
1-Methylimidazole as an Organic Catalyst for [3+3]-Cyclodimerization of Acylethynylpyrroles to Bis(acylmethylidene)dipyrrolo[1,2-a:1′,2′-d]pyrazines

Abstract
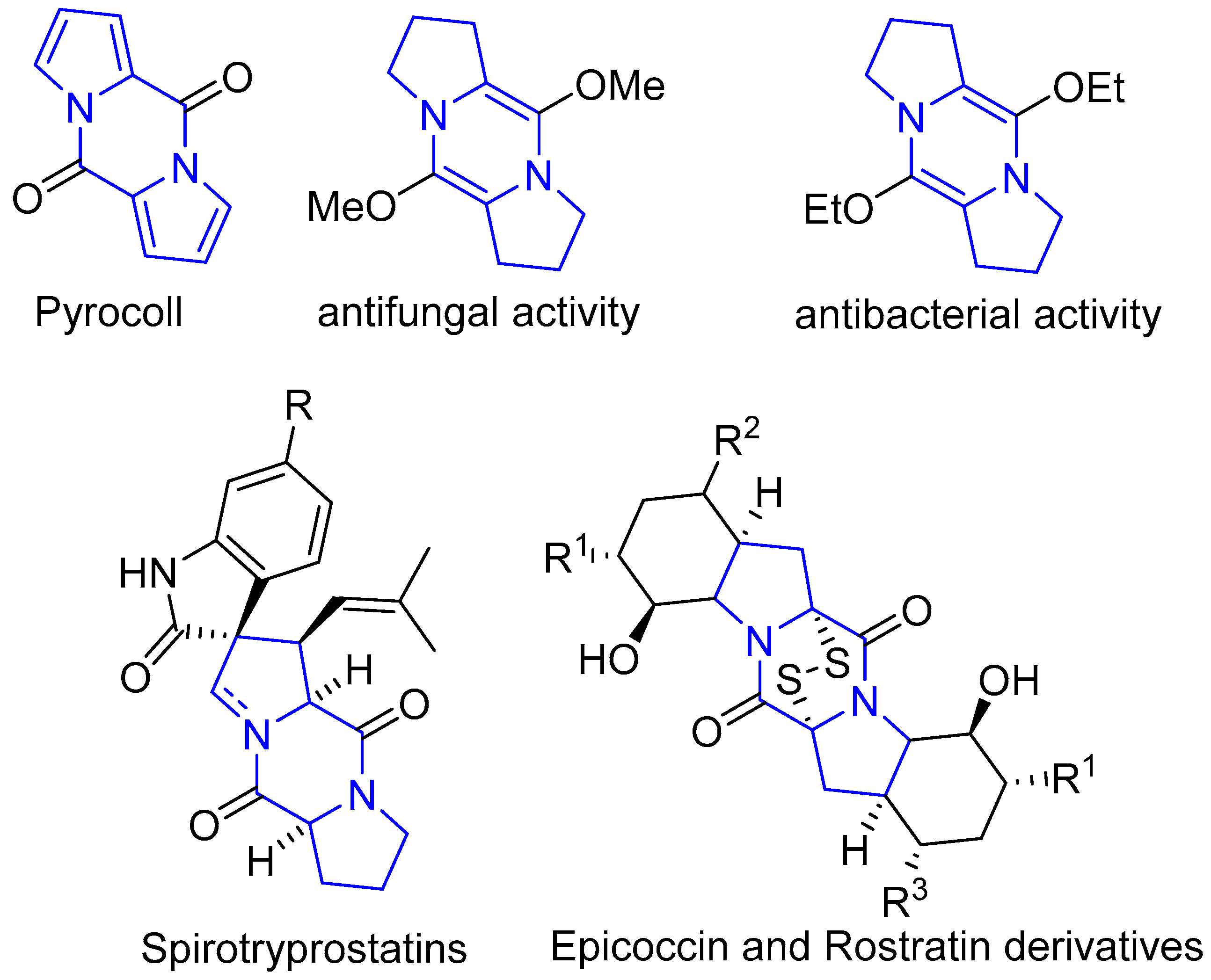
:1. Introduction
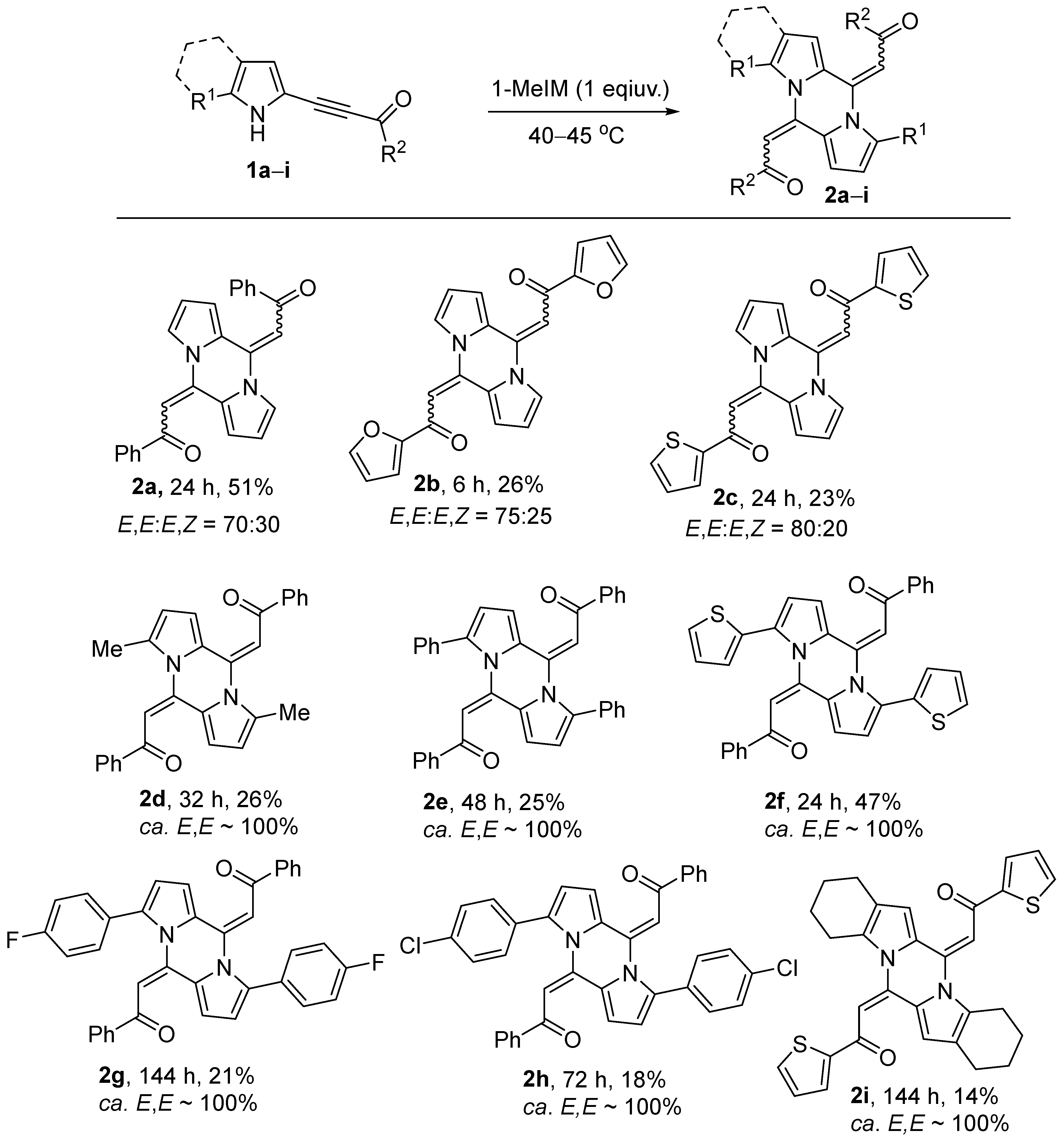
2. Results
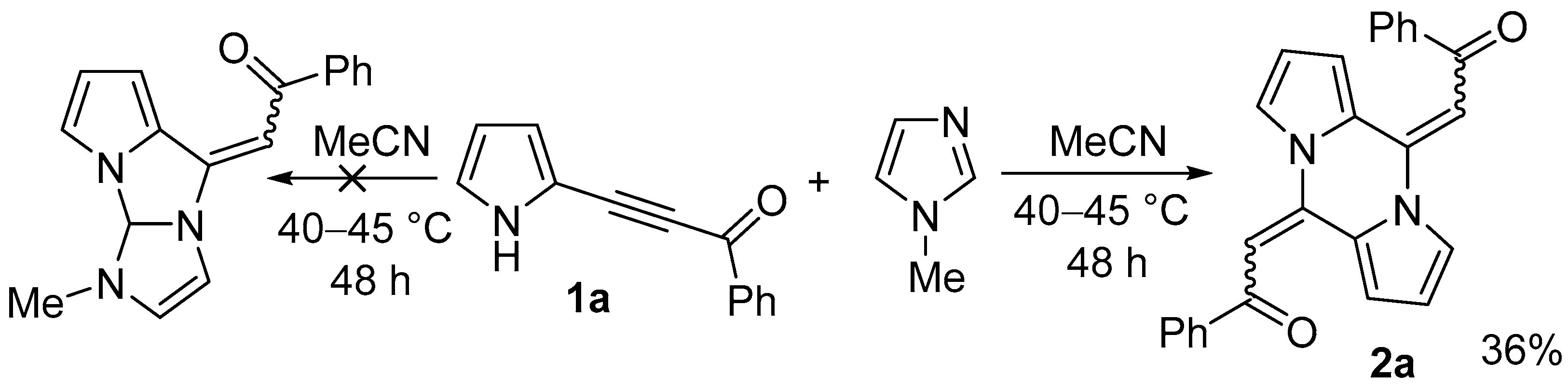
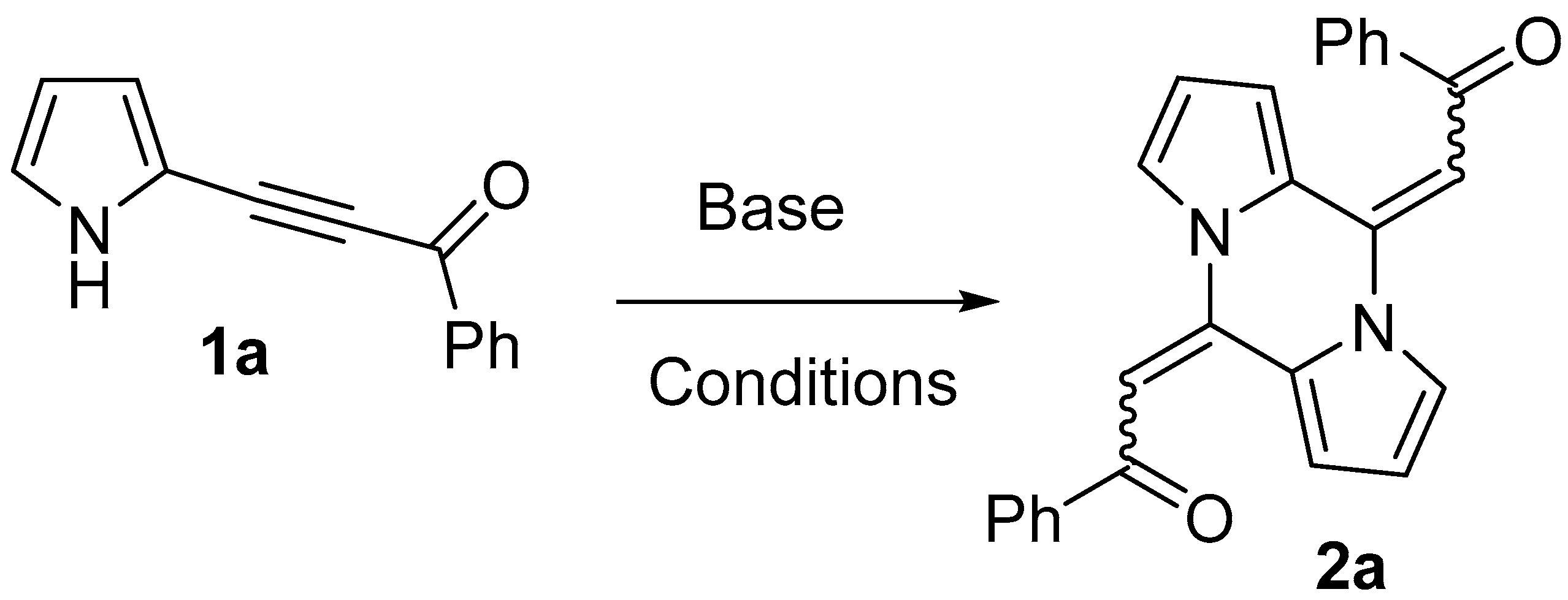
2.1. Optimization of the Reaction Conditions
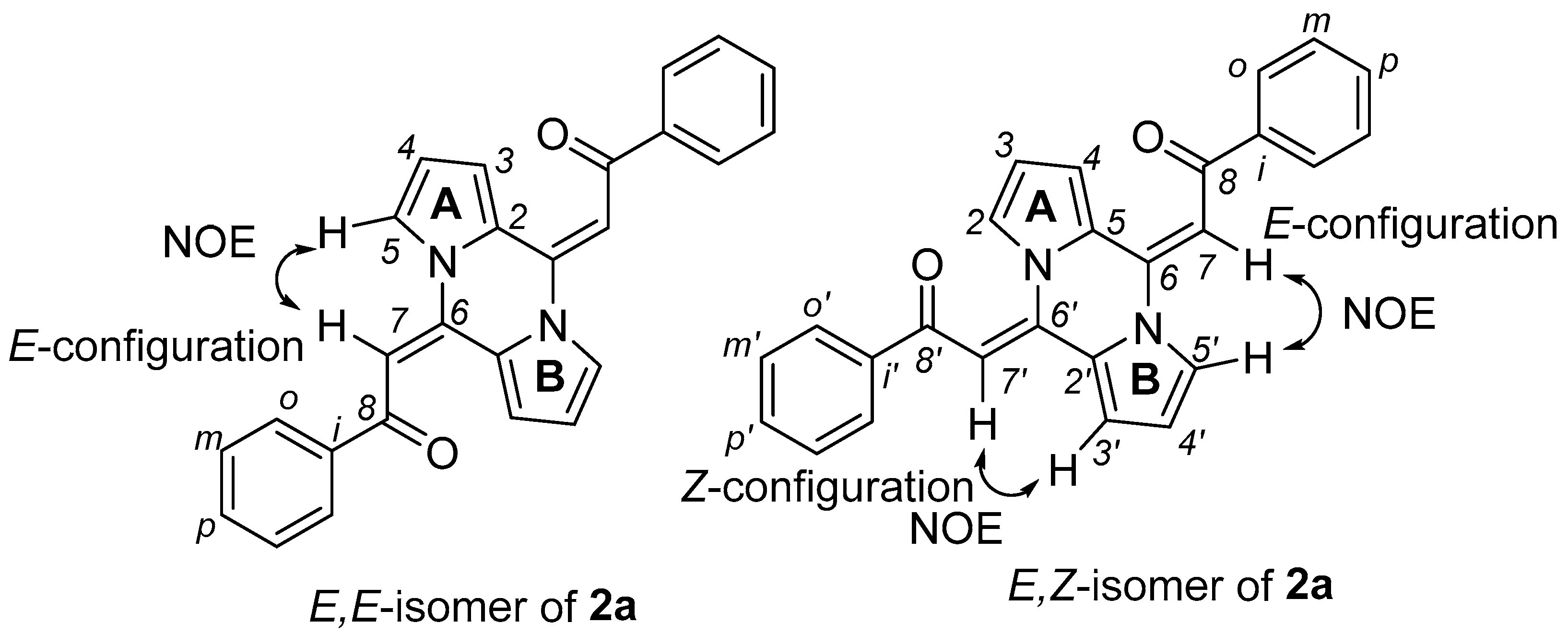
2.2. Study on Substrate Scope
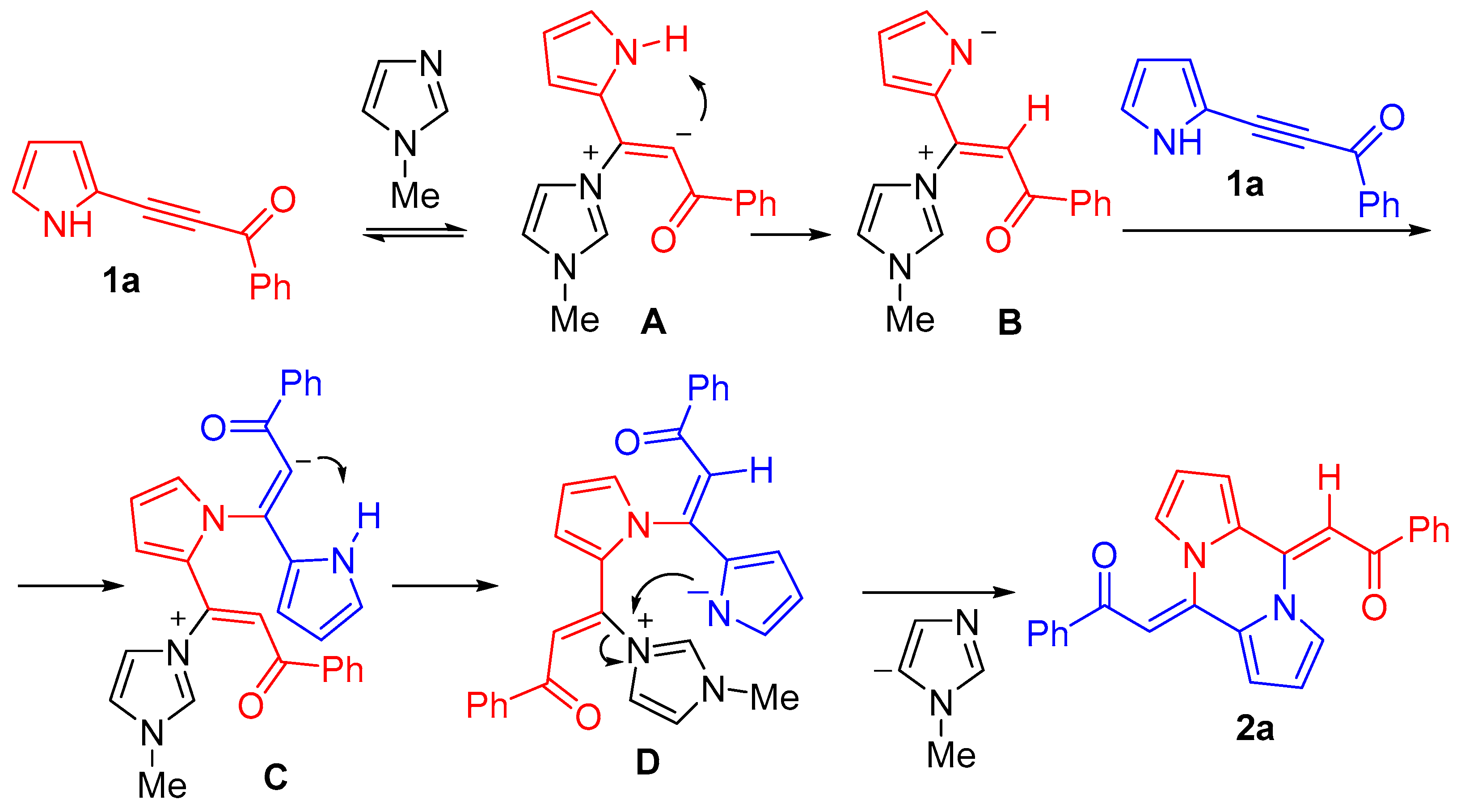
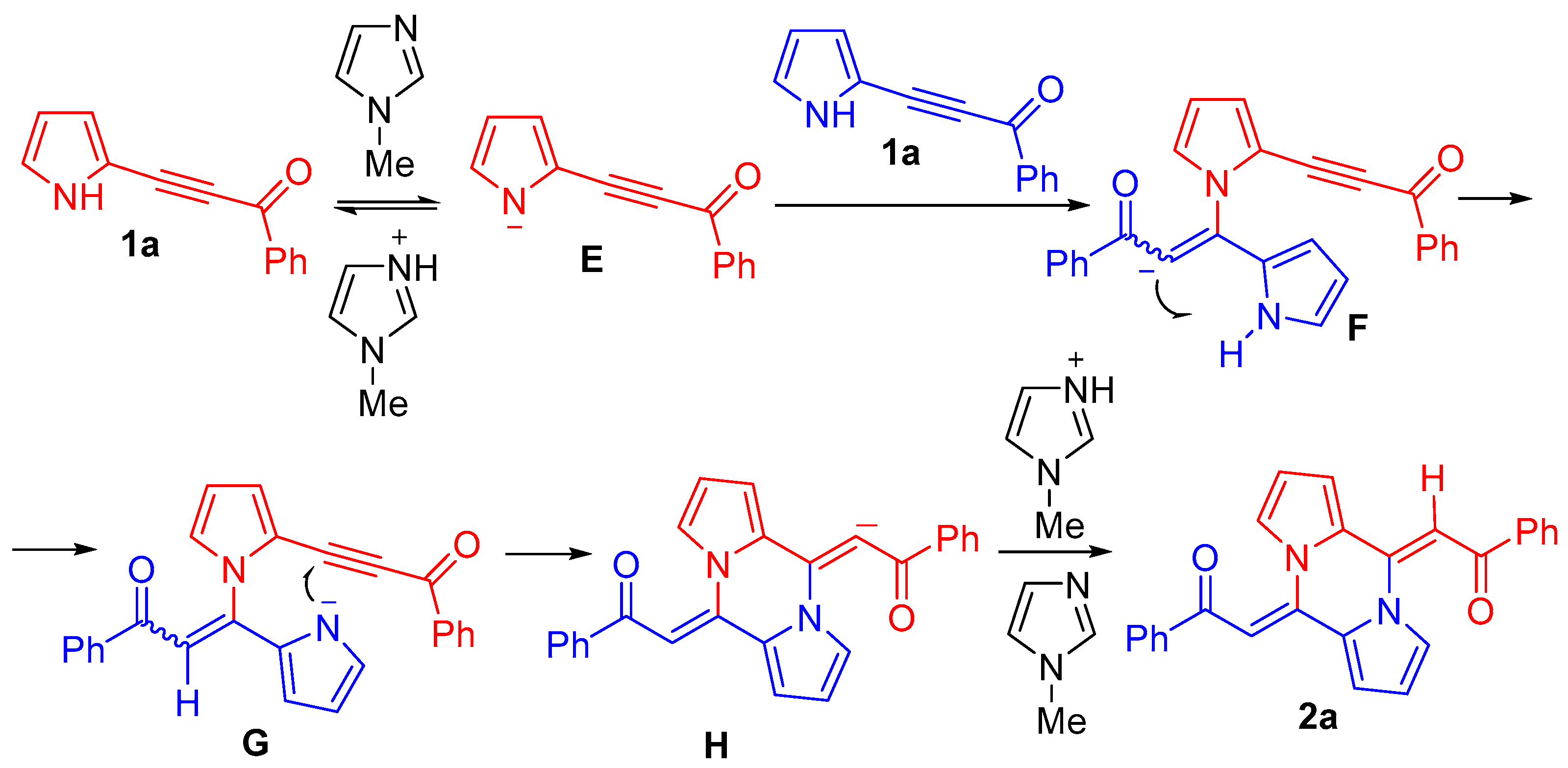
3. Discussion
4. Materials and Methods
4.1. General Considerations
4.2. General Procedure for the Synthesis of Pyrazines 2
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bhardwaj, V.; Gumber, D.; Abbot, V.; Dhiman, S.; Sharma, P. Pyrrole: A resourceful small molecule in key medicinal hetero-aromatics. RSC Adv. 2015, 5, 15233–15266. [Google Scholar] [CrossRef]
- Gholap, S.S. Pyrrole: An emerging scaffold for construction of valuable therapeutic agents. Eur. J. Med. Chem. 2016, 110, 13–31. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.; Rani, V.; Abbot, V.; Kapoor, Y.; Konar, D.; Kumar, K. Recent synthetic and medicinal perspectives of pyrroles: An overview. J. Pharm. Chem. Chem. Sci. 2017, 1, 17–32. [Google Scholar]
- Tzankova, D.; Vladimirova, S.; Peikova, L.; Georgieva, M. Synthesis of pyrrole and substituted pyrroles (review). J. Chem. Technol. Metall. 2018, 53, 451–464. [Google Scholar]
