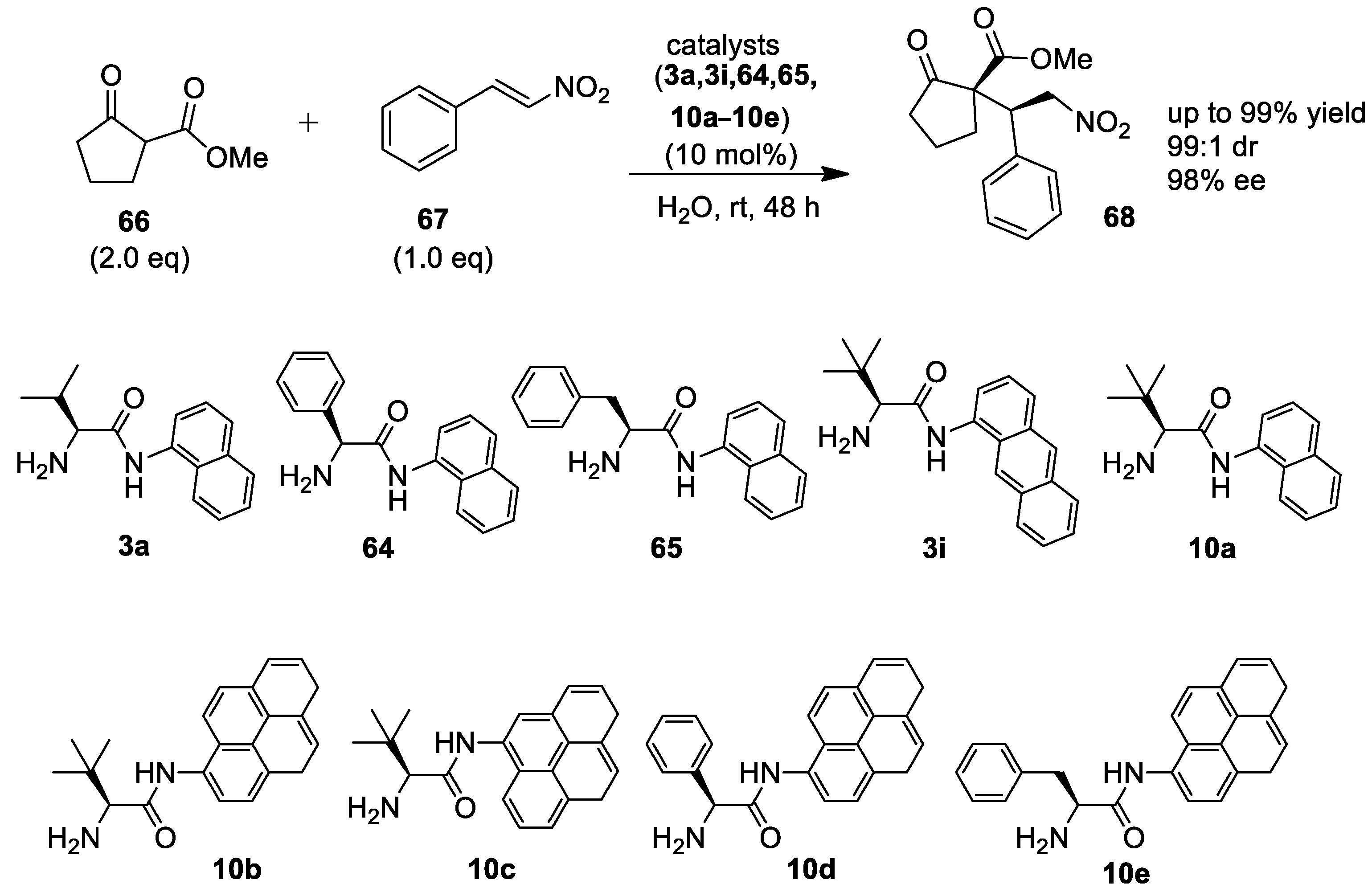{kind=link}
{kind=link}
{kind=link}
{kind=link}
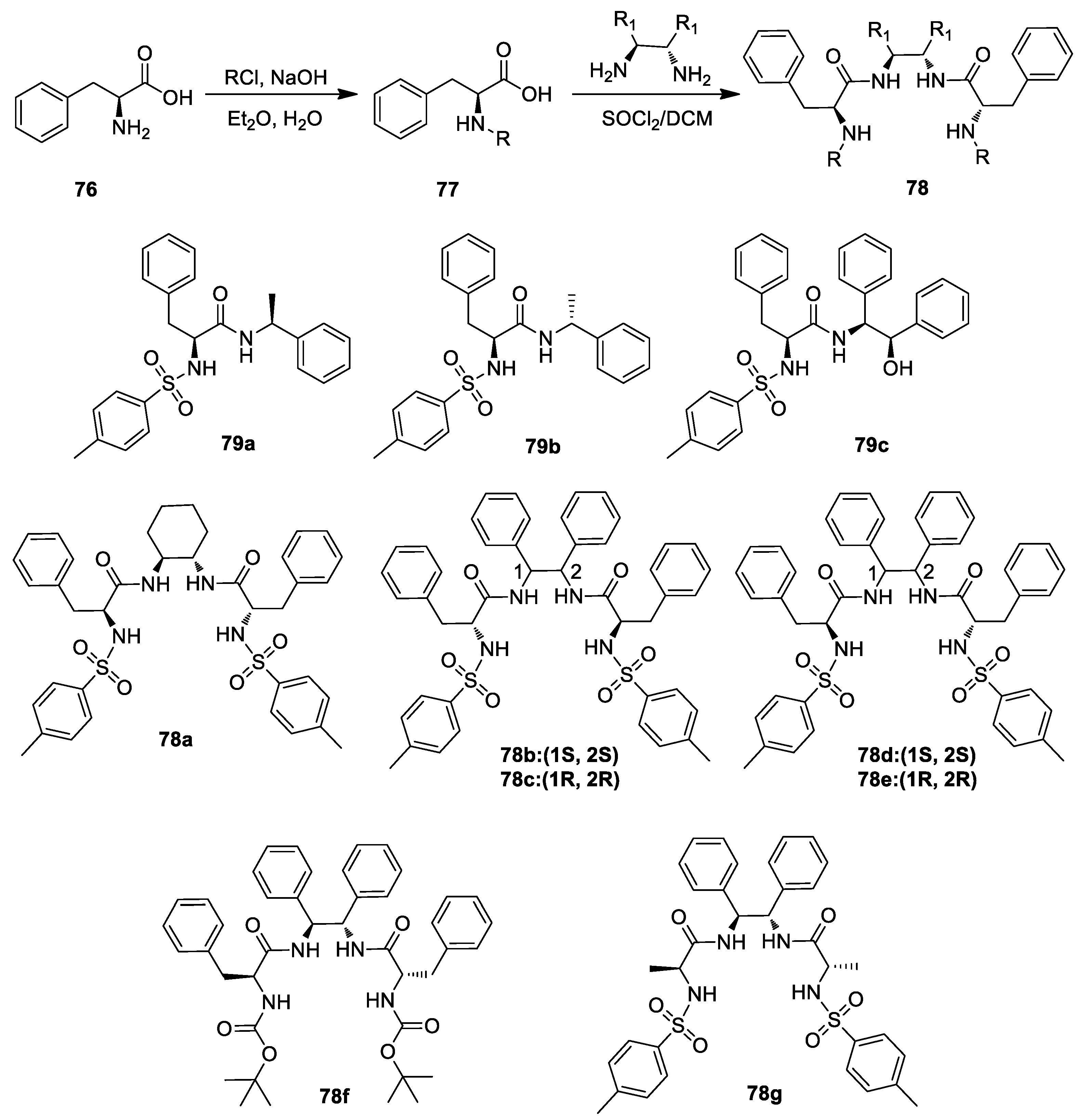{kind=link}
{kind=link}
{kind=link}
{kind=link}
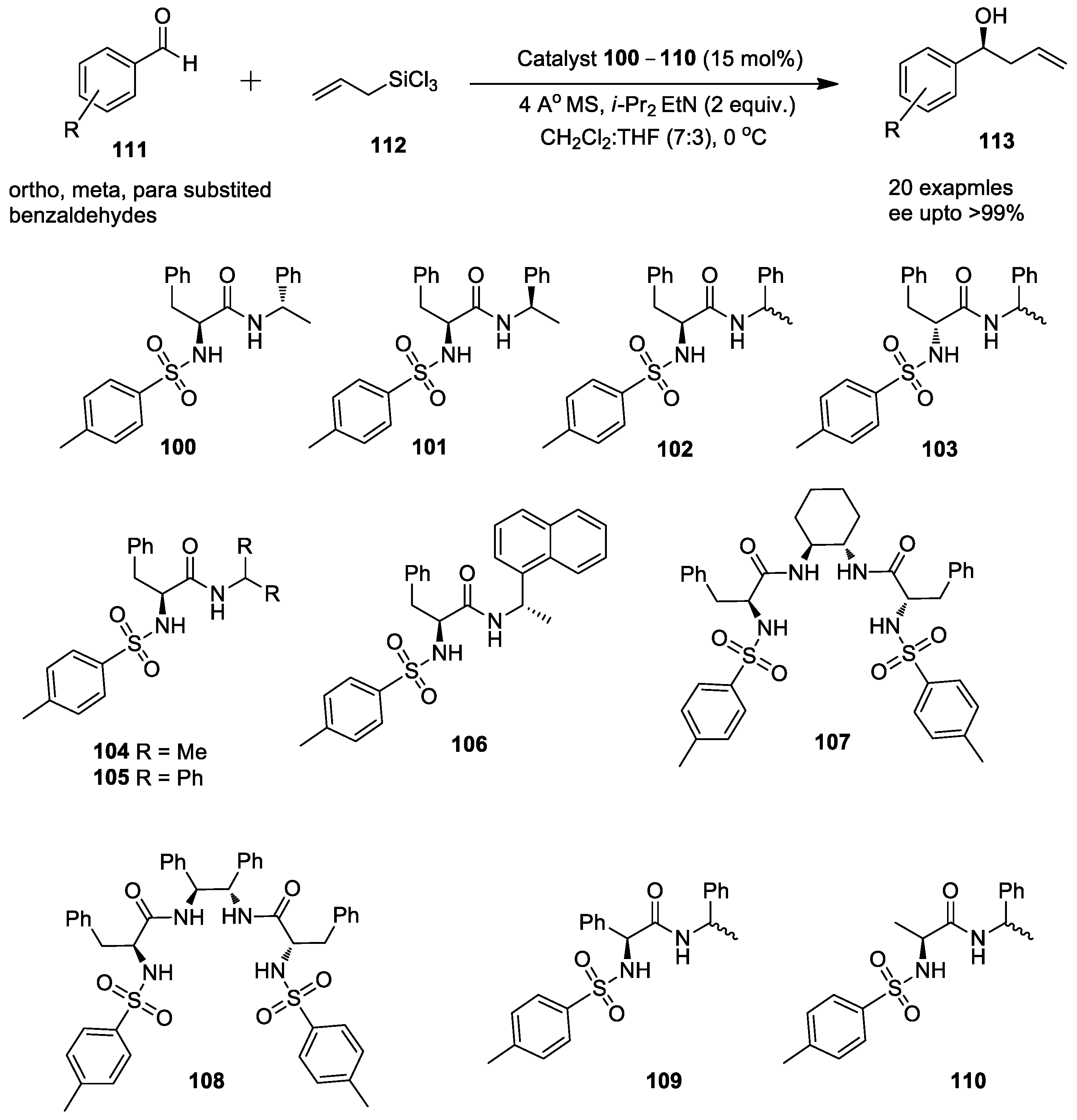{kind=link}
{kind=link}
{kind=link}
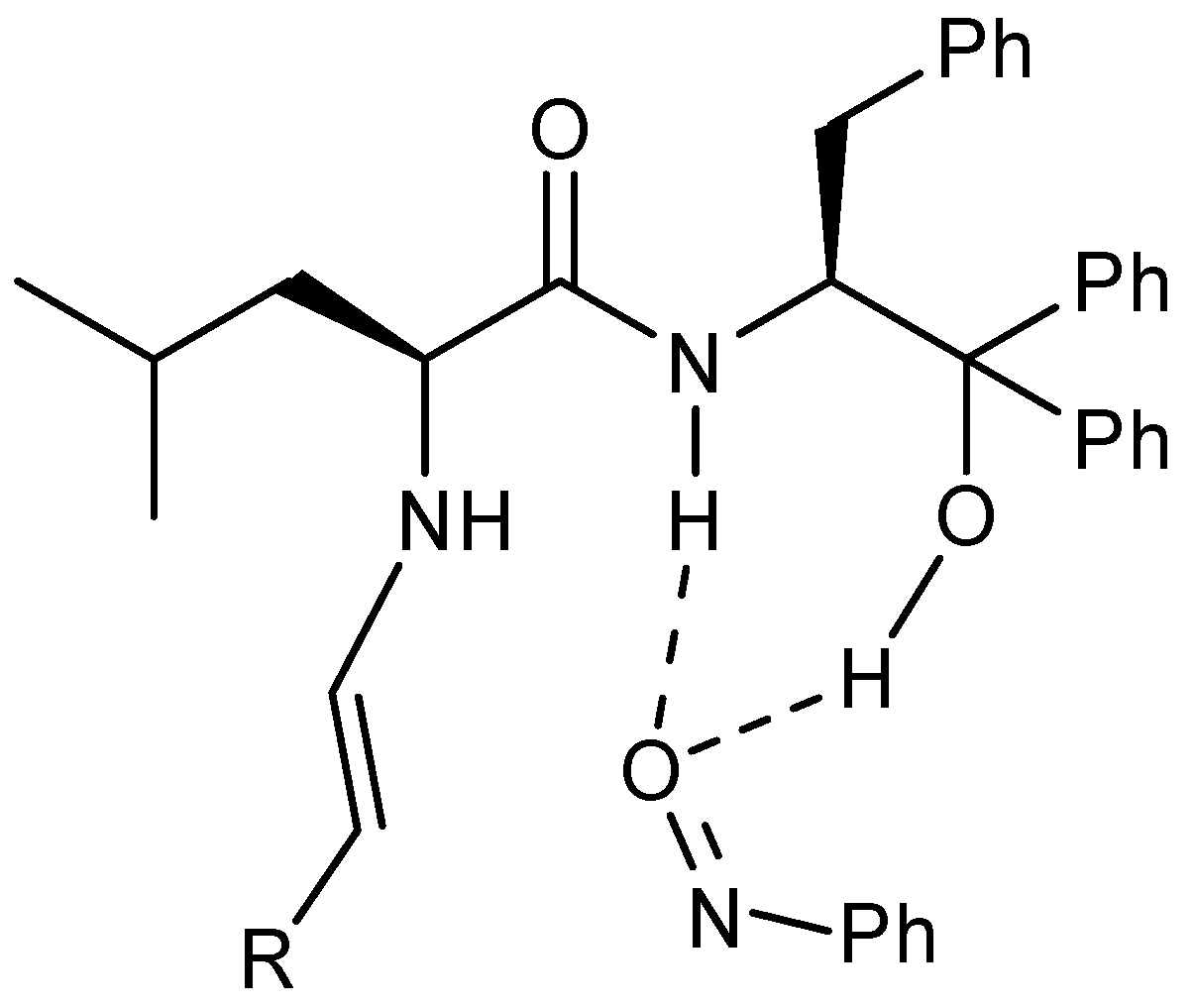{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
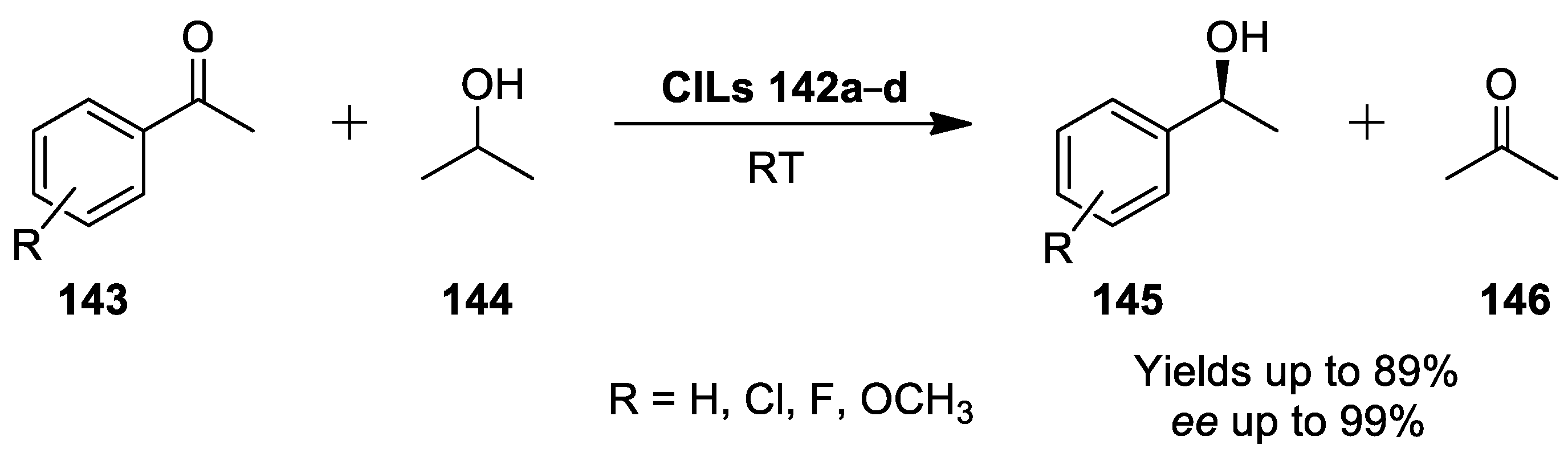{kind=link}
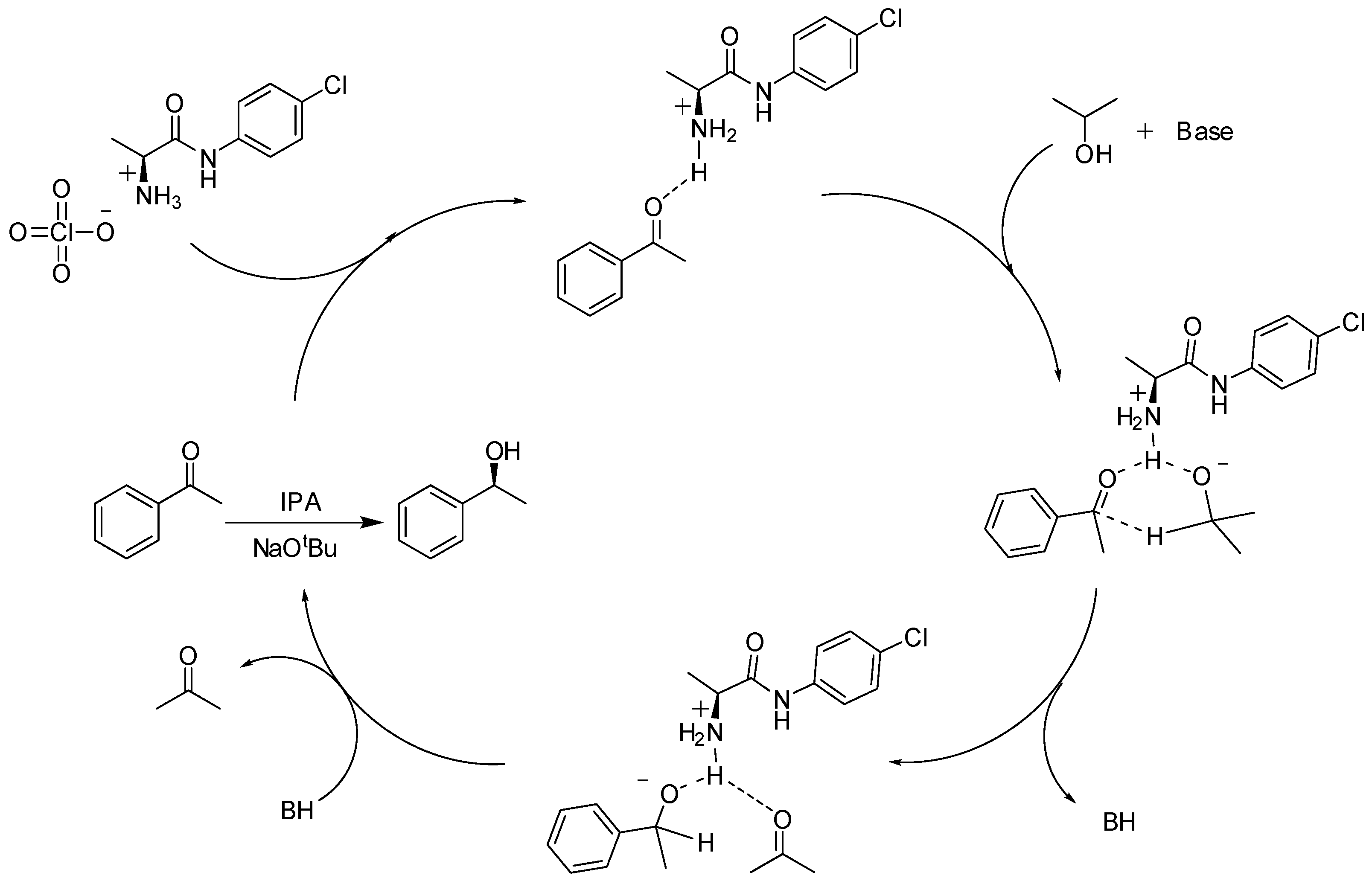{kind=link}
1. Introduction
2. Primary α-Amino-Amide-Catalyzed Asymmetric Organic Transformations
2.1. Asymmetric Aldol Reaction
2.2. Asymmetric Michael Reaction
2.3. Asymmetric Strecker Reaction
2.4. Enantioselective Allylation of Aldehydes
2.5. Hydrosilylation of 1,4-Benzooxazines
2.6. Asymmetric N-Specific Reaction of Nitrosobenzene with Aldehydes
2.7. Opening of Epoxide
2.8. Asymmetric Reduction of N-Aryl Imines
2.9. Asymmetric Hydrogen Transfer of Acetophenone
3. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, L.; Liu, X.Q.; Jiang, H.L.; Sun, L.B. Metal-Organic Frameworks for Heterogeneous Basic Catalysis. Chem. Rev. 2017, 117, 8129–8176. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Park, W.J.; Jun, C.H. Metal-Organic Cooperative Catalysis in C-H and C-C Bond Activation. Chem. Rev. 2017, 117, 8977–9015. [Google Scholar] [CrossRef] [PubMed]
- Afewerki, S.; Córdova, A. Combinations of Aminocatalysts and Metal Catalysts: A Powerful Cooperative Approach in Selective Organic Synthesis. Chem. Rev. 2016, 116, 13512–13570. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniyam, R.; Prasenjit, G. The Developing Concept of Bifunctional Catalysis with Transition Metal N-Heterocyclic Carbene Complexes. Eur. J. Inorg. Chem. 2016, 2016, 1448–1465. [Google Scholar] [CrossRef]
- Parmar, D.; Sugiono, E.; Raja, S.; Rueping, M. Addition and Correction to Complete Field Guide to Asymmetric BINOL-Phosphate Derived Brønsted Acid and Metal Catalysis: History and Classification by Mode of Activation; Brønsted Acidity, Hydrogen Bonding, Ion Pairing, and Metal Phosphates. Chem. Rev. 2017, 117, 10608–10620. [Google Scholar] [CrossRef] [Green Version]
- Parmar, D.; Sugiono, E.; Raja, S.; Rueping, M. Complete Field Guide to Asymmetric BINOL-Phosphate Derived Brønsted Acid and Metal Catalysis: History and Classification by Mode of Activation; Brønsted Acidity, Hydrogen Bonding, Ion Pairing, and Metal Phosphates. Chem. Rev. 2014, 114, 9047–9153. [Google Scholar] [CrossRef]
- Newton, C.G.; Wang, S.G.; Oliveira, C.C.; Cramer, N. Catalytic Enantioselective Transformations Involving C–H Bond Cleavage by Transition-Metal Complexes. Chem. Rev. 2017, 117, 8908–8976. [Google Scholar] [CrossRef]
- Paull, D.H.; Abraham, C.J.; Scerba, M.T.; Danforth, E.A.A.; Lectka, T. Bifunctional Asymmetric Catalysis: Cooperative Lewis Acid/Base Systems. Accounts Chem. Res. 2008, 41, 655–663. [Google Scholar] [CrossRef] [Green Version]
- Pellissier, H. Recent Developments in Enantioselective Metal-Catalyzed Domino Reactions. Adv. Synth. Catal. 2016, 358, 2194–2259. [Google Scholar] [CrossRef]
- Jin, H.; Kim, S.T.; Hwang, G.S.; Ryu, D.H. l-Proline Derived Bifunctional Organocatalysts: Enantioselective Michael Addition of Dithiomalonates to trans-β-Nitroolefins. J. Org. Chem. 2016, 81, 3263–3274. [Google Scholar] [CrossRef]
