

Influence of Components Deposition Order on Silver Species Formation in Bimetallic Ag-Fe System Supported on Mordenite
 ,
,  , , ,
, , ,  , , , and
, , , and
Abstract
:1. Introduction
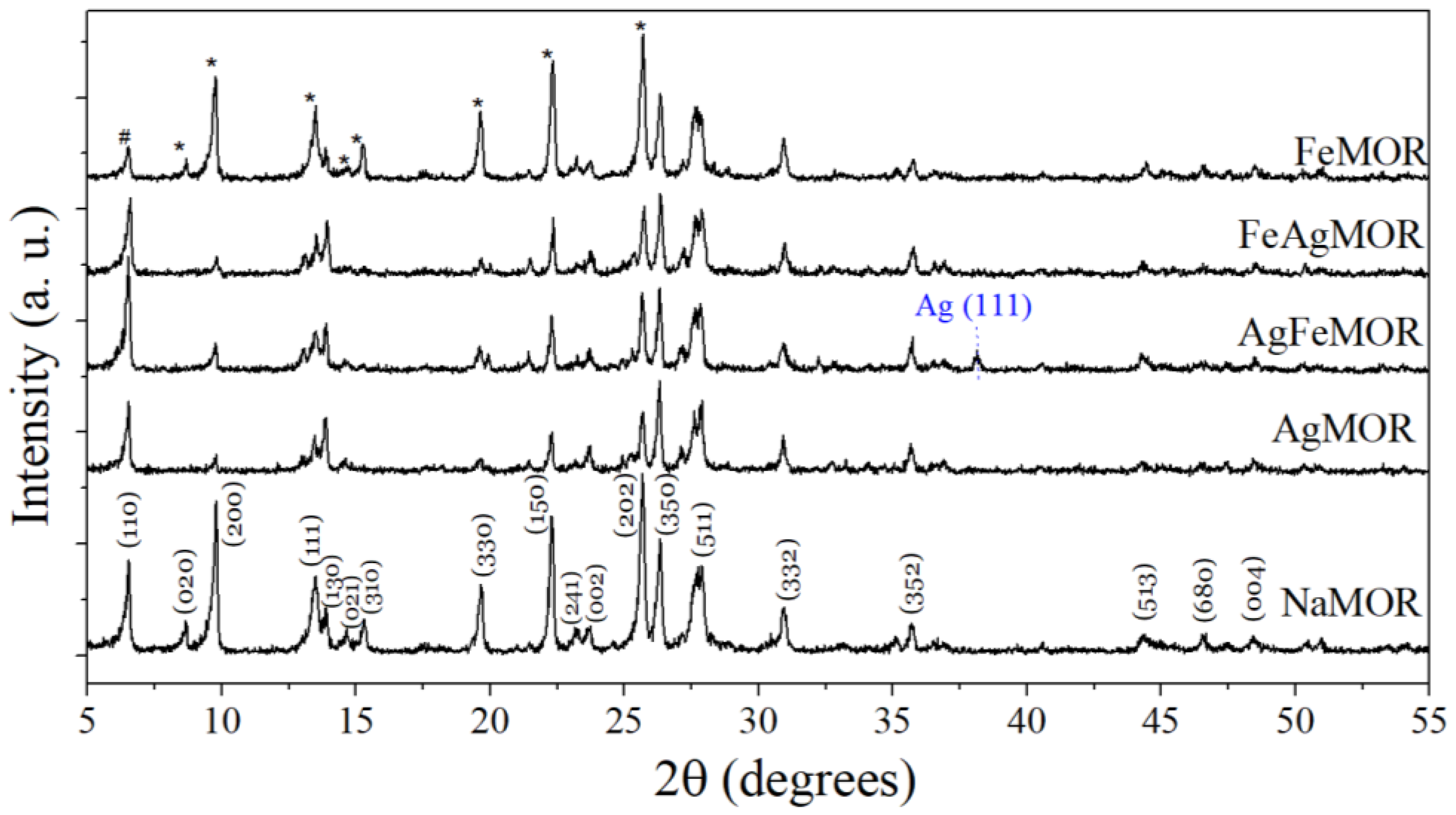
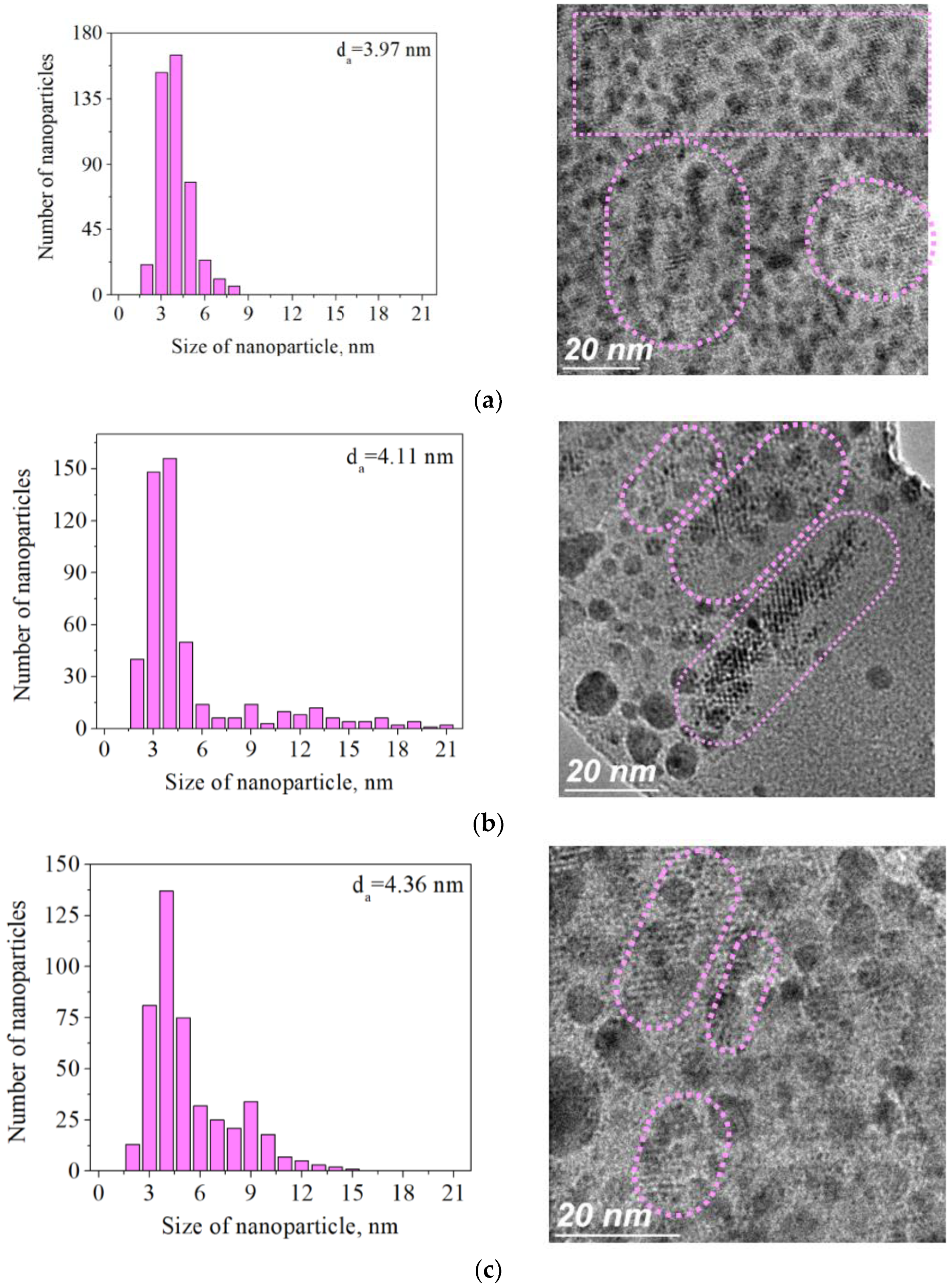
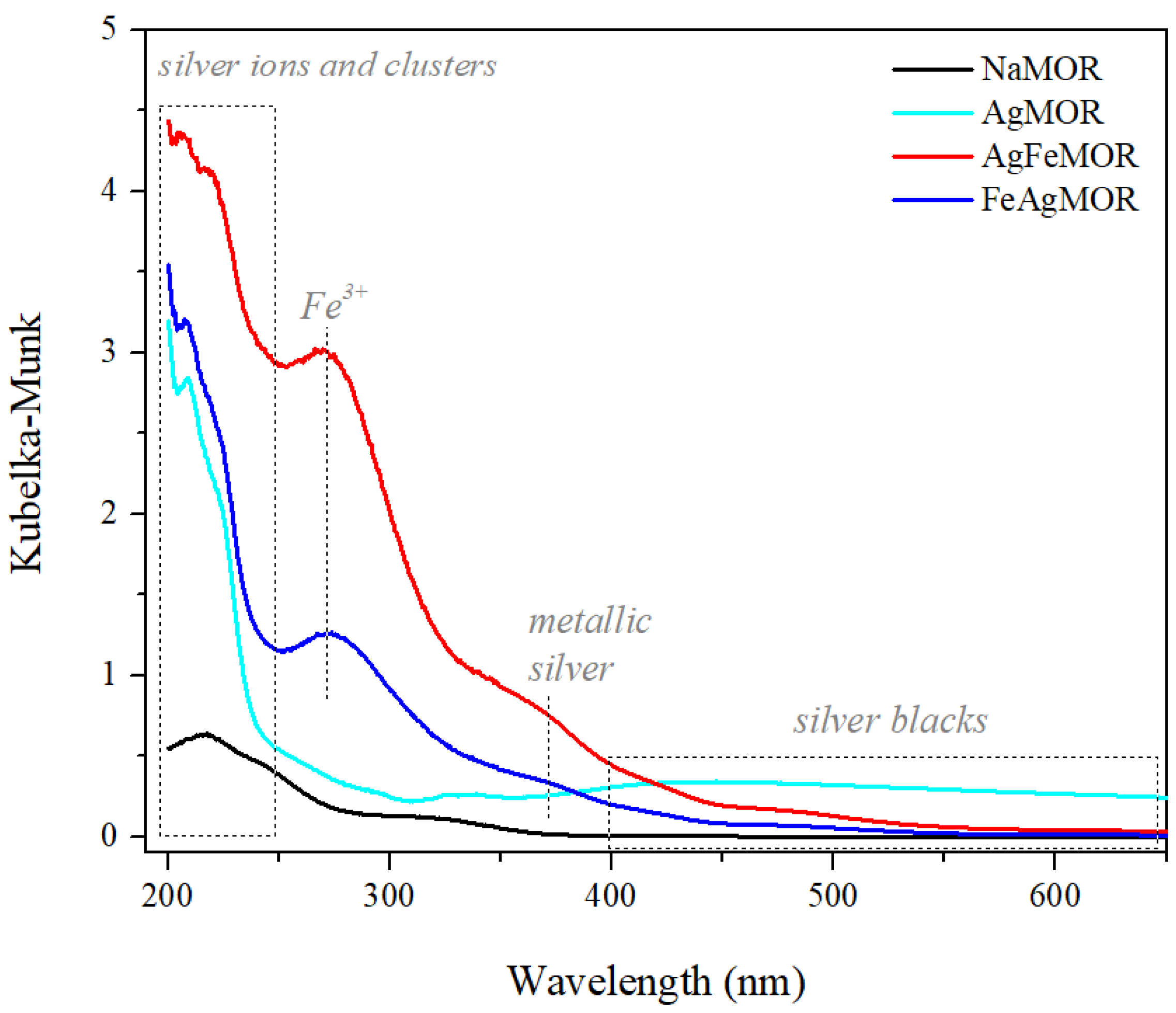
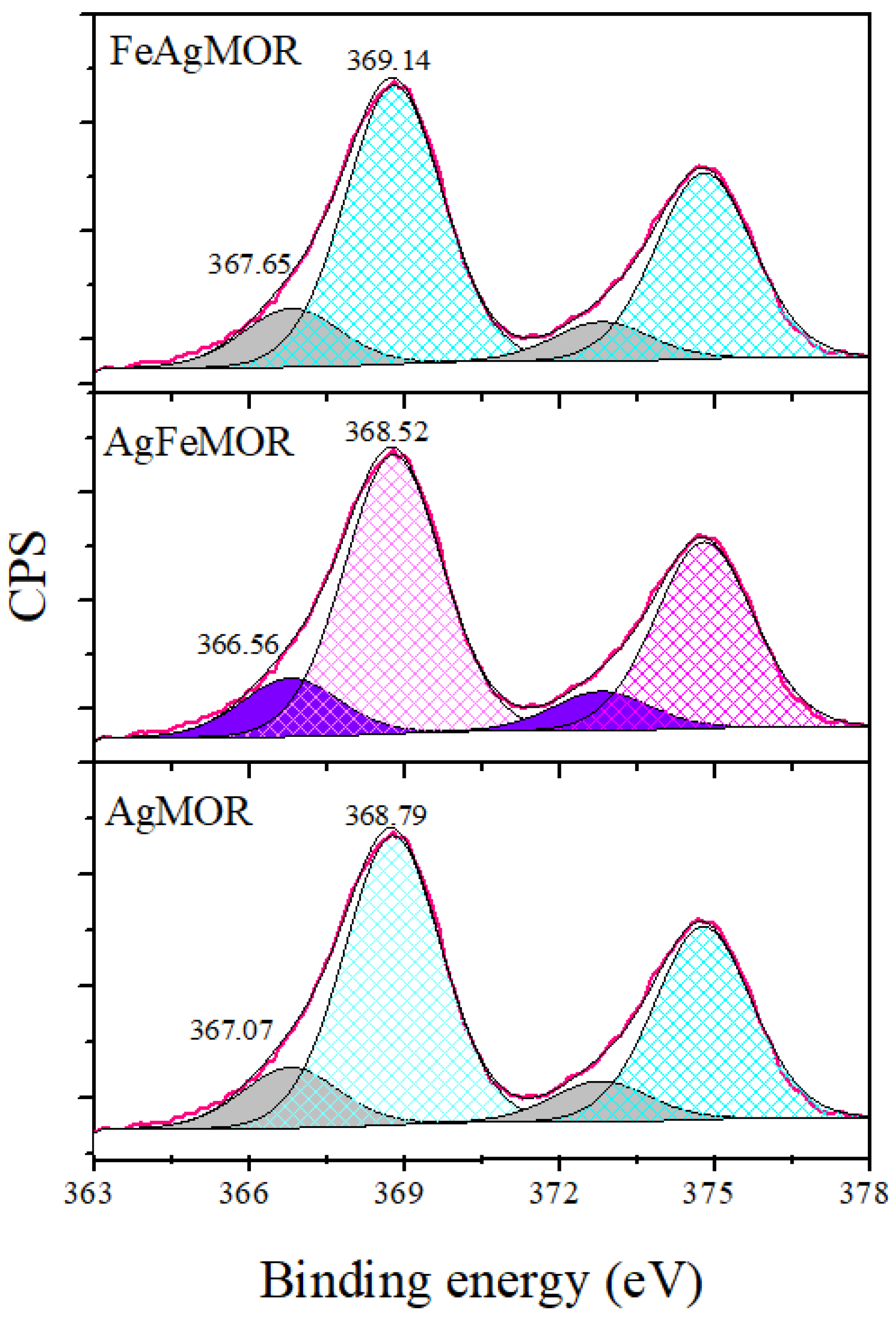
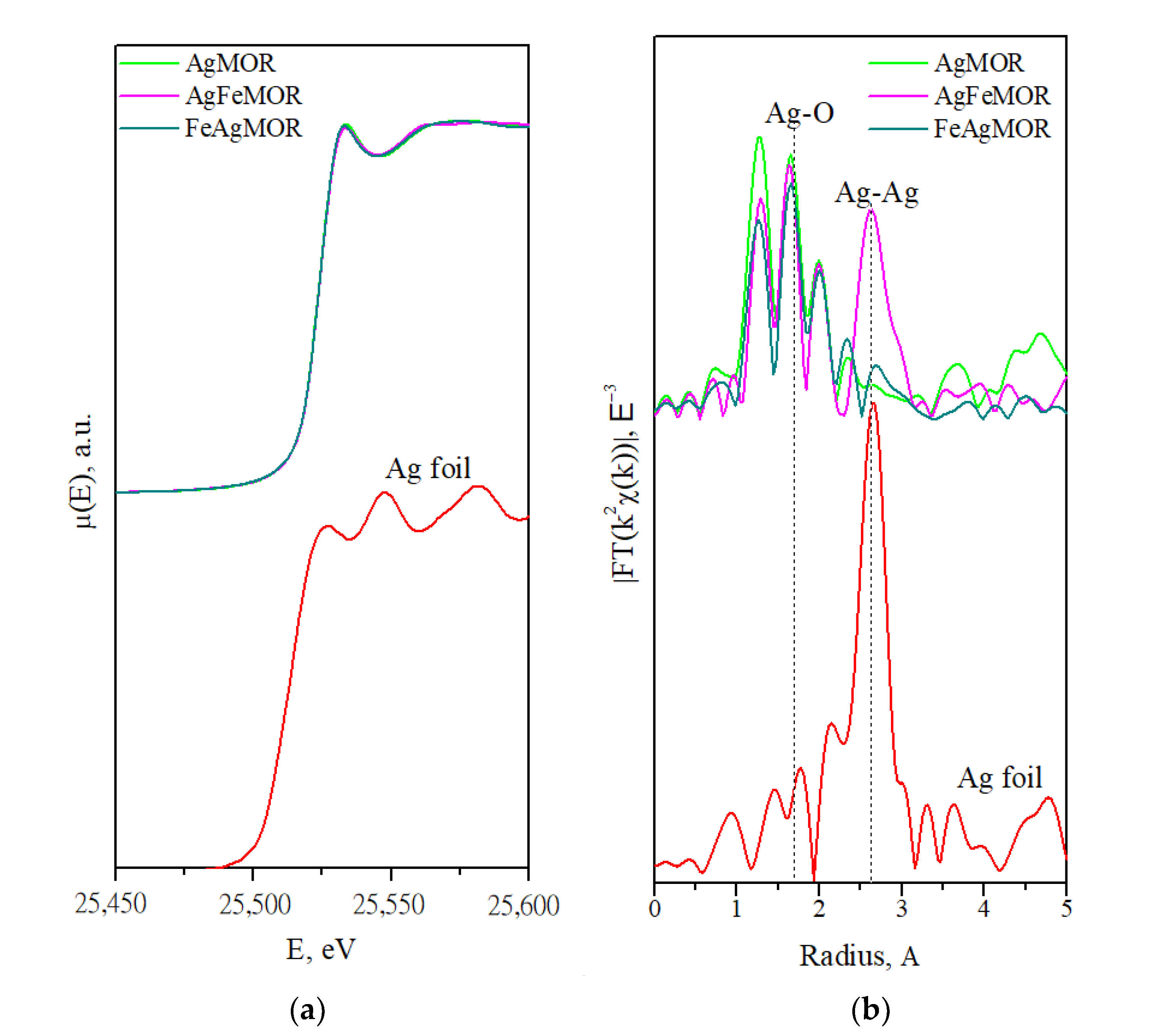
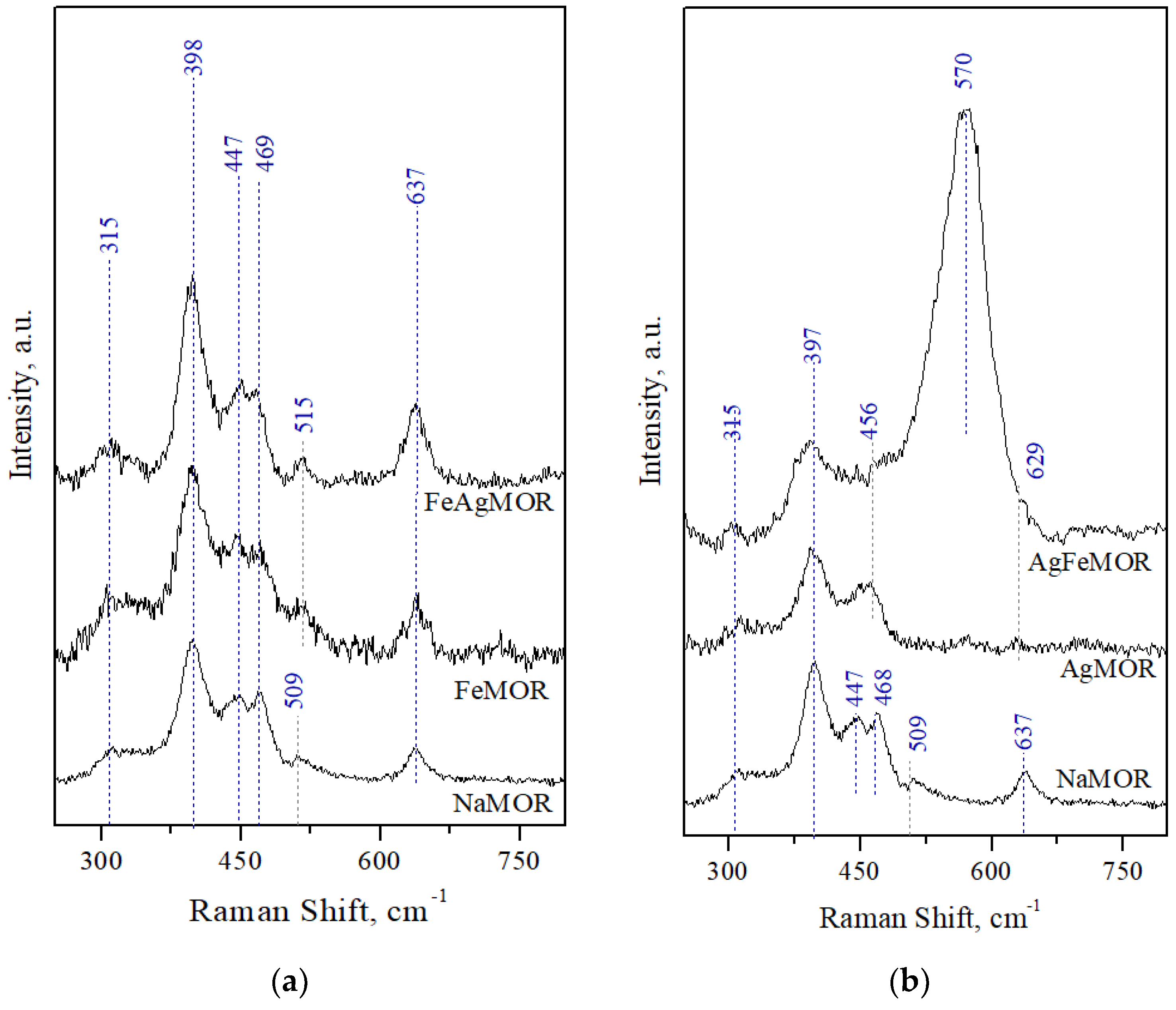
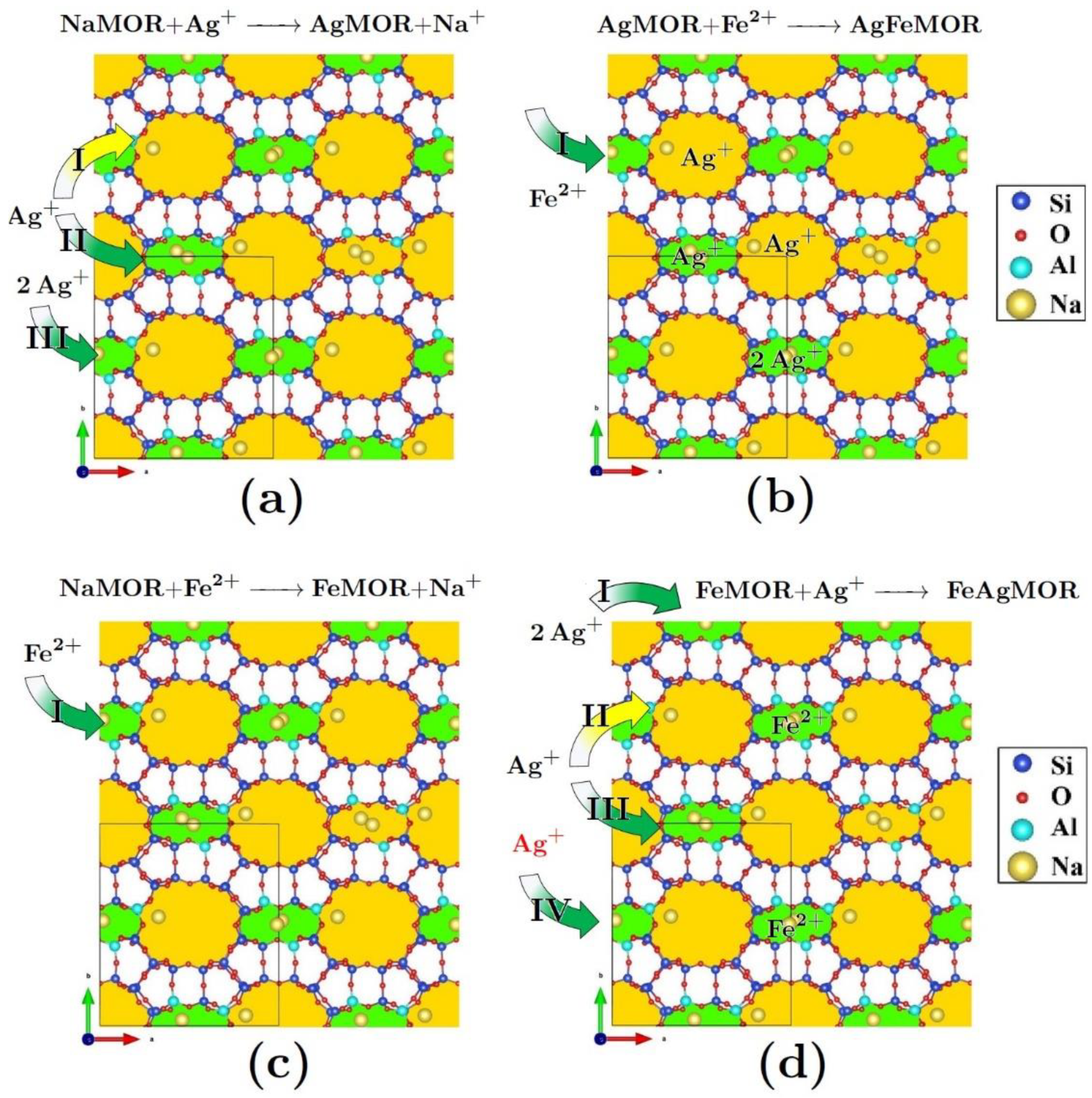
2. Results and Discussion
3. Materials and Methods
3.1. Samples Preparation
3.2. Characterization Methods
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, L.; Corma, A. Confining isolated atoms and clusters in crystalline porous materials for catalysis. Nat. Rev. Mater. 2020, 6, 244–263. [Google Scholar] [CrossRef]
- Sánchez-López, P.; Kotolevich, Y.; Yocupicio-Gaxiola, R.I.; Antúnez-García, J.; Chowdari, R.K.; Petranovskii, V.; Fuentes-Moyado, S. Recent Advances in Catalysis Based on Transition Metals Supported on Zeolites. Front. Chem. 2021, 9, 716745. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Gao, S.; Yu, J. Metal Sites in Zeolites: Synthesis, Characterization, and Catalysis. Chem. Rev. 2022. [Google Scholar] [CrossRef] [PubMed]