- Baumann, M.; Baxendale, I.R. An overview of the synthetic routes to the best selling drugs containing 6-membered heterocycles. Beilstein J. Org. Chem. 2013, 9, 2265–2319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathew, B.V.; Arikkatt, S.D.; Sindhu, T.J.; Chanran, M.; Bhat, A.R.; Krishnakumar, K.A. A review of biological potential of pyrazine and related heterocyclic compounds. World J. Pharm. Pharm. Sci. 2014, 3, 1124–1132. [Google Scholar]
- Dehnavi, F.; Alizadeh, S.R.; Ebrahimzadeh, M.A. Pyrrolopyrazine derivatives: Synthetic approaches and biological activities, Med. Chem. Res. 2021, 30, 1981–2006. [Google Scholar] [CrossRef]
- Kim, J.; Park, M.; Choi, J.; Singh, D.K.; Kwon, H.J.; Kim, S.H.; Kim, I. Design, synthesis, and biological evaluation of novel pyrrolo[1,2-a]pyrazine derivatives. Bioorg. Med. Chem. Lett. 2019, 29, 1350–1356. [Google Scholar] [CrossRef]
- Garzan, A.; Willby, M.J.; Ngo, H.X.; Gajadeera, C.S.; Green, K.D.; Holbrook, S.Y.L.; Hou, C.; Posey, J.E.; Tsodikov, O.V.; Garneau-Tsodikova, S. Combating enhanced intracellular survival (Eis)-mediated kanamycin resistance of Mycobacterium tuberculosis by novel pyrrolo[1,5-a]pyrazine-based Eis inhibitors. ACS Infect. Dis. 2017, 3, 302–309. [Google Scholar] [CrossRef]