- Albrecht, L.; Jiang, H.; Jørgensen, K.A. Hydrogen-Bonding in Aminocatalysis: From Proline and Beyond. Chem. Eur. J. 2014, 20, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Kumarswamyreddy, N.; Kesavan, V. Enantioselective Synthesis of Dihydrospiro[indoline-3,4′-pyrano[2,3-c]pyrazole] Derivatives via Michael/Hemiketalization Reaction. Org. Lett. 2016, 18, 1354–1357. [Google Scholar] [CrossRef] [PubMed]
- Kizirian, J.C. Chiral Tertiary Diamines in Asymmetric Synthesis. Chem. Rev. 2008, 108, 140–205. [Google Scholar] [CrossRef] [PubMed]
- Gualandi, A.; Grilli, S.; Savoia, D. Octa-1,7-diene-4,5-diamine Derivatives: Useful Intermediates for the Stereoselective Synthesis of Nitrogen Heterocycles and Ligands for Asymmetric Catalysis. Eur. J. Org. Chem. 2016, 2016, 3143–3156. [Google Scholar] [CrossRef]
- Notz, W.; Tanaka, F.; Barbas, C.F., III. Enamine-Based Organocatalysis with Proline and Diamines: The Development of Direct Catalytic Asymmetric Aldol, Mannich, Michael, and Diels−Alder Reactions. Acc. Chem. Res. 2004, 37, 580–591. [Google Scholar] [CrossRef]
- Melchiorre, P. Cinchona-based Primary Amine Catalysis in the Asymmetric Functionalization of Carbonyl Compounds. Angew. Chem. Int. Ed. 2012, 51, 9748–9770. [Google Scholar] [CrossRef]
- Kacprzak, K.J.; Gawronski, J. Resolution of Racemates and Enantioselective Analytics by Cinchona Alkaloids and Their Derivatives. In Cinchona Alkaloids in Synthesis and Catalysis: Ligands, Immobilization and Organocatalysis; Wiley-VCH: Weinheim, Germany, 2009; pp. 419–469. [Google Scholar]
- Tian, S.K.; Chen, Y.; Hang, J.; Tang, L.; McDaid, P.; Deng, L. Asymmetric Organic Catalysis with Modified Cinchona Alkaloids. Acc. Chem. Res. 2004, 37, 621–631. [Google Scholar] [CrossRef]
- Zheng, S.; Schienebeck, C.M.; Zhang, W.; Wang, H.Y.; Tang, W. Cinchona Alkaloids as Organocatalysts in Enantioselective Halofunctionalization of Alkenes and Alkynes. Asia. J. Org. Chem. 2014, 3, 366–376. [Google Scholar] [CrossRef]
- Subba Reddy, U.V.; Chennapuram, M.; Seki, C.; Kwon, E.; Okuyama, Y.; Hiroto Nakano, H. Catalytic Efficiency of Primary β-Amino Alcohols and Their Derivatives in Organocatalysis. Eur. J. Org. Chem. 2016, 2016, 4124–4143. [Google Scholar] [CrossRef]
- Madarasz, A.; Dosa, Z.; Varga, S.; Soos, T.; Csampai, A.; Papai, I. Thiourea Derivatives as Brønsted Acid Organocatalysts. ACS Catal. 2016, 6, 4379–4387. [Google Scholar] [CrossRef]
- Kotke, M.; Schreiner, P.R. (Thio)urea Organocatalysts. In Hydrogen Bonding in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2009; pp. 141–351. [Google Scholar]
- Jakab, G.; Schreiner, P.R. Brønsted Acids: Chiral (Thio)urea Derivatives. In Comprehensive Enantioselective Organocatalysis: Catalysts, Reactions and Applications; Wiley-VCH: Weinheim, Germany, 2013; pp. 315–341. [Google Scholar]
- Chauhan, P.; Mahajan, S.; Kaya, U.; Hack, D.; Enders, D. Bifunctional Amine-Squaramides: Powerful Hydrogen-Bonding Organocatalysts for Asymmetric Domino/Cascade Reactions. Adv. Synth. Catal. 2015, 357, 253–281. [Google Scholar] [CrossRef]
