
Solvent Coordination Effect on Copper-Based Molecular Catalysts for Controlled Radical Polymerization
 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
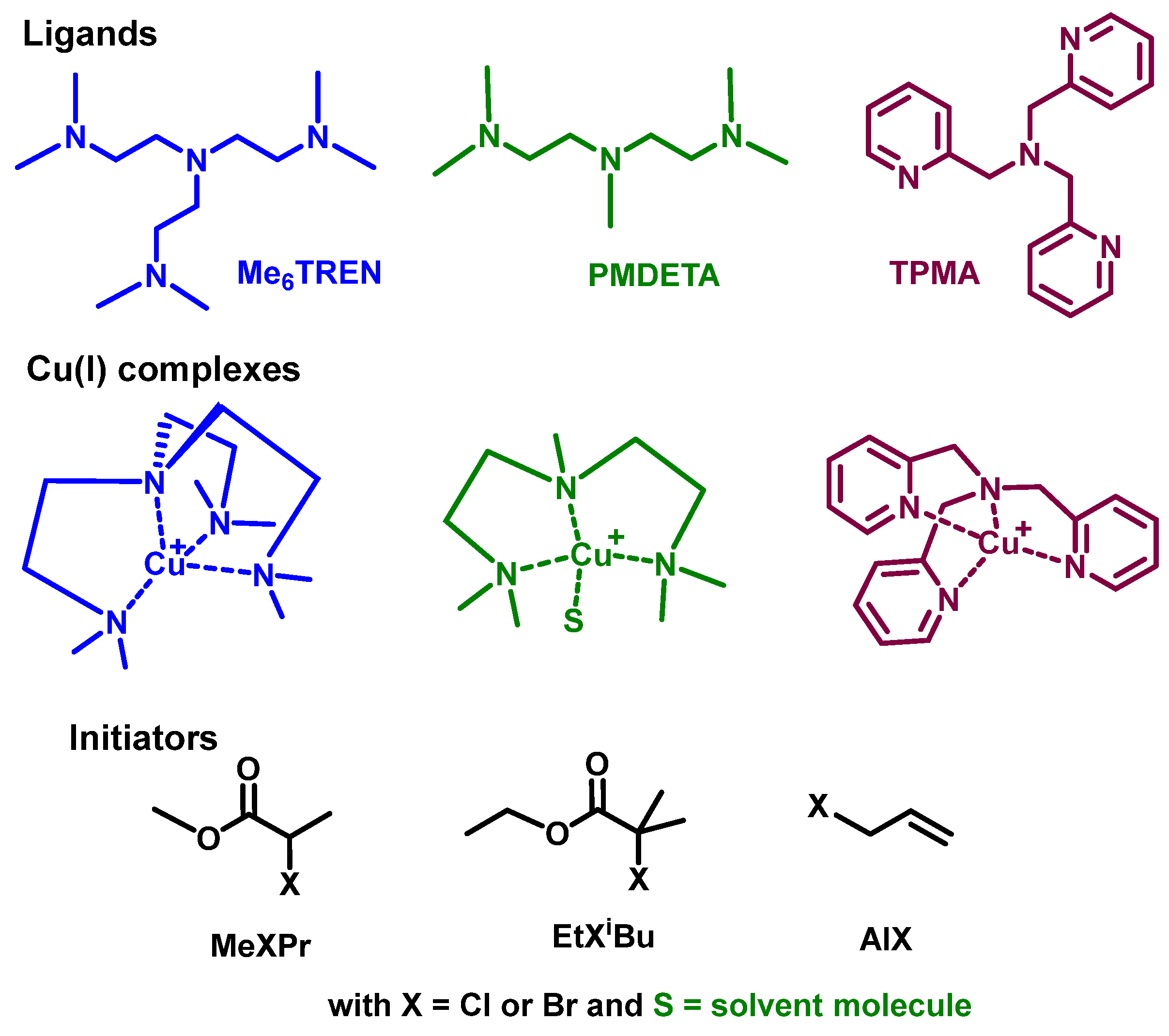
2.1. Oxidation of CuI Complexes by the X• Radical
2.2. Solvent Coordination to Copper Complexes
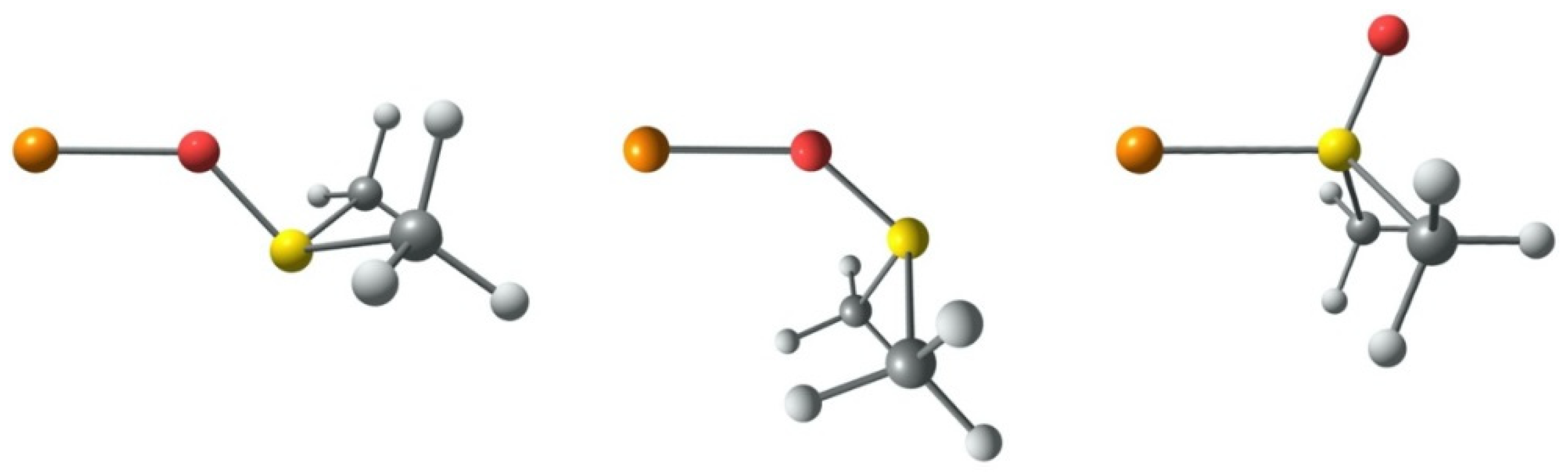
2.2.1. Analysis of Solvent Coordination to Copper(I/II) Centers
2.2.2. CuI/IIL(S)]+/2+ Complexes
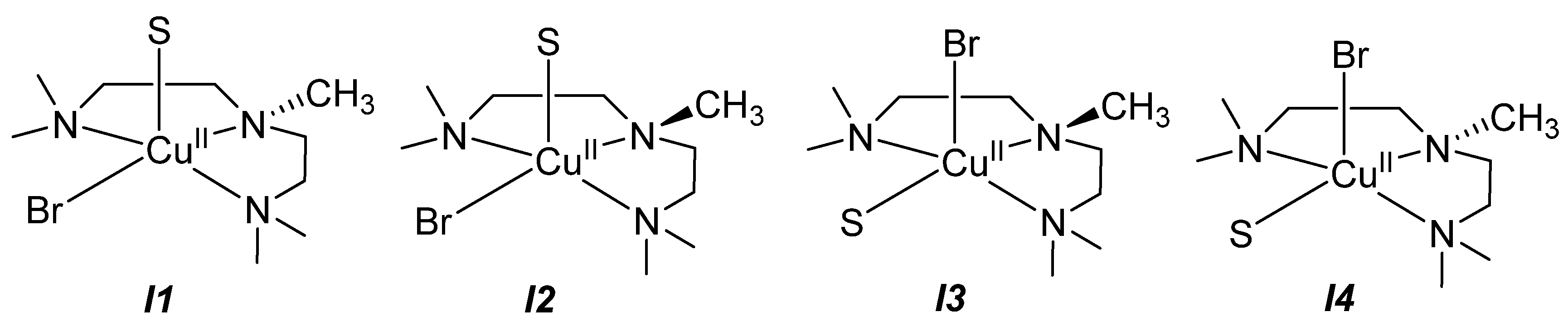
2.2.3. Analysis of the Isomers [CuII(PMDETA)X(S)]+
2.3. Reactions including the Explicit Solvent Coordination
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Matyjaszewski, K.; Xia, J. Atom Transfer Radical Polymerization. Chem. Rev. 2001, 101, 2921–2990. [Google Scholar] [CrossRef] [PubMed]
- Matyjaszewski, K. Atom Transfer Radical Polymerization (ATRP): Current Status and Future Perspectives. Macromolecules 2012, 45, 4015–4039. [Google Scholar] [CrossRef]
- Matyjaszewski, K. Advanced Materials by Atom Transfer Radical Polymerization. Adv. Mater. 2018, 30, 1706441. [Google Scholar] [CrossRef] [PubMed]
- Ribelli, T.G.; Lorandi, F.; Fantin, M.; Matyjaszewski, K. Atom Transfer Radical Polymerization: Billion Times More Active Catalysts and New Initiation Systems. Macromol. Rapid Commun. 2019, 40, 1800616. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Coote, M.L.; Gennaro, A. Ab Initio Evaluation of the Thermodynamic and Electrochemical Properties of Alkyl Halides and Radicals and Polymerization. J. Am. Chem. Soc. 2008, 130, 12762–12774. [Google Scholar] [CrossRef] [PubMed]
- De Paoli, P.; Isse, A.A.; Bortolamei, N.; Gennaro, A. New Insights into the Mechanism of Activation of Atom Transfer Radical Polymerization by Cu(I) Complexes. Chem. Commun. 2011, 47, 3580–3582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isse, A.A.; Bortolamei, N.; De Paoli, P.; Gennaro, A. On the Mechanism of Activation of Copper-Catalyzed Atom Transfer Radical Polymerization. Electrochim. Acta 2013, 110, 655–662. [Google Scholar] [CrossRef]
- Tang, W.; Tsarevsky, N.V.; Matyjaszewski, K. Determination of Equilibrium Constants for Atom Transfer Radical Polymerization. J. Am. Chem. Soc. 2006, 128, 1598–1604. [Google Scholar] [CrossRef]
- Tang, W.; Kwak, Y.; Braunecker, W.; Tsarevsky, N.V.; Coote, M.L.; Matyjaszewski, K. Understanding Atom Transfer Radical Polymerization: Effect of Ligand and Initiator Structures on the Equilibrium Constants. J. Am. Chem. Soc. 2008, 130, 10702–10713. [Google Scholar] [CrossRef]
- Pan, X.; Fantin, M.; Yuan, F.; Matyjaszewski, K. Externally Controlled Atom Transfer Radical Polymerization. Chem. Soc. Rev. 2018, 47, 5457–5490. [Google Scholar] [CrossRef]