- Xue, K.; Mo, Y.; Long, B.; Wei, W.; Shan, C.; Guo, S.; Niu, L. Single-atom catalysts supported on ordered porous materials: Synthetic strategies and applications. InfoMat 2022, 4, e12296. [Google Scholar] [CrossRef]
- Luo, W.; Cao, W.; Bruijnincx, P.C.A.; Lin, L.; Wang, A.; Zhang, T. Zeolite-supported metal catalysts for selective hydrodeoxygenation of biomass-derived platform molecules. Green Chem. 2019, 21, 3744–3768. [Google Scholar] [CrossRef]
- Dalena, F.; Giglio, E.; Marino, A.; Aloise, A.; Giorgianni, G.; Migliori, M.; Giordano, G. Steam Reforming of Bioethanol Using Metallic Catalysts on Zeolitic Supports: An Overview. Catalysts 2022, 12, 617. [Google Scholar] [CrossRef]
- Liu, H.; You, C.; Wang, H. Experimental and Density Functional Theory Studies on the Zeolite-Based Fe–Ni–W Trimetallic Catalyst for High-Temperature NOx Selective Catalytic Reduction: Identification of Active Sites Suppressing Ammonia Over-oxidation. ACS Catal. 2021, 11, 1189–1201. [Google Scholar] [CrossRef]