- Madhu, C.; Subba Reddy, U.V.; Seki, C.; Okuyama, C.; Kwon, E.; Uwai, K.; Tokiwa, M.; Takeshita, M.; Nakano, H. Hybrid-Type Squaramide-Fused Amino Alcohol Organocatalysts for Enantioselective Nitro-Aldol Reaction of Nitromethane with Isatins. Eur. J. Org. Chem. 2017, 2017, 1638–1646. [Google Scholar] [CrossRef]
- Madhu, C.; Subba Reddy, U.V.; Seki, C.; Okuyama, Y.; Kwon, E.; Uwai, K.; Tokiwa, M.; Takeshita, M.; Nakano, H. Hybrid-Type Squaramide-Fused Amino Alcohol Organocatalysts for Enantioselective Diels–Alder Reactions of 3-Hydroxy-2-Pyridones with Maleimides. Eur. J. Org. Chem. 2017, 2017, 4633–4641. [Google Scholar] [CrossRef]
- Andres, J.M.; Losada, J.; Maestro, A.; Ferrer, P.R.; Pedrosa, R. Supported and Unsupported Chiral Squaramides as Organocatalysts for Stereoselective Michael Additions: Synthesis of Enantiopure Chromenes and Spirochromanes. J. Org. Chem. 2017, 82, 8444–8454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, K.; Akiyama, T. Brønsted Acids: Chiral Phosphoric Acid Catalysts in Asymmetric Synthesis. In Comprehensive Enantioselective Organocatalysis: Catalysts, Reactions, and Applications; Wiley-VCH: Weinheim, Germany, 2013; pp. 289–314. [Google Scholar]
- Yamamoto, H.; Cheon, C.H. Chiral Lewis Acids and Brønsted Acids in Asymmetric Synthesis. In Catalytic Asymmetric Synthesis, 3rd ed; Wiley-VCH: Weinheim, Germany, 2010; pp. 119–161. [Google Scholar]
- Lu, N.; Li, R.; Wei, Z.; Cao, J.; Liang, D.; Lin, Y.; Duan, H. Enantio- and Diastereoselective Nitro-Mannich Reaction of α-Aryl Nitromethanes with Amidosulfones Catalyzed by Phase-Transfer Catalysts. J. Org. Chem. 2017, 82, 4668–4676. [Google Scholar] [CrossRef]
- Wang, B.; Liu, Y.; Sun, C.; Wei, Z.; Cao, J.; Liang, D.; Lin, Y.; Duan, H. Asymmetric Phase-Transfer Catalysts Bearing Multiple Hydrogen-Bonding Donors: Highly Efficient Catalysts for Enantio- and Diastereoselective Nitro-Mannich Reaction of Amidosulfones. Org. Lett. 2014, 16, 6432–6435. [Google Scholar] [CrossRef]
- Yamamoto, H.; Payette, J.N. Brønsted Acids, H-Bond Donors, and Combined Acid Systems in Asymmetric Catalysis. In Hydrogen Bonding in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2009; pp. 73–140. [Google Scholar]
- Freund, M.; Tsogoeva, S.B. Peptides for Asymmetric Catalysis. Catalytic Methods in Asymmetric Synthesis: Advanced Materials, Techniques and Applications; Wiley-VCH: Weinheim, Germany, 2011; pp. 529–578. [Google Scholar] [CrossRef]
- Davie, E.A.C.; Mennen, S.M.; Xu, Y.; Miller, S.J. Asymmetric Catalysis Mediated by Synthetic Peptides. Chem. Rev. 2007, 107, 5759–5812. [Google Scholar] [CrossRef]
- Liu, X.; Lin, L.; Feng, X. X. Amide-based bifunctional organocatalysts in asymmetric reactions. Chem. Commun. 2009, 6145–6158. [Google Scholar] [CrossRef]
- Panday, S.K. Advances in the chemistry of proline and its derivatives: An excellent amino acid with versatile applications in asymmetric synthesis. Tetrahedron Asymmetry 2011, 22, 1817–1847. [Google Scholar] [CrossRef]
- Yadav, G.D.; Singh, S. Prolinamide-Catalysed Asymmetric Organic Transformations. ChemistrySelect 2019, 4, 5591–5618. [Google Scholar] [CrossRef]