- Min, K.; Gao, H.; Matyjaszewski, K. Use of Ascorbic Acid as Reducing Agent for Synthesis of Well-Defined Polymers by ARGET ATRP. Macromolecules 2007, 40, 1789–1791. [Google Scholar] [CrossRef]
- Simakova, A.; Averick, S.E.; Konkolewicz, D.; Matyjaszewski, K. Aqueous ARGET ATRP. Macromolecules 2012, 45, 6371–6379. [Google Scholar] [CrossRef]
- Mendonça, P.V.; Ribeiro, J.P.M.; Abreu, C.M.R.; Guliashvili, T.; Serra, A.C.; Coelho, J.F.J. Thiourea Dioxide as a Green and Affordable Reducing Agent for the ARGET ATRP of Acrylates, Methacrylates, Styrene, Acrylonitrile, and Vinyl Chloride. ACS Macro Lett. 2019, 8, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Abreu, C.M.R.; Serra, A.C.; Popov, A.V.; Matyjaszewski, K.; Guliashvili, T.; Coelho, J.F.J. Ambient Temperature Rapid SARA ATRP of Acrylates and Methacrylates in Alcohol–Water Solutions Mediated by a Mixed Sulfite/Cu(II)Br2 Catalytic System. Polym. Chem. 2013, 4, 5629–5636. [Google Scholar] [CrossRef]
- Mendes, J.P.; Branco, F.; Abreu, C.M.R.; Mendonça, P.V.; Serra, A.C.; Popov, A.V.; Guliashvili, T.; Coelho, J.F.J. Sulfolane: An Efficient and Universal Solvent for Copper-Mediated Atom Transfer Radical (Co)Polymerization of Acrylates, Methacrylates, Styrene, and Vinyl Chloride. ACS Macro Lett. 2014, 3, 858–861. [Google Scholar] [CrossRef]
- Lorandi, F.; Fantin, M.; Isse, A.A.; Gennaro, A. RDRP in the Presence of Cu0: The Fate of Cu(I) Proves the Inconsistency of SET-LRP Mechanism. Polymer 2015, 72, 238–245. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Jakubowski, W.; Min, K.; Tang, W.; Huang, J.; Braunecker, W.A.; Tsarevsky, N.V. Diminishing Catalyst Concentration in Atom Transfer Radical Polymerization with Reducing Agents. Proc. Natl. Acad. Sci. USA 2006, 103, 15309–15314. [Google Scholar] [CrossRef] [Green Version]
- Konkolewicz, D.; Magenau, A.J.D.; Averick, S.E.; Simakova, A.; He, H.; Matyjaszewski, K. ICAR ATRP with Ppm Cu Catalyst in Water. Macromolecules 2012, 45, 4461–4468. [Google Scholar] [CrossRef]
- Tasdelen, M.A.; Uygun, M.; Yagci, Y. Photoinduced Controlled Radical Polymerization. Macromol. Rapid Commun. 2011, 32, 58–62. [Google Scholar] [CrossRef]
- Konkolewicz, D.; Schröder, K.; Buback, J.; Bernhard, S.; Matyjaszewski, K. Visible Light and Sunlight Photoinduced ATRP with Ppm of Cu Catalyst. ACS Macro Lett. 2012, 1, 1219–1223. [Google Scholar] [CrossRef]