- Kumar, S.; Kumar, N.; Roy, P.; Sondhi, S.M. Synthesis, anti-inflammatory, and antiproliferative activity evaluation of isoindole, pyrrolopyrazine, benzimidazoisoindole, and benzimidazopyrrolopyrazine derivatives. Mol. Divers. 2013, 17, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.M.; Wei, Y.; Yang, J.; Li, H.H.; Liu, M.D.; Huang, N.Y. Synthesis and in vitro anti-inflammatory activity of pyrrolo[1,2-a]pyrazines via Pd-catalyzed intermolecular cyclization reaction. Adv. Mater. Res. 2014, 830, 115–118. [Google Scholar] [CrossRef]
- Reker, D.; Seet, M.; Pillong, M.; Koch, C.P.; Schneider, P.; Witschel, M.C.; Rottmann, M.; Freymond, C.; Brun, R.; Schweizer, B.; et al. Deorphaning pyrrolopyrazines as potent multi-target antimalarial agents. Angew. Chem. Int. Ed. 2014, 53, 7079–7084. [Google Scholar] [CrossRef] [PubMed]
- Kiran, G.S.; Priyadharsini, S.; Sajayan, A.; Ravindran, A.; Selvin, J. An antibiotic agent pyrrolo[1,2-a]pyrazine-1,4-dione,hexahydro isolated from a marine bacteria Bacillus tequilensis MSI45 effectively controls multi-drug resistant Staphylococcus aureus. RSC Adv. 2018, 8, 17837–17846. [Google Scholar] [CrossRef] [PubMed]
- Dawidowski, M.; Wilczek, M.; Kubica, K.; Skolmowski, M.; Turło, J. Structure–activity relationships of the aromatic site in novel anticonvulsant pyrrolo[1,2-a]pyrazine derivatives. Bioorg. Med. Chem. Lett. 2013, 23, 6106–6110. [Google Scholar] [CrossRef]