- Casiraghi, G.; Baltistan, L.; Curti, C.; Rassu, G.; Canard, F. The Vinylogous Aldol and Related Addition Reactions: Ten Years of Progress. Chem. Rev. 2011, 111, 3076–3154. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, F.; Barbas, C.F., III. Antibody-catalyzed Aldol Reactions. In Modern Aldol Reactions; Marsala, R., Ed.; Wiley-VCH: Berlin, Germany, 2004. [Google Scholar]
- Palomo, C.; Oiarbide, M.; Garciak, J.M. Current progress in the asymmetric aldol addition reaction. Chem. Soc. Rev. 2004, 33, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.Y.; Wang, Y.Z.; Cun, L.F.; Gong, L.Z. l-Proline amides catalyze direct asymmetric aldol reactions of aldehydes with methylthioacetone and fluoroacetone. Tetrahedron Asymmetry 2007, 18, 237–242. [Google Scholar] [CrossRef]
- Bisai, V.; Bisai, A.; Singh, V.K. Enantioselective organocatalytic aldol reaction using small organic molecules. Tetrahedron 2012, 68, 4541–4580. [Google Scholar] [CrossRef]
- Guillena, G. Modern Methods in Stereoselective Aldol Reactions; Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar]
- Scheffler, U.; Mahrwald, R. Recent Advances in Organocatalytic Methods for Asymmetric C-C Bond Formation. Chem. Eur. J. 2013, 19, 14346–14396. [Google Scholar] [CrossRef] [PubMed]
- Eder, U.; Sauer, G.; Wiechert, R. New Type of Asymmetric Cyclization to Optically Active Steroid CD Partial Structures. Angew. Chem. Int. Ed. Engl. 1971, 10, 496–497. [Google Scholar] [CrossRef]
- Hajos, Z.G.; Parrish, D.R. Asymmetric synthesis of bicyclic intermediates of natural product chemistry. J. Org. Chem. 1974, 39, 1615–1621. [Google Scholar] [CrossRef]
- List, B.; Lerner, R.A.; Barbas, C.F., III. Proline-Catalyzed Direct Asymmetric Aldol Reactions. J. Am. Chem. Soc. 2000, 122, 2395–2396. [Google Scholar] [CrossRef]
- Notz, W.; List, B. Catalytic Asymmetric Synthesis of anti-1,2-Diols. J. Am. Chem. Soc. 2000, 122, 7386–7387. [Google Scholar] [CrossRef]
- Giacalone, F.; Gruttadauria, M.; Agrigento, P.; Meo, P.L.; Noto, R. Advances towards Highly Active and Stereoselective Simple and Cheap Proline-Based Organocatalysts. Eur. J. Org. Chem. 2010, 2010, 5696–5704. [Google Scholar] [CrossRef]
- Armstrong, A.; Bhonoah, Y.; White, A.J.P. Constrained β-Proline Analogues in Organocatalytic Aldol Reactions: The Influence of Acid Geometry. J. Org. Chem. 2009, 74, 5041–5048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montroni, E.; Sanap, S.P.; Lombardo, M.; Quintavalla, A.; Trombini, C.; Dhavale, D.D. A New Robust and Efficient Ion-Tagged Proline Catalyst Carrying an Amide Spacer for the Asymmetric Aldol Reaction. Adv. Synth. Catal. 2011, 353, 3234–3240. [Google Scholar] [CrossRef]
- Bassan, A.; Zou, W.; Reyes, E.; Himo, F.; Córdova, A. The Origin of Stereoselectivity in Primary Amino Acid Catalyzed Intermolecular Aldol Reactions. Angew. Chem. Int. Ed. 2005, 44, 7190–7194. [Google Scholar] [CrossRef]
- Amedjkouh, M. Aqua-organocatalyzed direct asymmetric aldol reaction with acyclic amino acids and organic bases with control of diastereo- and enantioselectivity. Tetrahedron Asymmetry 2007, 18, 390–395. [Google Scholar] [CrossRef]
- Wu, C.L.; Long, X.Q.; Li, S.; Fu, X. Simple and inexpensive threonine-based organocatalysts as highly active and recoverable catalysts for large-scale asymmetric direct stoichiometric aldol reactions on water. Tetrahedron Asymmetry 2012, 23, 315–328. [Google Scholar] [CrossRef]