- Anastasaki, A.; Nikolaou, V.; Zhang, Q.; Burns, J.; Samanta, S.R.; Waldron, C.; Haddleton, A.J.; McHale, R.; Fox, D.; Percec, V.; et al. Copper(II)/Tertiary Amine Synergy in Photoinduced Living Radical Polymerization: Accelerated Synthesis of ω-Functional and α,ω-Heterofunctional Poly(Acrylates). J. Am. Chem. Soc. 2014, 136, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Magenau, A.J.D.; Strandwitz, N.C.; Gennaro, A.; Matyjaszewski, K. Electrochemically Mediated Atom Transfer Radical Polymerization. Science 2011, 332, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Fantin, M.; Isse, A.A.; Gennaro, A.; Matyjaszewski, K. Understanding the Fundamentals of Aqueous ATRP and Defining Conditions for Better Control. Macromolecules 2015, 48, 6862–6875. [Google Scholar] [CrossRef]
- Lorandi, F.; Fantin, M.; Isse, A.A.; Gennaro, A. Electrochemically Mediated Atom Transfer Radical Polymerization of N-Butyl Acrylate on Non-Platinum Cathodes. Polym. Chem. 2016, 7, 5357–5365. [Google Scholar] [CrossRef]
- Chmielarz, P.; Fantin, M.; Park, S.; Isse, A.A.; Gennaro, A.; Magenau, A.J.D.; Sobkowiak, A.; Matyjaszewski, K. Electrochemically Mediated Atom Transfer Radical Polymerization (eATRP). Prog. Polym. Sci. 2017, 69, 47–78. [Google Scholar] [CrossRef]
- Isse, A.A.; Gennaro, A. Electrochemistry for Atom Transfer Radical Polymerization. Chem. Rec. 2021, 21, 2203–2222. [Google Scholar] [CrossRef]
- Zaborniak, I.; Chmielarz, P. Ultrasound-Mediated Atom Transfer Radical Polymerization (ATRP). Materials 2019, 12, 3600. [Google Scholar] [CrossRef] [Green Version]
- Kaur, A.; Ribelli, T.G.; Schröder, K.; Matyjaszewski, K.; Pintauer, T. Properties and ATRP Activity of Copper Complexes with Substituted Tris(2-Pyridylmethyl)Amine-Based Ligands. Inorg. Chem. 2015, 54, 1474–1486. [Google Scholar] [CrossRef]
- Melville, J.N.; Bernhardt, P.V. Electrochemical Exploration of Active Cu-Based Atom Transfer Radical Polymerization Catalysis through Ligand Modification. Inorg. Chem. 2021, 60, 9709–9719. [Google Scholar] [CrossRef]
- Wang, Y.; Kwak, Y.; Buback, J.; Buback, M.; Matyjaszewski, K. Determination of ATRP Equilibrium Constants under Polymerization Conditions. ACS Macro Lett. 2012, 1, 1367–1370. [Google Scholar] [CrossRef]
- Morick, J.; Buback, M.; Matyjaszewski, K. Effect of Pressure on Activation–Deactivation Equilibrium Constants for ATRP of Methyl Methacrylate. Macromol. Chem. Phys. 2012, 213, 2287–2292. [Google Scholar] [CrossRef]
- Horn, M.; Matyjaszewski, K. Solvent Effects on the Activation Rate Constant in Atom Transfer Radical Polymerization. Macromolecules 2013, 46, 3350–3357. [Google Scholar] [CrossRef]
- Smolne, S.; Buback, M. Kinetic Investigations of Cu-Mediated ATRP in Aqueous Solution. Macromol. Chem. Phys. 2015, 216, 894–902. [Google Scholar] [CrossRef]