- Dawidowski, M.; Chońska, J.; Mika, W.; Turło, J. Novel fluorinated pyrrolo[1,2-a]pyrazine-2, 6-dione derivatives: Synthesis and anticonvulsant evaluation in animal models of epilepsy. Bioorg. Med. Chem. 2014, 22, 5410–5427. [Google Scholar] [CrossRef] [PubMed]
- Dieter, A.; Hamm, A.; Fiedler, H.-P.; Goodfellow, M.; Müller, W.E.G.; Brun, R.; Beil, W.; Bringmann, G.J. Pyrocoll, an antibiotic, antiparasitic and antitumor compound produced by a novel alkaliphilic streptomyces strain. J. Antibiot. 2003, 56, 639–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edmondson, S.; Danishefsky, S.J.; Sepp-Lorenzino, L.; Rosen, N. Total synthesis of spirotryprostatin A, leading to the discovery of some biologically promising analogues. J. Am. Chem. Soc. 1999, 121, 2147–2155. [Google Scholar] [CrossRef]
- Xi, Y.-K.; Zhang, H.; Li, R.-X.; Kang, S.-Y.; Li, J.; Li, Y. Total synthesis of spirotryprostatins through organomediated intramolecular umpolung cyclization. Chem. Eur. J. 2019, 25, 3005–3009. [Google Scholar] [CrossRef]
- Jansen, R.; Sood, S.; Mohr, K.I.; Kunze, B.; Irschik, H.; Stadler, M.; Müller, R. Nannozinones and Sorazinones, unprecedented pyrazinones from myxobacteria. J. Nat. Prod. 2014, 77, 2545–2552. [Google Scholar] [CrossRef]
- Li, H.; Liu, L.; Zhang, S.; Cui, W.; Lv, J. Identification of antifungal compounds produced by lactobacillus casei AST18. Curr. Microb. 2012, 65, 156–161. [Google Scholar] [CrossRef]
