
A Combined Computational–Experimental Study on the Substrate Binding and Reaction Mechanism of Salicylic Acid Decarboxylase
 , , , and
, , , and 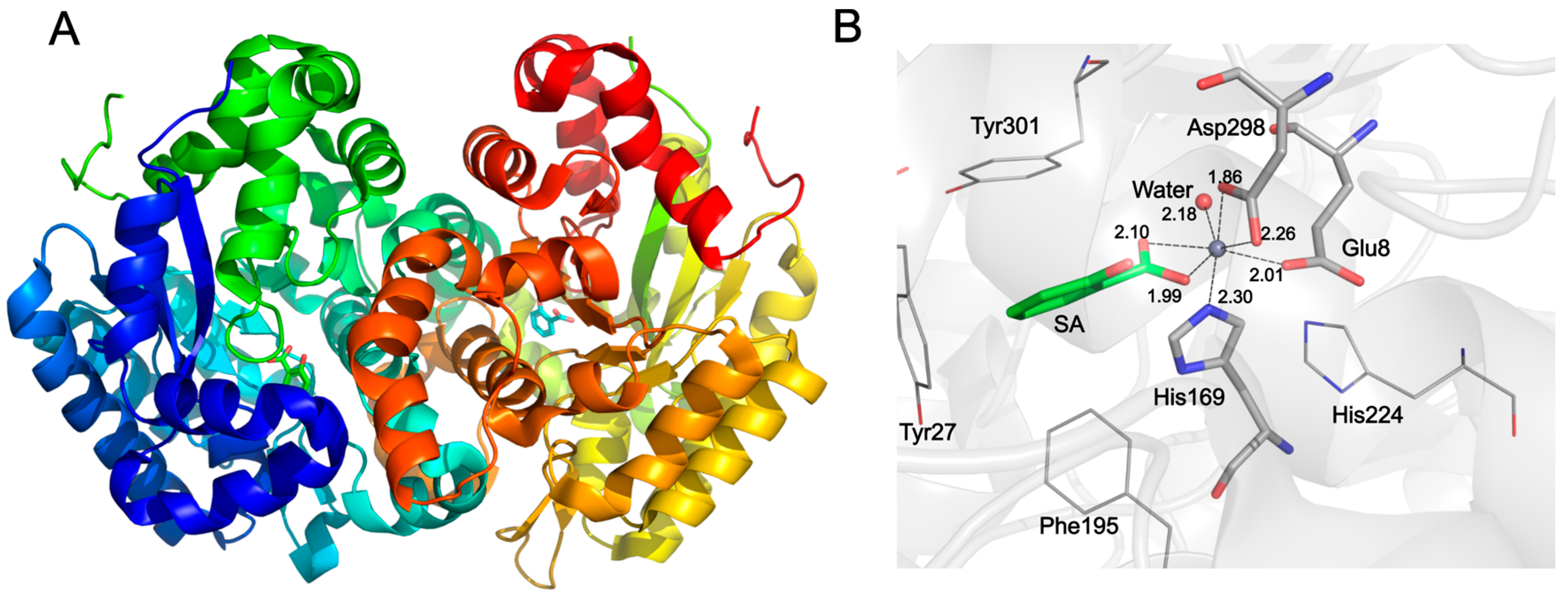{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
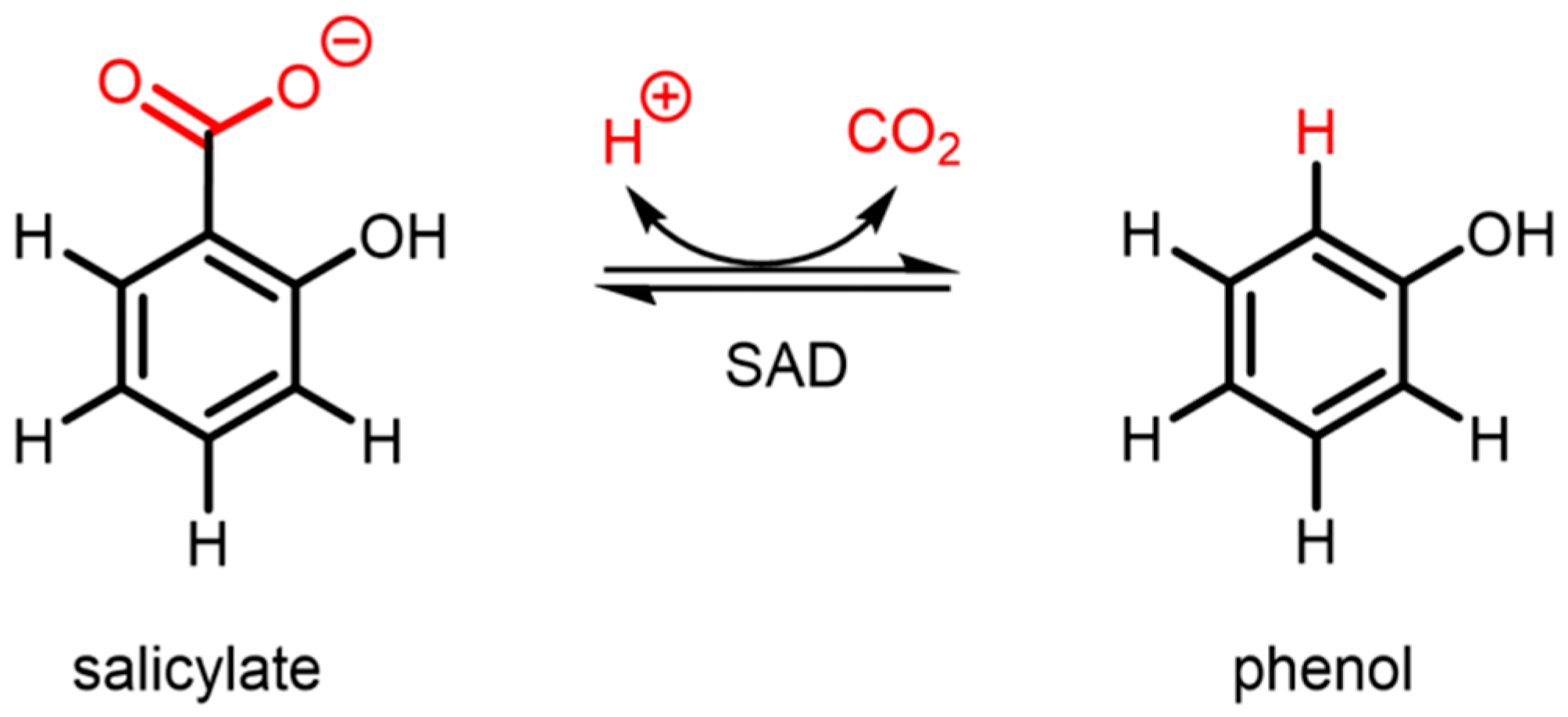
:1. Introduction
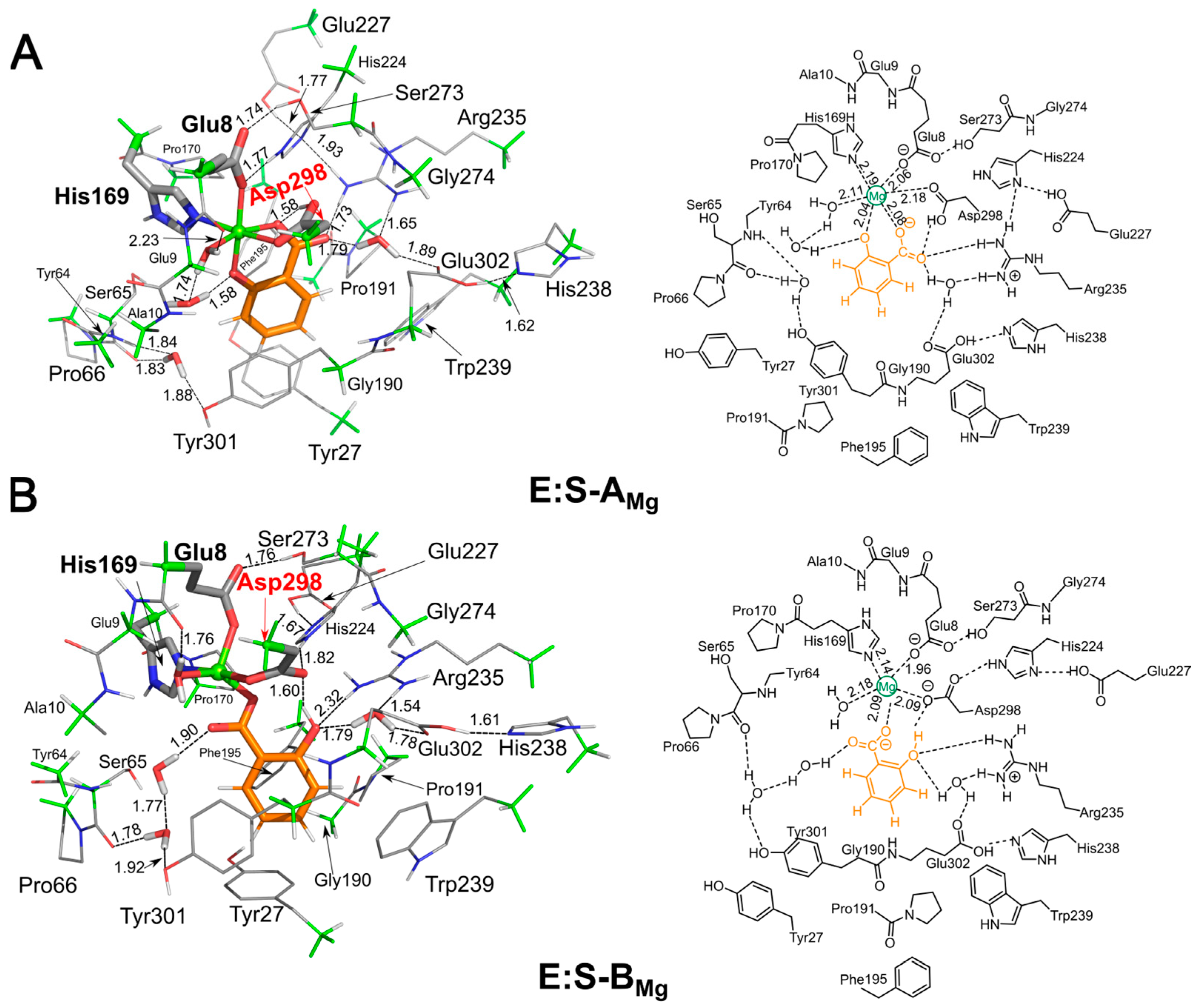
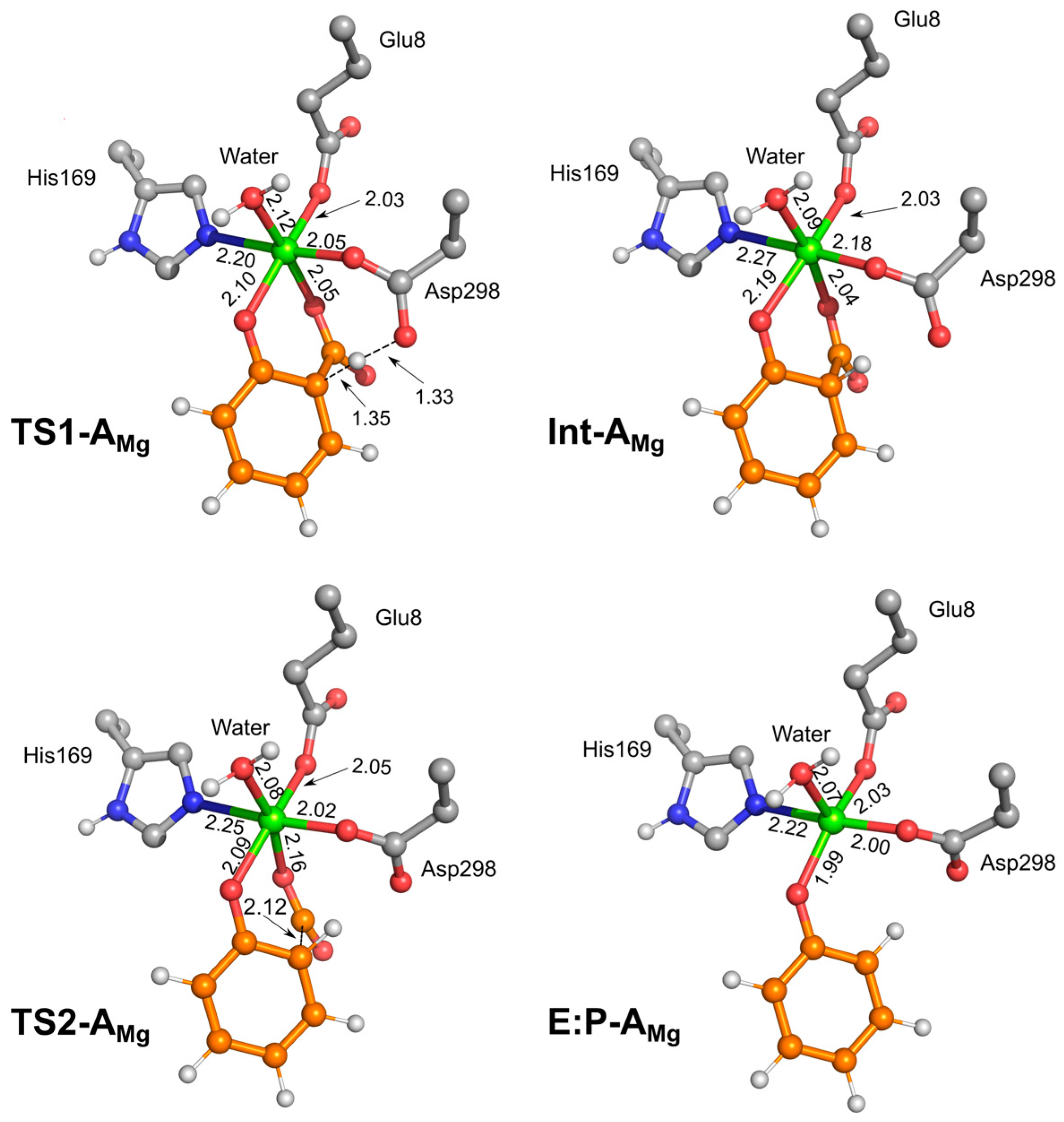
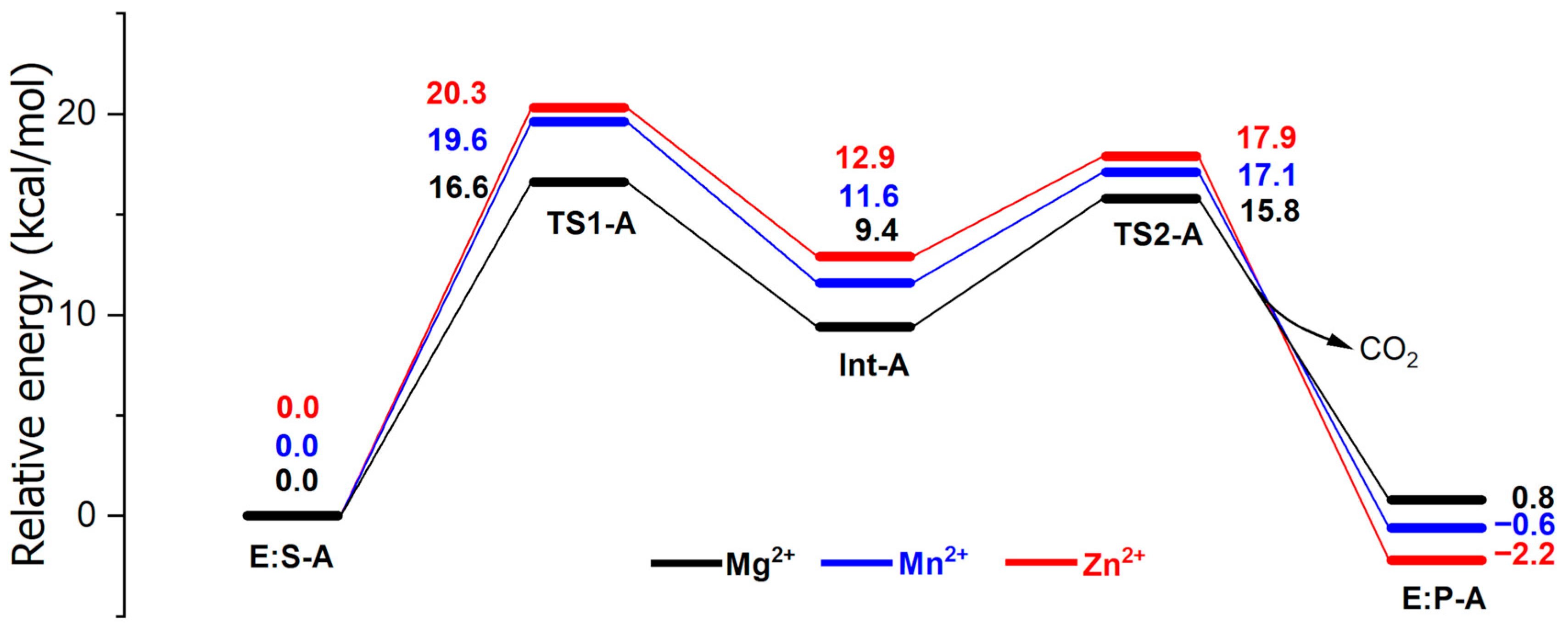
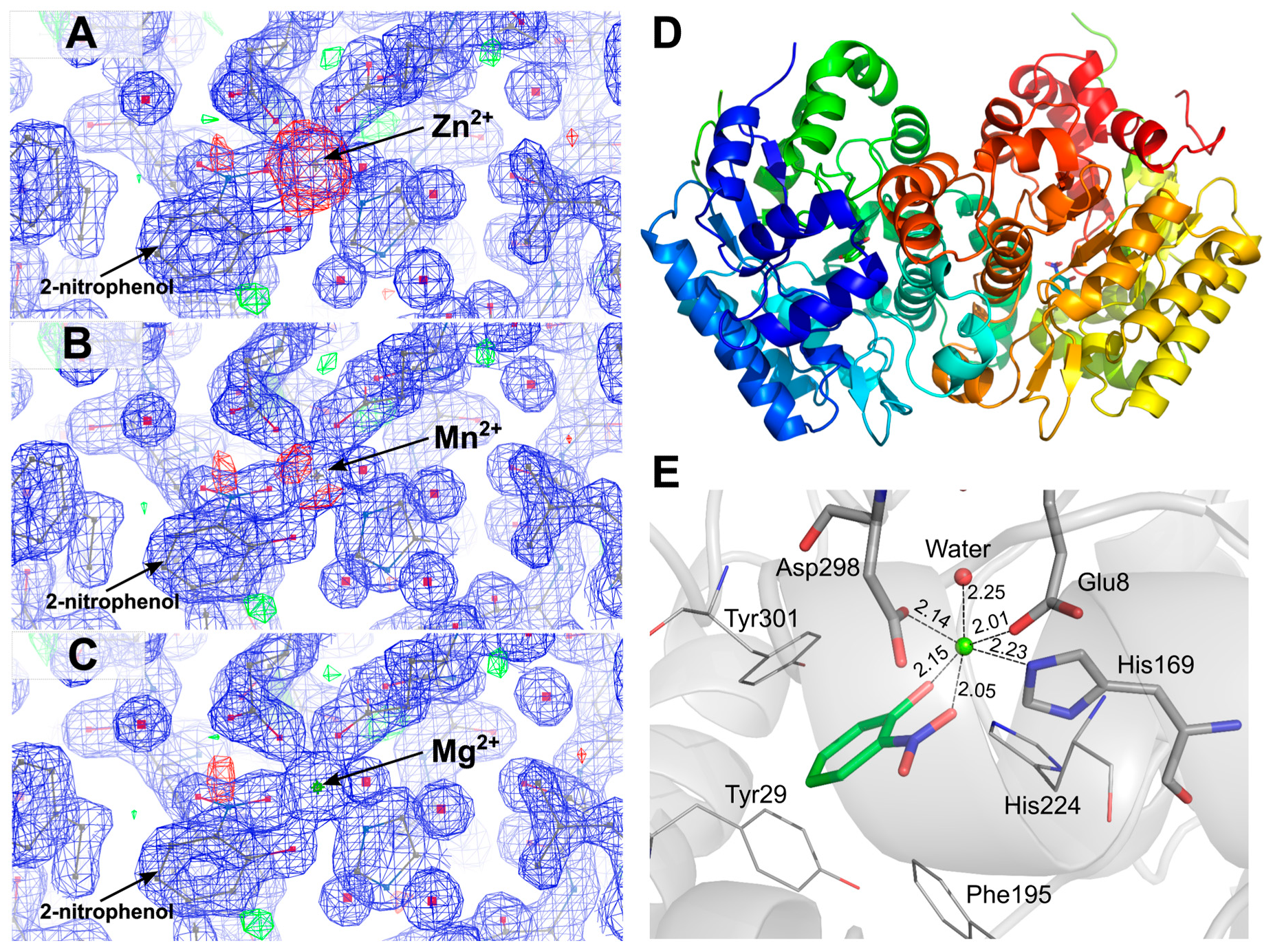
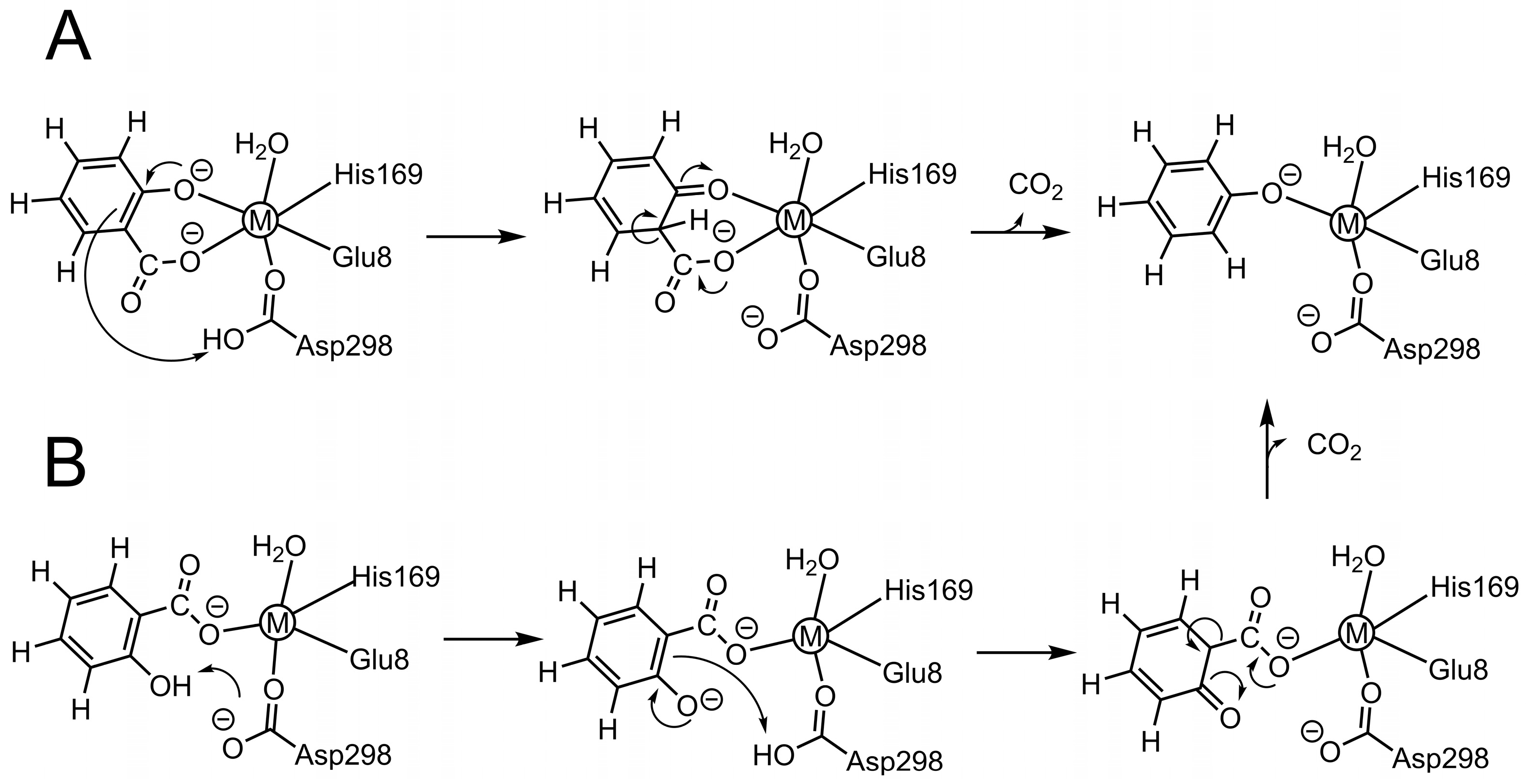
2. Results and Discussion
3. Computational and Experimental Details
3.1. Computational Details
3.2. Active Site Model
3.3. Cloning and Protein Purification
3.4. Crystallization, Data Collection, Structure Determination, and Refinement
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kirimura, K.; Gunji, H.; Wakayama, R.; Hattori, T.; Ishii, Y. Enzymatic Kolbe–Schmitt Reaction to Form Salicylic Acid from Phenol: Enzymatic Characterization and Gene Identification of a Novel Enzyme, Trichosporon Moniliiforme Salicylic Acid Decarboxylase. Biochem. Biophys. Res. Commun. 2010, 394, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, A.S.; Jeskey, H. The Kolbe-Schmitt Reaction. Chem. Rev. 1957, 57, 583–620. [Google Scholar] [CrossRef]