- Pavan, P.; Lorandi, F.; De Bon, F.; Gennaro, A.; Isse, A.A. Enhancement of the Rate of Atom Transfer Radical Polymerization in Organic Solvents by Addition of Water: An Electrochemical Study. ChemElectroChem 2021, 8, 2450–2458. [Google Scholar] [CrossRef]
- Coullerez, G.; Malmström, E.; Jonsson, M. Solvent Effects on the Redox Properties of Cu Complexes Used as Mediators in Atom Transfer Radical Polymerization. J. Phys. Chem. A 2006, 110, 10355–10360. [Google Scholar] [CrossRef]
- Gillies, M.B.; Matyjaszewski, K.; Norrby, P.-O.; Pintauer, T.; Poli, R.; Richard, P. A DFT Study of R−X Bond Dissociation Enthalpies of Relevance to the Initiation Process of Atom Transfer Radical Polymerization. Macromolecules 2003, 36, 8551–8559. [Google Scholar] [CrossRef]
- Braunecker, W.A.; Brown, W.C.; Morelli, B.C.; Tang, W.; Poli, R.; Matyjaszewski, K. Origin of Activity in Cu-, Ru-, and Os-Mediated Radical Polymerization. Macromolecules 2007, 40, 8576–8585. [Google Scholar] [CrossRef]
- Isse, A.A.; Gennaro, A.; Lin, C.Y.; Hodgson, J.L.; Coote, M.L.; Guliashvili, T. Mechanism of Carbon−Halogen Bond Reductive Cleavage in Activated Alkyl Halide Initiators Relevant to Living Radical Polymerization: Theoretical and Experimental Study. J. Am. Chem. Soc. 2011, 133, 6254–6264. [Google Scholar] [CrossRef]
- Ribelli, T.G.; Wahidur Rahaman, S.M.; Daran, J.C.; Krys, P.; Matyjaszewski, K.; Poli, R. Effect of Ligand Structure on the CuII–R OMRP Dormant Species and Its Consequences for Catalytic Radical Termination in ATRP. Macromolecules 2016, 49, 7749–7757. [Google Scholar] [CrossRef]
- Braunecker, W.A.; Tsarevsky, N.V.; Gennaro, A.; Matyjaszewski, K. Thermodynamic Components of the Atom Transfer Radical Polymerization Equilibrium: Quantifying Solvent Effects. Macromolecules 2009, 42, 6348–6360. [Google Scholar] [CrossRef]
- Lanzalaco, S.; Fantin, M.; Scialdone, O.; Galia, A.; Isse, A.A.; Gennaro, A.; Matyjaszewski, K. Atom Transfer Radical Polymerization with Different Halides (F, Cl, Br, and I): Is the Process “Living” in the Presence of Fluorinated Initiators? Macromolecules 2017, 50, 192–202. [Google Scholar] [CrossRef]
- Hildenbrand, D.L.; Lau, K.H. Dissociation Energies of the Cu and Ag Monohalides and of Ni Monofluoride. J. Phys. Chem. A 2006, 110, 11886–11889. [Google Scholar] [CrossRef] [PubMed]
- Isse, A.A.; Lin, C.Y.; Coote, M.L.; Gennaro, A. Estimation of Standard Reduction Potentials of Halogen Atoms and Alkyl Halides. J. Phys. Chem. B 2011, 115, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Zerk, T.J.; Bernhardt, P.V. Solvent Dependent Anion Dissociation Limits Copper(I) Catalysed Atom Transfer Reactions. Dalton Trans. 2013, 42, 11683–11694. [Google Scholar] [CrossRef] [Green Version]