- Revelou, P.; Kokotos, C.G.; Minakakis, P.M. Novel prolinamide–ureas as organocatalysts for the asymmetric aldol reaction. Tetrahedron 2012, 68, 8732–8738. [Google Scholar] [CrossRef]
- Maycock, C.D.; Ventura, M.R. New organocatalysts derived from tartaric and glyceric acids for direct aldol reactions. Tetrahedron Asymmetry 2012, 23, 1262–1271. [Google Scholar] [CrossRef]
- Kokotos, C.G. Construction of Tertiary Alcohols Bearing Perfluoroalkyl Chains Catalyzed by Prolinamide-Thioureas. J. Org. Chem. 2012, 77, 1131–1135. [Google Scholar] [CrossRef]
- Cabrera, H.R.; Huelgas, G.; Perez, J.M.H.; Walsh, P.J.; Somanathan, R.; Parrodi, C.A.D. Homochiral l-prolinamido-sulfonamides and their use as organocatalysts in aldol reactions. Tetrahedron Asymmetry 2015, 26, 163–172. [Google Scholar] [CrossRef]
- Robak, M.T.; Herbage, M.A.; Ellman, J.A. Development of an N-sulfinyl prolinamide for the asymmetric aldol reaction. Tetrahedron 2011, 67, 4412–4416. [Google Scholar] [CrossRef]
- Hara, N.; Tamura, R.; Funahashi, Y.; Nakamura, S. N-(Heteroarenesulfonyl)prolinamides-Catalyzed Aldol Reaction between Acetone and Aryl Trihalomethyl Ketones. Org. Lett. 2011, 13, 1662–1665. [Google Scholar] [CrossRef] [PubMed]
- Da, C.S.; Che, L.P.; Guo, Q.P.; Wu, F.C.; Ma, X.; Jia, Y.N. 2,4-Dinitrophenol as an Effective Cocatalyst: Greatly Improving the Activities and Enantioselectivities of Primary Amine Organocatalysts for Asymmetric Aldol Reactions. J. Org. Chem. 2009, 74, 2541–2546. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, J.; Peddinti, R.K. Glucosamine-Based Primary Amines as Organocatalysts for the Asymmetric Aldol Reaction. J. Org. Chem. 2011, 76, 3502–3505. [Google Scholar] [CrossRef]
- Xu, B.; Li, L.; Gou, S. A chiral primary-tertiary-1,2-diamine as an efficient catalyst in asymmetric aldehyde–ketone or ketone–ketone aldol reactions. Tetrahedron Asymmetry 2013, 24, 1556–1561. [Google Scholar] [CrossRef]
- Karmakar, A.; Maji, T.; Wittmann, S.; Reiser, O. L-Proline/CoCl2-Catalyzed Highly Diastereo- and Enantioselective Direct Aldol Reactions. Chem. Eur. J. 2011, 17, 11024–11029. [Google Scholar] [CrossRef] [PubMed]
- Ichibakase, T.; Nakajima, M. Direct Enantioselective Aldol−Tishchenko Reaction Catalyzed by Chiral Lithium Diphenylbinaphtholate. Org. Lett. 2011, 13, 1579–1581. [Google Scholar] [CrossRef] [PubMed]
- Montroni, E.; Lombardo, M.; Quintavalla, A.; Trombini, C.; Gruttadauria, M.; Giacalone, F. A Liquid–Liquid Biphasic Homogeneous Organocatalytic Aldol Protocol Based on the Use of a Silica Gel Bound Multilayered Ionic Liquid Phase. ChemCatChem 2012, 4, 1000–1006. [Google Scholar] [CrossRef]
- Maria, E.D.H.; Sunil, K.P.; Elisabeth, C.; Rikke, A.; Steffen, V.F.H.; Trond, U. A protocol for amide bond formation with electron deficient amines and sterically hindered substrates. Org. Biomol. Chem. 2016, 14, 430–433. [Google Scholar] [CrossRef] [Green Version]
- Okuyama, Y.; Nakano, H.; Watanabe, Y.; Makabe, M.; Takeshita, M.; Uwai, K.; Kabuto, C.; Kwon, E. Organocatalytic activity of 4-hydroxy-prolinamide alcohol with different noncovalent coordination sites in asymmetric Michael and direct aldol reactions. Tetrahedron Lett. 2009, 50, 193–197. [Google Scholar] [CrossRef]