- Luo, J.; Larrosa, I. C−H Carboxylation of Aromatic Compounds through CO2 Fixation. ChemSusChem 2017, 10, 3317–3332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plasch, K.; Hofer, G.; Keller, W.; Hay, S.; Heyes, D.J.; Dennig, A.; Glueck, S.M.; Faber, K. Pressurized CO2 as a Carboxylating Agent for the Biocatalytic Ortho-Carboxylation of Resorcinol. Green Chem. 2018, 20, 1754–1759. [Google Scholar] [CrossRef] [Green Version]
- Boullard, O.; Leblanc, H.; Besson, B. Salicylic Acid. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2000; p. a23_477. ISBN 978-3-527-30673-2. [Google Scholar]
- Madan, R.K.; Levitt, J. A Review of Toxicity from Topical Salicylic Acid Preparations. J. Am. Acad. Dermatol. 2014, 70, 788–792. [Google Scholar] [CrossRef] [PubMed]
- Seibert, C.M.; Raushel, F.M. Structural and Catalytic Diversity within the Amidohydrolase Superfamily. Biochemistry 2005, 44, 6383–6391. [Google Scholar] [CrossRef]
- Song, M.; Zhang, X.; Liu, W.; Feng, J.; Cui, Y.; Yao, P.; Wang, M.; Guo, R.-T.; Wu, Q.; Zhu, D. 2,3-Dihydroxybenzoic Acid Decarboxylase from Fusarium Oxysporum: Crystal Structures and Substrate Recognition Mechanism. ChemBioChem 2020, 21, 2950–2956. [Google Scholar] [CrossRef]
- Xu, S.; Li, W.; Zhu, J.; Wang, R.; Li, Z.; Xu, G.-L.; Ding, J. Crystal Structures of Isoorotate Decarboxylases Reveal a Novel Catalytic Mechanism of 5-Carboxyl-Uracil Decarboxylation and Shed Light on the Search for DNA Decarboxylase. Cell Res. 2013, 23, 1296–1309. [Google Scholar] [CrossRef] [Green Version]