- Parham, A.H.; Pflumm, C.; Jatsch, A.; Eberle, T.; Stoessel, P.; Kroeber, J.V.; Kaiser, J. Organic electroluminescent devices using mixtures of electron transport materials and methods for producing the devices and the mixtures and formulations containing them, World Intellectual Property Organization, WO2014094964 A1 20140626. Chem. Abstr. 2014, 161, 146357. [Google Scholar]
- Choi, D.S. Preparation of fused indole derivatives as organic electronic device materials. Patent Repub. Korea 2020, KR 2175379 B1 20201109. Chem. Abstr. 2020, 174, 31116. [Google Scholar]
- Liu, Y.; Jiang, D.; Guo, Y.; Dai, W.; Li, Y.; Deng, D.; Zhang, L.; Gao, W.; Niu, J. N-Heterobiphenyl organic compound and application thereof for organic light emitting diode. China, CN111892607 A 20201106. Chem. Abstr. 2014, 175, 621328. [Google Scholar]
- Watanabe, M.; Kobayashi, T.; Kajigaeshi, S.; Kanemasa, S. Azafulvenes 1. A novel generative method of 6-amino-1-azafulvene and its cycloaddition reaction with isocyanate and isothiocyanate. Chem. Lett. 1975, 4, 607–610. [Google Scholar] [CrossRef] [Green Version]
- Nevolina, T.A.; Shcherbinin, V.A.; Shpuntov, P.M.; Butin, A.V. Protolytic opening of the furan ring in the synthesis of 5H,10H-dipyrrolo[1,2-a:1′,2′-d]pyrazines. Chem. Heterocycl. Compd. 2012, 48, 828–830. [Google Scholar] [CrossRef]