- Lu, Y.; Zhang, Z.; Lin, F.; Wang, H.; Wang, Y. Single-atom Automobile Exhaust Catalysts. ChemNanoMat 2020, 6, 1659–1682. [Google Scholar] [CrossRef]
- Pappas, D.K.; Kvande, K.; Kalyva, M.; Dyballa, M.; Lomachenko, K.A.; Arstad, B.; Borfecchia, E.; Bordiga, S.; Olsbye, U.; Beato, P.; et al. Influence of Cu-speciation in mordenite on direct methane to methanol conversion: Multi-Technique characterization and comparison with NH3 selective catalytic reduction of NOx. Catal. Today 2021, 369, 105–111. [Google Scholar] [CrossRef]
- Xu, G.; Wang, H.; Yu, Y.; He, H. Role of silver species in H2-NH3-SCR of NOx over Ag/Al2O3 catalysts: Operando spectroscopy and DFT calculations. J. Catal. 2021, 395, 1–9. [Google Scholar] [CrossRef]
- Antúnez-García, J.; Galván, D.H.; Petranovskii, V.; Murrieta-Rico, F.N.; Yocupicio-Gaxiola, R.I.; Shelyapina, M.G.; Fuentes-Moyado, S. Aluminum distribution in mordenite-zeolite framework: A new outlook based on density functional theory calculations. J. Solid State Chem. 2022, 306, 122725. [Google Scholar] [CrossRef]