- Goto, M.; Hayashi, H.; Miyahara, I.; Hirotsu, K.; Yoshida, M.; Oikawa, T. Crystal Structures of Nonoxidative Zinc-Dependent 2,6-Dihydroxybenzoate (γ-Resorcylate) Decarboxylase from Rhizobium Sp. Strain MTP-10005. J. Biol. Chem. 2006, 281, 34365–34373. [Google Scholar] [CrossRef] [Green Version]
- Vladimirova, A.; Patskovsky, Y.; Fedorov, A.A.; Bonanno, J.B.; Fedorov, E.V.; Toro, R.; Hillerich, B.; Seidel, R.D.; Richards, N.G.J.; Almo, S.C.; et al. Substrate Distortion and the Catalytic Reaction Mechanism of 5-Carboxyvanillate Decarboxylase. J. Am. Chem. Soc. 2016, 138, 826–836. [Google Scholar] [CrossRef]
- Sheng, X.; Zhu, W.; Huddleston, J.; Xiang, D.F.; Raushel, F.M.; Richards, N.G.J.; Himo, F. A Combined Experimental-Theoretical Study of the LigW-Catalyzed Decarboxylation of 5-Carboxyvanillate in the Metabolic Pathway for Lignin Degradation. ACS Catal. 2017, 7, 4968–4974. [Google Scholar] [CrossRef]
- Hofer, G.; Sheng, X.; Braeuer, S.; Payer, S.E.; Plasch, K.; Goessler, W.; Faber, K.; Keller, W.; Himo, F.; Glueck, S.M. Metal Ion Promiscuity and Structure of 2,3-Dihydroxybenzoic Acid Decarboxylase of Aspergillus Oryzae. ChemBioChem 2021, 22, 652–656. [Google Scholar] [CrossRef] [PubMed]