- Kimura, J.; Subba Reddy, U.V.; Kohari, Y.; Seki, C.; Mawatari, Y.; Uwai, K.; Okuyama, Y.; Kwon, E.; Tokiwa, M.; Takeshita, M.; et al. Simple Primary Amino Amide Organocatalyst for Enantioselective Aldol Reactions of Isatins with Ketones. Eur. J. Org. Chem. 2016, 2016, 3748–3756. [Google Scholar] [CrossRef]
- Owolabi, I.A.; Subba Reddy, U.V.; Chennapuram, M.; Seki, C.; Okuyama, Y.; Kwon, E.; Uwai, K.; Tokiwa, M.; Takeshita, M.; Nakano, H. A new type of amino amide organocatalyzed enantioselective crossed aldol reaction of ketones with aromatic aldehydes. Tetrahedron 2018, 74, 4705–4711. [Google Scholar] [CrossRef]
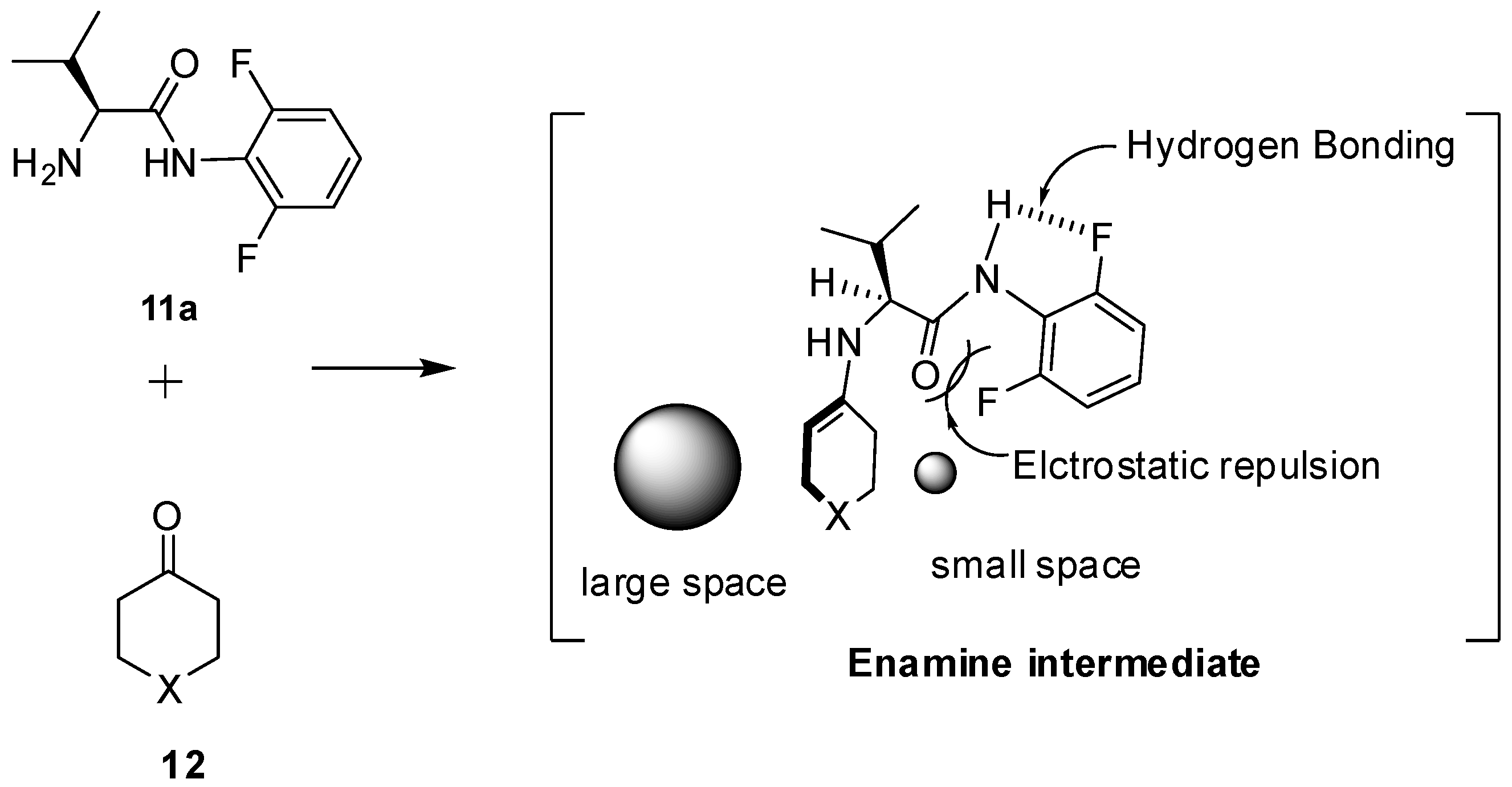
- Tanimura, Y.; Yasunaga, K.; Ishimaru, K. Electrostatic Repulsion and Hydrogen-Bonding Interactions in a Simple N-Aryl-L-valinamide Organocatalyst Control the Stereoselectivity in Asymmetric Aldol Reactions. Eur. J. Org. Chem. 2013, 2013, 6535–6539. [Google Scholar] [CrossRef]
- Wang, J.; Liu, Q.; Hao, Q.; Sun, Y.; Luo, Y.; Yang, H. Enantioselective Aldol Reaction Between Isatins and Cyclohexanone Catalyzed by Amino Acid Sulphonamides. Chirality 2015, 27, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Demuynck, A.L.W.; Vanderleyden, J.; Sels, B.F. Direct Asymmetric syn-Aldol Reactions of Linear Aliphatic Ketones with Primary Amino Acid-Derived Diamines. Adv. Synth. Catal. 2010, 352, 2421–2426. [Google Scholar] [CrossRef]
- Bisticha, A.; Triandafillidi, I.; Kokotos, C.G. tert-Butyl esters of peptides as organocatalysts for the asymmetric aldol reaction. Tetrahedron Asymmetry 2015, 26, 102–108. [Google Scholar] [CrossRef]
- Larionova, N.A.; Kucherenko, A.S.; Siyutkin, D.E.; Zlotin, S.G. (S)-Threonine/α,α-(S)-diphenylvalinol-derived chiral ionic liquid: An immobilized organocatalyst for asymmetric syn-aldol reactions. Tetrahedron 2011, 67, 1948–1954. [Google Scholar] [CrossRef]
- Gerasimchuk, V.V.; Kucherenko, A.S.; Fakhrutdinov, A.N.; Medvedev, M.G.; Nelyubina, Y.V.; Zlotin, S.G. Towards Sustainable Amino Acid Derived Organocatalysts for Asymmetric syn-Aldol Reactions. Eur. J. Org. Chem. 2017, 2017, 2540–2544. [Google Scholar] [CrossRef]
- Sarkar, D.; Harman, K.; Ghosh, S.; Headley, A.D. Chiral amine organocatalysts for the syn-aldol reaction involving substituted benzaldehydes and hydroxyacetone. Tetrahedron Asymmetry 2011, 22, 1051–1054. [Google Scholar] [CrossRef]
- Gavendova, M.; Lennon, C.M.; Coffey, L.; Manesiotis, P.; Kinsella, M. Novel β-amino Amide Organocatalysts for the Synthesis of Pharmaceutically Relevant Oxindoles. ChemistrySelect 2019, 4, 8246–8254. [Google Scholar] [CrossRef]
- Tsogoeva, S.B. Recent Advances in Asymmetric Organocatalytic 1,4-Conjugate Additions. Eur. J. Org. Chem. 2007, 2007, 1701–1716. [Google Scholar] [CrossRef]
- Almasi, D.; Alonso, D.A.; Najera, C. Organocatalytic asymmetric conjugate additions. Tetrahedron Asymmetry 2007, 18, 299–365. [Google Scholar] [CrossRef]
- Vicario, J.L.; Badia, D.; Carrillo, L. Organocatalytic Enantioselective Michael and Hetero-Michael Reactions. Synthesis 2007, 2007, 2065–2092. [Google Scholar] [CrossRef]