- Sobenina, L.N.; Trofimov, B.A. Recent strides in the transition metal-free cross-coupling of haloacetylenes with electron-rich heterocycles in solid media. Molecules 2020, 25, 2490. [Google Scholar] [CrossRef] [PubMed]
- Sobenina, L.N.; Sagitova, E.F.; Ushakov, I.A.; Trofimov, B.A. Transition-metal-free synthesis of pyrrolo[1,2-a]pyrazines via intramolecular cyclization of N-propargyl(pyrrolyl)enaminones. Synthesis 2017, 49, 4065–4081. [Google Scholar] [CrossRef] [Green Version]
- Oparina, L.A.; Belyaeva, K.V.; Kolyvanov, N.A.; Ushakov, I.A.; Gotsko, M.D.; Sobenina, L.N.; Vashchenko, A.V.; Trofimov, B.A. Catalyst-free annulation of acylethynylpyrroles with 1-pyrrolines: A straightforward access to tetrahydrodipyrrolo[1,2-a:1′,2′-c]imidazoles. J. Org. Chem. 2022, 87, 9518–9531. [Google Scholar] [CrossRef]
- Sobenina, L.N.; Tomilin, D.N.; Trofimov, B.A. C-Ethynylpyrroles: Synthesis and reactivity. Russ. Chem. Rev. 2014, 83, 475–501. [Google Scholar] [CrossRef]
- Ushakov, I.A.; Afonin, A.V.; Voronov, V.K.; Stepanova, Z.V.; Sobenina, L.N.; Mikhaleva, A.I. NMR study of the steric and electronic structure of 2-(2-acylethenyl)pyrroles. Russ. J. Org. Chem. 2003, 39, 1318–1324. [Google Scholar] [CrossRef]
- Afonin, A.V.; Ushakov, I.A.; Vashchenko, A.V.; Kondrashov, E.V.; Rulev, A.Y. GIAO, DFT, AIM and NBO analysis of the N-H···O intramolecular hydrogen-bond influence on the 1J(N,H) coupling constant in push–pull