- Sánchez-López, P.; Kotolevich, Y.; Miridonov, S.; Chávez-Rivas, F.; Fuentes, S.; Petranovskii, V. Bimetallic AgFe Systems on Mordenite: Effect of Cation Deposition Order in the NO Reduction with C3H6/CO. Catalysts 2019, 9, 58. [Google Scholar] [CrossRef] [Green Version]
- Yeom, Y.; Li, M.; Sachtler, W.; Weitz, E.W. Low-temperature NOx reduction with ethanol over Ag/Y: A comparison with Ag/γ-Al2O3 and BaNa/Y. J. Catal. 2007, 246, 413–427. [Google Scholar] [CrossRef]
- Bartolomeu, R.; Mendes, A.N.; Fernandes, A.; Henriques, C.; da Costa, P.; Ribeiro, M.F. NOx SCR with decane using Ag–MFI catalysts: On the effect of silver content and co-cation presence. Catal. Sci. Technol. 2015, 6, 3038–3048. [Google Scholar] [CrossRef]
- Rodríguez-Iznaga, I.; Petranovskii, V.; Chávez-Rivas, F.; Shelyapina, M.G. Bimetallic Copper-Silver Systems Supported on Natural Clinoptilolite: Long-Term Changes in Nanospecies’ Composition and Stability. Inorganics 2022, 10, 34. [Google Scholar] [CrossRef]
- Liu, Q.; Bian, C.; Ming, S.; Guo, L.; Zhang, S.; Pang, L.; Liu, P.; Chen, Z.; Li, T. The opportunities and challenges of iron-zeolite as NH3-SCR catalyst in purification of vehicle exhaust. Appl. Catal. A Gen. 2020, 607, 117865. [Google Scholar] [CrossRef]