- Thirumalaikumar, M. Enantioselective Michael Addition Reactions. Org. Prep. Proced. Int. 2011, 43, 67–129. [Google Scholar] [CrossRef]
- Yong, Z.; Wei, W. Recent advances in organocatalytic asymmetric Michael reactions. Catal. Sci. Technol. 2012, 2, 42–53. [Google Scholar] [CrossRef]
- Nising, C.F.; Brase, S. Recent developments in the field of oxa-Michael reactions. Chem. Soc. Rev. 2012, 41, 988–999. [Google Scholar] [CrossRef]
- Tokoroyama, T. Discovery of the Michael Reaction. Eur. J. Org. Chem. 2010, 2010, 2009–2016. [Google Scholar] [CrossRef]
- O’Reilly, R.A. The Stereoselective Interaction of Warfarin and Metronidazole in Man. N. Engl. J. Med. 1976, 295, 354–357. [Google Scholar] [CrossRef]
- Wingard, L.B.; O’Reilly, R.A.; Levy, G. Pharmacokinetics of warfarin enantiomers: A search for intrasubject correlations. Clin. Pharmacol. Ther. 1978, 23, 212–217. [Google Scholar] [CrossRef]
- Rang, H.P.; Dale, M.M.; Ritter, J.M.; Gardner, P. Pharmacology, 4th ed.; Churchill Livingstone: London, UK, 2001. [Google Scholar]
- Kumagai, J.; Kohari, Y.; Seki, C.; Uwai, K.; Okuyama, Y.; Kwon, E.; Nakano, H. Chiral Primary Amino Amide Alcohol Organocatalyst for the Asymmetric Michael Addition of 4-Hydroxycoumarin with α, β-Unsaturated Ketones. Heterocycles 2015, 90, 1124–1134. [Google Scholar] [CrossRef]
- Owolabi, I.A.; Chennapuram, M.; Seki, C.; Okuyama, Y.; Kwon, E.; Uwai, K.; Tokiwa, M.; Takeshita, M.; Nakano, H. Amino amide organocatalysts for asymmetric michael addition of β-Keto esters with β-nitroolefins. Bull. Chem. Soc. Jpn. 2019, 92, 696–701. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.D. Organocatalytic asymmetric Michael addition of aldehydes and ketones to nitroalkenes catalyzed by adamantoyl l-prolinamide. RSC Adv. 2015, 5, 5863–5874. [Google Scholar] [CrossRef]
- Strecker, A. Ueber die künstliche Bildung der Milchsäure und einen neuen, dem Glycocoll homologen Körper. Chem. Pharm. 1850, 75, 27–45. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Liu, X.; Feng, X. Asymmetric Strecker Reactions. Chem. Rev. 2011, 111, 6947–6983. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.H.; Saravanan, S.; Kureshy, R.I.; Abdi, S.H.R.; Bajaj, H.C. Mn(III) salen complexes-catalyzed enantioselective addition of trimethyl silylcyanide to N-benzylimines in the presence of 4-phenyl pyridine-N-oxide as an additive. Tetrahedron Asymmetry 2010, 21, 2076–2080. [Google Scholar] [CrossRef]
- Khan, N.H.; Saravanan, S.; Kureshy, R.I.; Abdi, S.H.R.; Sadhukhan, A.; Bajaj, H.C. Asymmetric addition of trimethylsilylcyanide to N-benzylimines catalyzed by recyclable chiral dimeric V(V) salen complex. J. Organomet. Chem. 2010, 695, 1133–1137. [Google Scholar] [CrossRef]
- Seayad, A.M.; Ramalingam, B.; Yoshinaga, K.; Nagata, T.; Chai, C.L.L. Highly Enantioselective Titanium-Catalyzed Cyanation of Imines at Room Temperature. Org. Lett. 2010, 12, 264–267. [Google Scholar] [CrossRef]
- Merino, P.; López, M.E.M.; Tejero, T.; Herrera, R.P. Organocatalyzed Strecker reactions. Tetrahedron 2009, 65, 1219–1234. [Google Scholar] [CrossRef]