- Zerk, T.J.; Martinez, M.; Bernhardt, P.V. A Kinetico-Mechanistic Study on CuII Deactivators Employed in Atom Transfer Radical Polymerization. Inorg. Chem. 2016, 55, 9848–9857. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Odarich, I.A.; Pavlishchuk, A.V.; Kalibabchuk, V.A.; Haukka, M. Crystal Structure of μ-Oxalodi hydroxamato-Bis [(2,2′-Bipyrid yl)(Di methyl Sulfoxide-ΚO)Copper(II)] Bis (Perchlorate). Acta Crystallogr. Sect. Crystallogr. Commun. 2016, 72, 147–150. [Google Scholar] [CrossRef] [Green Version]
- Giziroglu, E.; Aygün, M.; Sarikurkcu, C.; Kazar, D.; Orhan, N.; Firinci, E.; Soyleyici, H.C.; Gokcen, C. Synthesis, Characterization and Antioxidant Activity of New Dibasic Tridentate Ligands: X-ray Crystal Structures of DMSO Adducts of 1,3-Dimethyl-5-Acetyl-Barbituric Acid o-Hydroxybenzoyl Hydrazone Copper(II) Complex. Inorg. Chem. Commun. 2013, 36, 199–205. [Google Scholar] [CrossRef]
- Pintauer, T.; Matyjaszewski, K. Structural Aspects of Copper Catalyzed Atom Transfer Radical Polymerization. Coord. Chem. Rev. 2005, 249, 1155–1184. [Google Scholar] [CrossRef]
- Scott, M.J.; Holm, R.H. Molecular Assemblies Containing Linear and Bent [FeIII-CN-CuII] Bridge Units: Synthesis, Structures, and Relevance to Cyanide-Inhibited Heme-Copper Oxidases. J. Am. Chem. Soc. 1994, 116, 11357–11367. [Google Scholar] [CrossRef]
- Olguín, J.; Bernès, S.; Gasque, L. Fluoride Ion as Ligand and Hydrogen Bond Acceptor: Crystal Structures of Two Dinuclear Cuii Complexes Built on a Diazecine Template. Crystals 2012, 2, 1357–1365. [Google Scholar] [CrossRef] [Green Version]
- Margraf, G.; Bats, J.W.; Wagner, M.; Lerner, H.-W. Copper(II) PMDTA and Copper(II) TMEDA Complexes: Precursors for the Synthesis of Dinuclear Dicationic Copper(II) Complexes. Inorg. Chim. Acta 2005, 358, 1193–1203. [Google Scholar] [CrossRef]
- Marenich, A.V.; Olson, R.M.; Kelly, C.P.; Cramer, C.J.; Truhlar, D.G. Self-Consistent Reaction Field Model for Aqueous and Nonaqueous Solutions Based on Accurate Polarized Partial Charges. J. Chem. Theory Comput. 2007, 3, 2011–2033. [Google Scholar] [CrossRef] [PubMed]
- Harvey, J.N.; Himo, F.; Maseras, F.; Perrin, L. Scope and Challenge of Computational Methods for Studying Mechanism and Reactivity in Homogeneous Catalysis. ACS Catal. 2019, 9, 6803–6813. [Google Scholar] [CrossRef]
- Gaussian 09 Citation|Gaussian.Com. Available online: https://gaussian.com/g09citation/ (accessed on 25 May 2022).