- Gao, F. Fe-Exchanged Small-Pore Zeolites as Ammonia Selective Catalytic Reduction (NH3-SCR) Catalysts. Catalysts 2020, 10, 1324. [Google Scholar] [CrossRef]
- Zhu, N.; Lian, Z.; Zhang, Y.; Shan, W.; He, H. Improvement of low-temperature catalytic activity over hierarchical Fe-Beta catalysts for selective catalytic reduction of NO with NH3. Chin. Chem. Lett. 2019, 30, 867–870. [Google Scholar] [CrossRef]
- Shelyapina, M.G.; Gurgul, J.; Łątka, K.; Sánchez-López, P.; Bogdanov, D.; Kotolevich, Y.; Petranovskii, V.; Fuentes, S. Mechanism of formation of framework Fe3+ in bimetallic Ag-Fe mordenites-Effective catalytic centers for deNOx reaction. Microporous Mesoporous Mater. 2020, 299, 109841. [Google Scholar] [CrossRef]
- Sánchez-López, P.; Kotolevich, Y.; Khramov, E.; Chowdari, R.K.; Estrada, M.A.; Berlier, G.; Zubavichus, Y.; Fuentes, S.; Petranovskii, V.; Chávez-Rivas, F. Properties of Iron-Modified-by-Silver Supported on Mordenite as Catalysts for NOx Reduction. Catalysts 2020, 10, 1156. [Google Scholar] [CrossRef]