diaminoenones. Magn. Reson. Chem. 2010, 48, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Trofimov, B.A.; Stepanova, Z.V.; Sobenina, L.N.; Mikhaleva, A.I.; Ushakov, I.A. Ethynylation of pyrroles with 1-acyl-2-bromoacetylenes on alumina: A formal inverse Sonogashira coupling. Tetrahedron Lett. 2004, 45, 6513–6516. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Imidazoles, mol% | Solvent | Temp., °C | Time, h | Conversion of 1a, % | Yield of 2a, % |
|---|---|---|---|---|---|---|
| 1 | - | MeCN | 40–45 | 48 | ~0 | 0 |
| 2 | 1-MeIM, 20 | MeCN | 20–25 | 48 | ~0 | 0 |
| 3 | 1-MeIM, 20 | MeCN | 40–45 | 48 | 99 | 36 |
| 4 | 1-MeIM, 20 | 1,4-dioxane | 40–45 | 48 | 85 | 20 |
| 5 | 1-MeIM, 100 | - | 40–45 | 24 | 95 | 51 |
| 6 | 1-MeIM, 500 | - | 40–45 | 24 | 99 | 42 |
| 7 | 1-MeIM, 100 | n-hexane | 40–45 | 8 | 99 | 38 |
| 8 | 1-PhIM, 100 | - | 40–45 | 48 | 96 | 26 |
| 9 | 1-MeBIM, 100 | MeCN | 40–45 | 24 | 24 | 0 |
| Entry | Base, mol% | Solvent | Temp., °C | Time, h | Conversion of 1a, % | Yield of 2a, % |
|---|---|---|---|---|---|---|
| 1 | KOH, 20 | MeCN | 40–45 | 8 | 97 | 32 |
| 2 | KOH, 20 | n-hexane | 40–45 | 8 | 0 | 0 |
| 3 | KOH, 20 | DMSO | 40–45 | 8 | 100 | 0 |
| 4 | KOH, 20 | THF | 40–45 | 8 | 0 | 0 |
| 5 | KOH, 20 | H2O | 40–45 | 48 | 100 | trace |
| 6 | KOH, 50 | MeCN | 40–45 | 4 | 100 | 29 |
| 7 | NaOH, 20 | MeCN | 40–45 | 8 | 85 | 20 |
| 8 | NaOH, 20 | MeCN | 20–25 | 24 | 99 | 5 |