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Skyner, R.E.; McDonagh, J.L.; Groom, C.R.; van Mourik, T.; Mitchell, J.B.O. A Review of Methods for the Calculation of Solution Free Energies and the Modelling of Systems in Solution. Phys. Chem. Chem. Phys. 2015, 17, 6174–6191. [Google Scholar] [CrossRef] [Green Version]
- O’boyle, N.M.; Tenderholt, A.L.; Langner, K.M. Cclib: A Library for Package-Independent Computational Chemistry Algorithms. J. Comput. Chem. 2008, 29, 839–845. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
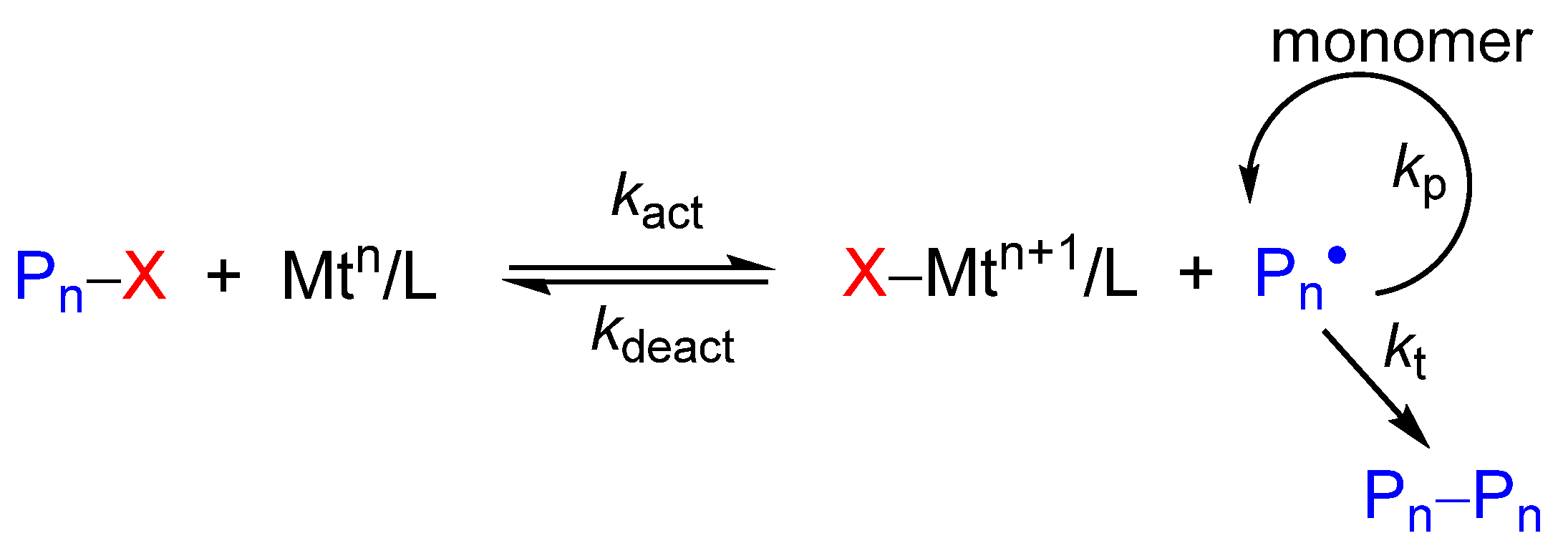{kind=link}
{kind=link}
| Reaction | ΔEr | ΔGr | ||
|---|---|---|---|---|
| Gas phase | Gas phase | In MeCN 1 | In DMSO 1 | |
| [CuI(Me6TREN)]+ + Cl• → [CuII(Me6TREN)Cl]+ | −62.2 | −51.8 | −59.2 | −59.3 |
| [CuI(Me6TREN)]+ + Br• → [CuII(Me6TREN)Br]+ | −47.1 | −37.2 | −43.8 | −43.9 |
| [CuI(PMDETA)]+ + Cl• → [CuII(PMDETA)Cl]+ | −59.8 | −49.6 | −58.5 | −58.6 |
| [CuI(PMDETA)]+ + Br• → [CuII(PMDETA)Br]+ | −44.9 | −35.5 | −43.3 | −43.4 |
| [CuI(TPMA)]+ + Cl• → [CuII(TPMA)Cl]+ | −59.1 | −48.9 | −57.1 | −57.1 |
| [CuI(TPMA)]+ + Br• → [CuII(TPMA)Br]+ | −44.5 | −34.6 | −42.0 | −42.0 |
| Reaction | ΔEr | ΔGr | ||
|---|---|---|---|---|
| Gas Phase | Gas Phase | In MeCN | In DMSO | |
| [CuI(Me6TREN)]+ + AlCl [CuII(Me6TREN)Cl]+ + Al• | 7.3 | 6.1 | 1.0 | 0.9 |
| [CuI(Me6TREN)]+ + AlBr [CuII(Me6TREN)Br]+ + Al• | 8.2 | 7.0 | 2.5 | 2.4 |
| [CuI(Me6TREN)]+ + EtCliBu [CuII(Me6TREN)Cl]+ + EtiBu• | 10.7 | 7.4 | 1.4 | 1.3 |
| [CuI(Me6TREN)]+ + EtBriBu [CuII(Me6TREN)Br]+ + EtiBu• | 11.1 | 7.5 | 2.0 | 2.0 |
| [CuI(Me6TREN)]+ + MeClPr [CuII(Me6TREN)Cl]+ + MePr• | 12.1 | 10.3 | 4.4 | 4.3 |
| [CuI(Me6TREN)]+ + MeBrPr [CuII(Me6TREN)Br]+ + MePr• | 13.3 | 11.4 | 6.1 | 6.0 |
| [CuI(PMDETA)]+ + AlCl [CuII(PMDETA)Cl]+ + Al• | 9.7 | 8.3 | 1.8 | 1.7 |
| [CuI(PMDETA)]+ + AlBr [CuII(PMDETA)Br]+ + Al• | 10.4 | 8.7 | 3.0 | 2.9 |
| [CuI(PMDETA)]+ + EtCliBu [CuII(PMDETA)Cl]+ + EtiBu• | 13.2 | 9.5 | 2.1 | 2.0 |
| [CuI(PMDETA)]+ + EtBriBu [CuII(PMDETA)Br]+ + EtiBu• | 13.3 | 9.2 | 2.6 | 2.5 |
| [CuI(PMDETA)]+ + MeClPr [CuII(PMDETA)Cl]+ + MePr• | 14.5 | 12.5 | 5.2 | 5.1 |
| [CuI(PMDETA)]+ + MeBrPr [CuII(PMDETA)Br]+ + MePr• | 15.5 | 13.0 | 6.6 | 6.5 |
| [CuI(TPMA)]+ + AlCl [CuII(TPMA)Cl]+ + Al• | 10.3 | 9.0 | 3.2 | 3.1 |
| [CuI(TPMA)]+ + AlBr [CuII(TPMA)Br]+ + Al• | 10.8 | 9.6 | 4.4 | 4.3 |
| [CuI(TPMA)]+ + EtCliBu [CuII(TPMA)Cl]+ + EtiBu• | 13.8 | 10.3 | 3.5 | 3.4 |
| [CuI(TPMA)]+ + EtBriBu [CuII(TPMA)Br]+ + EtiBu• | 13.7 | 10.0 | 4.0 | 3.9 |
| [CuI(TPMA)]+ + MeClPr [CuII(TPMA)Cl]+ + MePr• | 15.1 | 13.2 | 6.6 | 6.5 |