- Fuentes, S.; Petranovskii, V.; Kotolevich, Y.; Miridonov, S.; Sánchez-López, P.; Chávez-Rivas, F.; Machorro, R. Self-assembling of ordered domains of silver nanoparticles into the mordenite channel system. In Proceedings of the 13th International Symposium on Nanophotonics and Metamaterials, St. Petersburg, Russia, 4–8 June 2018; p. 194. [Google Scholar]
- Sánchez-Lopez, P.; Miridonov, S.; Kotolevich, Y.; Chávez-Rivas, F.; Machorro, R.; Shelyapina, M.; Petranovskii, V.; Fuentes-Moyado, S. Domains of ordered monosized Ag clusters stabilized in mordenite channels formed in bimetallic Fe-Ag system supported on mordenite. In Proceedings of the XV Congreso Mexicano de Catálisis y VI Congreso Internacional, Monterrey, Mexico, 1–6 October 2017. [Google Scholar]
- Heo, N.H.; Kim, Y.; Kim, J.J.; Seff, K. Surprising Intrazeolitic Chemistry of Silver. J. Phys. Chem. C 2016, 120, 5277–5287. [Google Scholar] [CrossRef]
- Ogden, J.S.; Bogdanchikova, N.E.; Corker, J.M.; Petranovskii, V.P. Structure of silver clusters embedded in erionite channels. Eur. Phys. J. D 1999, 9, 605–608. [Google Scholar] [CrossRef]