- Sheng, X.; Plasch, K.; Payer, S.E.; Ertl, C.; Hofer, G.; Keller, W.; Braeuer, S.; Goessler, W.; Glueck, S.M.; Himo, F.; et al. Reaction Mechanism and Substrate Specificity of Iso-Orotate Decarboxylase: A Combined Theoretical and Experimental Study. Front. Chem. 2018, 6, 608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, X.; Patskovsky, Y.; Vladimirova, A.; Bonanno, J.B.; Almo, S.C.; Himo, F.; Raushel, F.M. Mechanism and Structure of γ-Resorcylate Decarboxylase. Biochemistry 2018, 57, 3167–3175. [Google Scholar] [CrossRef] [PubMed]
- Sheng, X.; Himo, F. Mechanisms of Metal-Dependent Non-Redox Decarboxylases from Quantum Chemical Calculations. Comput. Struct. Biotechnol. J. 2021, 19, 3176–3186. [Google Scholar] [CrossRef]
- Wuensch, C.; Glueck, S.M.; Gross, J.; Koszelewski, D.; Schober, M.; Faber, K. Regioselective Enzymatic Carboxylation of Phenols and Hydroxystyrene Derivatives. Org. Lett. 2012, 14, 1974–1977. [Google Scholar] [CrossRef]
- Payer, S.E.; Faber, K.; Glueck, S.M. Non-Oxidative Enzymatic (De)Carboxylation of (Hetero)Aromatics and Acrylic Acid Derivatives. Adv. Synth. Catal. 2019, 361, 2402–2420. [Google Scholar] [CrossRef] [Green Version]