| 9 | NaOH, 20 | MeCN | 60–65 | 6 | 99 | 24 |
| 10 | t-BuOK, 20 | MeCN | 40–45 | 8 | 100 | 33 |
| 11 | DABCO, 20 | MeCN | 40–45 | 48 | 70 | 30 |
| 12 | Et3N, 20 | MeCN | 60–65 | 48 | 0 | 0 |
| 13 | Ph3P, 20 | MeCN | 70–75 | 96 | 95 | 0 |
| 14 | TMEDA, 20 | MeCN | 40–45 | 48 | 0 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belyaeva, K.V.; Nikitina, L.P.; Gen’, V.S.; Tomilin, D.N.; Sobenina, L.N.; Afonin, A.V.; Oparina, L.A.; Trofimov, B.A. 1-Methylimidazole as an Organic Catalyst for [3+3]-Cyclodimerization of Acylethynylpyrroles to Bis(acylmethylidene)dipyrrolo[1,2-a:1′,2′-d]pyrazines. Catalysts 2022, 12, 1604. https://doi.org/10.3390/catal12121604
Belyaeva KV, Nikitina LP, Gen’ VS, Tomilin DN, Sobenina LN, Afonin AV, Oparina LA, Trofimov BA. 1-Methylimidazole as an Organic Catalyst for [3+3]-Cyclodimerization of Acylethynylpyrroles to Bis(acylmethylidene)dipyrrolo[1,2-a:1′,2′-d]pyrazines. Catalysts. 2022; 12(12):1604. https://doi.org/10.3390/catal12121604
Chicago/Turabian StyleBelyaeva, Kseniya V., Lina P. Nikitina, Veronika S. Gen’, Denis N. Tomilin, Lyubov N. Sobenina, Andrei V. Afonin, Ludmila A. Oparina, and Boris A. Trofimov. 2022. "1-Methylimidazole as an Organic Catalyst for [3+3]-Cyclodimerization of Acylethynylpyrroles to Bis(acylmethylidene)dipyrrolo[1,2-a:1′,2′-d]pyrazines" Catalysts 12, no. 12: 1604. https://doi.org/10.3390/catal12121604