- Fiddy, S.G.; Bogdanchikova, N.E.; Petranovskii, V.P.; Ogden, J.S.; Avalos-Borja, M. EXAFS and optical spectroscopy characterisation of silver within zeolite matrices. Stud. Surf. Sci. Catal. 2002, 142, 1939–1946. [Google Scholar] [CrossRef]
- Fiddy, S.G.; Ogden, J.S.; Petranovskii, V.P. EXAFS and optical spectroscopy characterisation of reduction products of binary silver-copper ion mixture in mordenite. Eur. Phys. J. D–At. Mol. Opt. Phys. 2003, 24, 253–256. [Google Scholar] [CrossRef]
- Antúnez-García, J.; Galván, D.H.; Petranovskii, V.; Posada-Amarillas, A. A DFT study of copper-oxide clusters embedded in dry and water-immersed siliceous mordenite. Comput. Mater. Sci. 2015, 106, 140–148. [Google Scholar] [CrossRef]
- Bogdanchikova, N.E.; Petranovskii, V.P.; Machorro, M.R.; Sugi, Y.; Soto, G.V.M.; Fuentes, S. Stability of silver clusters in mordenites with different SiO2/Al2O3 molar ratio. Appl. Surf. Sci. 1999, 150, 58–64. [Google Scholar] [CrossRef]
- Gurin, V.S.; Petranovskii, V.P.; Bogdanchikova, N.E. Metal clusters and nanoparticles assembled in zeolites: An example of stable materials with controllable particle size. Mater. Sci. Eng. C 2002, 19, 327–331. [Google Scholar] [CrossRef]