| [CuI(TPMA)]+ + MeBrPr [CuII(TPMA)Br]+ + MePr• | 15.9 | 13.9 | 8.0 | 7.9 |
| S | CuI | CuII | ||
|---|---|---|---|---|
| Gas Phase | In Solvent 1 | Gas Phase | In Solvent 1 | |
| MeCN (end-on) | −51.7 | −24.8 | −161.6 | −38.7 |
| DMSO-κOα | −57.5 | −23.7 | −208.2 | −86.0 |
| DMSO-κOβ | −56.0 | −24.2 | - 2 | - 2 |
| DMSO-κS | −35.8 | −15.2 | −183.6 | −71.2 |
| Reaction | ΔEr Gas Phase | ΔGr Gas Phase | ΔGr In Solution 1 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| MeCN | DMSO-κO2 | DMSO-κS | MeCN | DMSO-κO2 | DMSO-κS | MeCN | DMSO-κO2 | DMSO-κS | |
| [CuI(Me6TREN)]+ + S [CuI(Me6TREN)(S)]+ | −17.9 | −20.8 | −15.2 | −7.5 | −9.4 | −2.6 | −4.4 | −2.2 | 0.4 |
| [CuI(PMDETA]+ + S [CuI(PMDETA)(S)]+ | −23.0 | −24.2 | −19.6 | −12.6 | −11.6 | −5.5 | −6.3 | −3.3 | −2.6 |
| [CuI(PMDETA)(S)]+ + S [CuI(PMDETA)(S)2]+ | −11.4 | −18.2 | - | −1.8 | −6.4 | - | 1.7 | 0.2 | - |
| [CuI(TPMA)]+ + S [CuI(TPMA)(S)]+ | −16.8 | −18.9 | −13.6 | −6.7 | −7.5 | −1.8 | −4.0 | −2.7 | −0.4 |
| [CuII(Me6TREN)]2+ + S [CuII(Me6TREN)(S)]2+ | −44.7 | −55.6 | −30.5 | −33.9 | −41.8 | −16.1 | −10.1 | −32.2 | −11.7 |
| [CuII(PMDETA)]2+ + S [CuII(PMDETA)(S)]2+ | −48.7 | −60.3 | - | −36.6 | −46.1 | - | −18.7 | −31.0 | - |
| [CuII(PMDETA)(S)]2+ + S [CuII(PMDETA)(S)2]2+ | −29.1 | −38.5 | - | −18.5 | −24.2 | - | −6.3 | −17.0 | - |
| [CuII(TPMA)]2+ + S [CuII(TPMA)(S)]2+ | −43.7 | −52.7 | −32.0 | −31.8 | −39.5 | −18.3 | −19.1 | −29.6 | −15.7 |
| Isomer | ||||||
|---|---|---|---|---|---|---|
| ΔEr Gas Phase | ΔGr Gas Phase | ΔGr In MeCN 1 | ΔEr Gas Phase | ΔGr Gas Phase | ΔGr In DMSO 1 | |
| I1 | −15.9 | −2.3 | 4.4 | −25.6 | −11.0 | −0.5 |
| I2 | −19.4 | −8.1 | −1.1 | −29.7 | −15.1 | −4.7 |
| I3 | −11.2 | 0.5 | 4.6 | −25.7 | −11.7 | −3.9 |
| I4 | −6.1 | 5.5 | 10.6 | −17.8 | −3.3 | 4.5 |
| Reaction | S = MeCN | S = DMSO | ||||
|---|---|---|---|---|---|---|
| ΔE Gas Phase | ΔGr Gas Phase | ΔGr 1 In Solution | ΔE Gas Phase | ΔGr Gas Phase | ΔGr 1 In Solution | |
| [CuI(Me6TREN)(S)]+ + AlCl [CuII(Me6TREN)Cl]+ + Al• + S | 25.2 | 13.6 | 4.7 | 28.1 | 15.4 | 3.8 |
| [CuI(Me6TREN)(S)]+ + AlBr [CuII(Me6TREN)Br]+ + Al• + S | 26.1 | 14.5 | 6.2 | 29.0 | 16.4 | 5.4 |
| [CuI(Me6TREN)(S)]+ + EtCliBu [CuII(Me6TREN)Cl]+ + EtiBu• + S | 28.6 | 14.9 | 5.1 | 31.5 | 16.7 | 4.2 |
| [CuI(Me6TREN)(S)]+ + EtBriBu [CuII(Me6TREN)Br]+ + EtiBu• + S | 29.0 | 15.0 | 5.8 | 31.9 | 16.9 | 4.9 |
| [CuI(Me6TREN)(S)]+ + MeClPr [CuII(Me6TREN)Cl]+ + MePr• + S | 30.0 | 17.8 | 8.1 | 32.9 | 19.6 | 7.3 |
| [CuI(Me6TREN)(S)]+ + MeBrPr [CuII(Me6TREN)Br]+ + MePr• + S | 31.2 | 18.8 | 9.8 | 34.1 | 20.7 | 9.0 |