- Ren, P.; Tan, Z.; Zhou, Y.; Tang, H.; Xu, P.; Liu, H.; Zhu, L. Biocatalytic CO2 Fixation Initiates Selective Oxidative Cracking of 1-Naphthol under Ambient Conditions. Green Chem. 2022, 24, 4766–4771. [Google Scholar] [CrossRef]
- Kirimura, K.; Yanaso, S.; Kosaka, S.; Koyama, K.; Hattori, T.; Ishii, Y. Production of P-Aminosalicylic Acid through Enzymatic Kolbe–Schmitt Reaction Catalyzed by Reversible Salicylic Acid Decarboxylase. Chem. Lett. 2011, 40, 206–208. [Google Scholar] [CrossRef]
- Kirimura, K.; Araki, M.; Ishihara, M.; Ishii, Y. Expanding Substrate Specificity of Salicylate Decarboxylase by Site-Directed Mutagenesis for Expansion of the Entrance Region Connecting to the Substrate Access Tunnel. Chem. Lett. 2019, 48, 58–61. [Google Scholar] [CrossRef]
- Ienaga, S.; Kosaka, S.; Honda, Y.; Ishii, Y.; Kirimura, K. P-Aminosalicylic Acid Production by Enzymatic Kolbe–Schmitt Reaction Using Salicylic Acid Decarboxylases Improved through Site-Directed Mutagenesis. Bull. Chem. Soc. Jpn. 2013, 86, 628–634. [Google Scholar] [CrossRef]
- Gao, X.; Wu, M.; Zhang, W.; Li, C.; Guo, R.-T.; Dai, Y.; Liu, W.; Mao, S.; Lu, F.; Qin, H.-M. Structural Basis of Salicylic Acid Decarboxylase Reveals a Unique Substrate Recognition Mode and Access Channel. J. Agric. Food Chem. 2021, 69, 11616–11625. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Grimme, S. Density Functional Theory with London Dispersion Corrections. WIREs Comput. Mol. Sci. 2011, 1, 211–228. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B Condens. Matter 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Bursch, M.; Caldeweyher, E.; Hansen, A.; Neugebauer, H.; Ehlert, S.; Grimme, S. Understanding and Quantifying London Dispersion Effects in Organometallic Complexes. Acc. Chem. Res. 2019, 52, 258–266. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for K to Au Including the Outermost Core Orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Lind, M.E.S.; Himo, F. Theoretical Study of Reaction Mechanism and Stereoselectivity of Arylmalonate Decarboxylase. ACS Catal. 2014, 4, 4153–4160. [Google Scholar] [CrossRef]
- Sheng, X.; Lind, M.E.S.; Himo, F. Theoretical Study of the Reaction Mechanism of Phenolic Acid Decarboxylase. FEBS J. 2015, 282, 4703–4713. [Google Scholar] [CrossRef]
- Sheng, X.; Himo, F. Mechanism of 3-Methylglutaconyl CoA Decarboxylase AibA/AibB: Pericyclic Reaction versus Direct Decarboxylation. Angew. Chem. Int. Ed. 2020, 59, 22973–22977. [Google Scholar] [CrossRef] [PubMed]
- Planas, F.; Sheng, X.; McLeish, M.J.; Himo, F. A Theoretical Study of the Benzoylformate Decarboxylase Reaction Mechanism. Front. Chem. 2018, 6, 205. [Google Scholar] [CrossRef] [PubMed]