- Saravanan, S.; Sadhukhan, A.; Khan, N.-U.H.; Kureshy, R.I.; Abdi, S.H.R.; Bajaj, H.C. C2-Symmetric Recyclable Organocatalyst for Enantioselective Strecker Reaction for the Synthesis of α-Amino Acid and Chiral Diamine- an Intermediate for APN Inhibitor. J. Org. Chem. 2012, 77, 4375–4384. [Google Scholar] [CrossRef]
- Saravanan, S.; Khan, N.H.; Kureshy, R.I.; Abdi, S.H.R.; Bajaja, H.C. Small Molecule as a Chiral Organocatalyst for Asymmetric Strecker Reaction. ACS Catal. 2013, 3, 2873–2880. [Google Scholar] [CrossRef]
- Ghosh, D.; Sahu, D.; Saravanan, S.; Abdi, S.H.R.; Ganguly, B.; Khan, N.H.; Kureshya, R.I.; Bajaja, H.C. Synthetically amenable amide derivatives of tosylated-amino acids as organocatalysts for enantioselective allylation of aldehydes: Computational rationale for enantioselectivity. Org. Biomol. Chem. 2013, 11, 3451–3460. [Google Scholar] [CrossRef]
- Liu, X.W.; Wang, C.; Yan, Y.; Wang, Y.Q.; Sun, J. An Organocatalyst Bearing Stereogenic Carbon and Sulfur Centers as an Efficient Promoter for Enantioselective Hydrosilylation of 1,4-Benzooxazines. J. Org. Chem. 2013, 78, 6276–6280. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Li, L.; Yi, L.; Da, C.S.; Zhou, Y.F. Direct asymmetric N-specific reaction of nitrosobenzene with aldehydes catalyzed by a chiral primary amine-based organocatalyst. Chirality 2011, 23, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Kureshy, R.I.; Saravanan, S.; Verma, S.; Jakhar, A.; Khan, N.H.; Abdi, S.H.R.; Bajaj, H.C. Unravelling a New Class of Chiral Organocatalyst for Asymmetric Ring-Opening Reaction of Meso Epoxides with Anilines. Org. Lett. 2014, 16, 2798–2801. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, C.; Zhoub, L.; Sun, J. l-Pipecolinic acid derived Lewis base organocatalyst for asymmetric reduction of N-aryl imines by trichlorosilane: Effects of the side amide group on catalytic performances. Org. Biomol. Chem. 2013, 11, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Javle, B.R.; Kinage, A.K. Chiral Amino-Acid-Amide Based Ionic Liquids as a Stereoselective Organocatalyst in Asymmetric Transfer Hydrogenation of Acetophenone at Room-Temperature. ChemistrySelect 2018, 3, 2623–2625. [Google Scholar] [CrossRef]























































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reddy, U.V.S.; Anusha, B.; Begum, Z.; Seki, C.; Okuyama, Y.; Tokiwa, M.; Tokiwa, S.; Takeshita, M.; Nakano, H. Catalytic Efficiency of Primary α-Amino Amides as Multifunctional Organocatalysts in Recent Asymmetric Organic Transformations. Catalysts 2022, 12, 1674. https://doi.org/10.3390/catal12121674
Reddy UVS, Anusha B, Begum Z, Seki C, Okuyama Y, Tokiwa M, Tokiwa S, Takeshita M, Nakano H. Catalytic Efficiency of Primary α-Amino Amides as Multifunctional Organocatalysts in Recent Asymmetric Organic Transformations. Catalysts. 2022; 12(12):1674. https://doi.org/10.3390/catal12121674
Chicago/Turabian StyleReddy, Ummareddy Venkata Subba, Bheemreddy Anusha, Zubeda Begum, Chigusa Seki, Yuko Okuyama, Michio Tokiwa, Suguru Tokiwa, Mitsuhiro Takeshita, and Hiroto Nakano. 2022. "Catalytic Efficiency of Primary α-Amino Amides as Multifunctional Organocatalysts in Recent Asymmetric Organic Transformations" Catalysts 12, no. 12: 1674. https://doi.org/10.3390/catal12121674