| [CuI(PMDETA)(S)]+ + AlCl [CuII(PMDETA)Cl]+ + Al• + S | 32.7 | 20.9 | 9.4 | 33.9 | 19.9 | 5.8 |
| [CuI(PMDETA)(S)]+ + AlBr [CuII(PMDETA)Br]+ + Al• + S | 33.4 | 21.4 | 10.5 | 34.6 | 20.3 | 7.0 |
| [CuI(PMDETA)(S)]+ + EtCliBu [CuII(PMDETA)Cl]+ + EtiBu• + S | 36.1 | 22.2 | 9.7 | 37.3 | 21.2 | 6.2 |
| [CuI(PMDETA)(S)]+ + EtBriBu [CuII(PMDETA)Br]+ + EtiBu• + S | 36.3 | 21.8 | 10.1 | 37.5 | 20.8 | 6.6 |
| [CuI(PMDETA)(S)]+ + MeClPr [CuII(PMDETA)Cl]+ + MePr• + S | 37.5 | 25.1 | 12.8 | 38.7 | 24.1 | 9.2 |
| [CuI(PMDETA)(S)]+ + MeBrPr [CuII(PMDETA)Br]+ + MePr• + S | 38.5 | 25.7 | 14.1 | 39.7 | 24.7 | 10.6 |
| [CuI(TPMA)(S)]+ + AlCl [CuII(TPMA)Cl]+ + Al• + S | 27.1 | 15.7 | 7.3 | 29.2 | 16.4 | 5.9 |
| [CuI(TPMA)(S)]+ + AlBr [CuII(TPMA)Br]+ + Al• + S | 27.6 | 16.3 | 8.5 | 29.7 | 17.0 | 7.1 |
| [CuI(TPMA)(S)]+ + EtCliBu [CuII(TPMA)Cl]+ + EtiBu• + S | 30.6 | 17.0 | 7.6 | 32.6 | 17.7 | 6.2 |
| [CuI(TPMA)(S)]+ + EtBriBu [CuII(TPMA)Br]+ + EtiBu• + S | 30.5 | 16.8 | 8.1 | 32.6 | 17.5 | 6.7 |
| [CuI(TPMA)(S)]+ + MeClPr [CuII(TPMA)Cl]+ + MePr• + S | 31.9 | 19.9 | 10.7 | 34.0 | 20.6 | 9.3 |
| [CuI(TPMA)(S)]+ + MeBrPr [CuII(TPMA)Cl]+ + MePr• + S | 32.7 | 20.6 | 12.1 | 34.8 | 21.3 | 10.7 |
| [CuI(PMDETA)(S)]+ + AlCl [CuII(PMDETA)Cl(S)]+ + Al• | 15.0 | 12.4 | 5.8 | 2.7 | 4.6 | −1.0 |
| [CuI(PMDETA)(S)]+ + AlBr [CuII(PMDETA)Br(S)]+ + Al• | 14.0 | 14.3 | 9.4 | 4.9 | 5.2 | 2.4 |
| [CuI(PMDETA)(S)]+ + EtCliBu [CuII(PMDETA)Cl(S)]+ + EtiBu• | 18.5 | 13.6 | 6.1 | 6.1 | 5.9 | −0.7 |
| [CuI(PMDETA)(S)]+ + EtBriBu [CuII(PMDETA)Br(S)]+ + EtiBu• | 16.9 | 13.8 | 9.0 | 7.8 | 5.7 | 2.0 |
| [CuI(PMDETA)(S)]+ + MeClPr [CuII(PMDETA)Cl(S)]+ + MePr• | 19.8 | 16.6 | 9.2 | 7.5 | 8.8 | 2.4 |
| [CuI(PMDETA)(S)]+ + MeBrPr [CuII(PMDETA)Br(S)]+ + MePr• | 19.1 | 17.6 | 13.0 | 10.0 | 9.6 | 6.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Racioppi, S.; Orian, L.; Tubaro, C.; Gennaro, A.; Isse, A.A. Solvent Coordination Effect on Copper-Based Molecular Catalysts for Controlled Radical Polymerization. Catalysts 2022, 12, 1656. https://doi.org/10.3390/catal12121656
Racioppi S, Orian L, Tubaro C, Gennaro A, Isse AA. Solvent Coordination Effect on Copper-Based Molecular Catalysts for Controlled Radical Polymerization. Catalysts. 2022; 12(12):1656. https://doi.org/10.3390/catal12121656
Chicago/Turabian StyleRacioppi, Stefano, Laura Orian, Cristina Tubaro, Armando Gennaro, and Abdirisak Ahmed Isse. 2022. "Solvent Coordination Effect on Copper-Based Molecular Catalysts for Controlled Radical Polymerization" Catalysts 12, no. 12: 1656. https://doi.org/10.3390/catal12121656