- Himo, F.; de Visser, S.P. Status Report on the Quantum Chemical Cluster Approach for Modeling Enzyme Reactions. Commun. Chem. 2022, 5, 29. [Google Scholar] [CrossRef]
- Siegbahn, P.E.M. A Quantum Chemical Approach for the Mechanisms of Redox-Active Metalloenzymes. RSC Adv. 2021, 11, 3495–3508. [Google Scholar] [CrossRef]
- Himo, F. Recent Trends in Quantum Chemical Modeling of Enzymatic Reactions. J. Am. Chem. Soc. 2017, 139, 6780–6786. [Google Scholar] [CrossRef] [Green Version]
- Blomberg, M.R.A.; Borowski, T.; Himo, F.; Liao, R.-Z.; Siegbahn, P.E.M. Quantum Chemical Studies of Mechanisms for Metalloenzymes. Chem. Rev. 2014, 114, 3601–3658. [Google Scholar] [CrossRef]
- Sheng, X.; Kazemi, M.; Planas, F.; Himo, F. Modeling Enzymatic Enantioselectivity using Quantum Chemical Methodology. ACS Catal. 2020, 10, 6430–6449. [Google Scholar] [CrossRef]
- Otwinowski, Z.; Minor, W. Processing of X-Ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997, 276, 307–326. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser Crystallographic Software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [Green Version]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef]
- Grosse-Kunstleve, R.W.; Afonine, P.V.; Moriarty, N.W.; Zwart, P.H.; Hung, L.-W.; Read, R.J. Iterative Model Building, Structure Refinement and Density Modification with the PHENIX AutoBuild Wizard. Acta Crystallogr. D Biol. Crystallogr. 2008, 64, 61–69. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Cowtan, K. Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brünger, A.T. Assessment of Phase Accuracy by Cross Validation: The Free R Value. Methods and Applications. Acta Crystallogr. Sect. D 1993, 49, 24–36. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, F.; Zhao, Y.; Zhang, C.; Wang, W.; Gao, J.; Li, Q.; Qin, H.; Dai, Y.; Liu, W.; Liu, F.; et al. A Combined Computational–Experimental Study on the Substrate Binding and Reaction Mechanism of Salicylic Acid Decarboxylase. Catalysts 2022, 12, 1577. https://doi.org/10.3390/catal12121577
Chen F, Zhao Y, Zhang C, Wang W, Gao J, Li Q, Qin H, Dai Y, Liu W, Liu F, et al. A Combined Computational–Experimental Study on the Substrate Binding and Reaction Mechanism of Salicylic Acid Decarboxylase. Catalysts. 2022; 12(12):1577. https://doi.org/10.3390/catal12121577
Chicago/Turabian StyleChen, Fuqiang, Yipei Zhao, Chenghua Zhang, Wei Wang, Jian Gao, Qian Li, Huimin Qin, Yujie Dai, Weidong Liu, Fufeng Liu, and et al. 2022. "A Combined Computational–Experimental Study on the Substrate Binding and Reaction Mechanism of Salicylic Acid Decarboxylase" Catalysts 12, no. 12: 1577. https://doi.org/10.3390/catal12121577