- Mulvaney, P. Surface Plasmon Spectroscopy of Nanosized Metal Particles. Langmuir 1996, 12, 788–800. [Google Scholar] [CrossRef]
- Gurin, V.S.; Petranovskii, V.P.; Hernandez, M.-A.; Bogdanchikova, N.E.; Alexeenko, A.A. Silver and copper clusters and small particles stabilized within nanoporous silicate-based materials. Mater. Sci. Eng. A 2005, 391, 71–76. [Google Scholar] [CrossRef]
- Mogensen, K.B.; Kneipp, K. Size-Dependent Shifts of Plasmon Resonance in Silver Nanoparticle Films Using Controlled Dissolution: Monitoring the Onset of Surface Screening Effects. J. Phys. Chem. C 2014, 118, 28075–28083. [Google Scholar] [CrossRef]
- Lysenko, V.S.; Mal’nev, A.F. Optical characteristics of metal blacks. J. Appl. Spectrosc. 1969, 10, 566–570. [Google Scholar] [CrossRef]
- Rodríguez-Iznaga, I.; Petranovskii, V.; Castillón-Barraza, F.; Concepción-Rosabal, B. Copper-Silver Bimetallic System on Natural Clinoptilolite: Thermal Reduction of Cu2+ and Ag+ Exchanged. J. Nanosci. Nanotechnol. 2011, 11, 5580–5586. [Google Scholar] [CrossRef]
- Santhosh Kumar, M.; Schwidder, M.; Grunert, W.; Bentrup, U.; Bruckner, A. Selective reduction of NO with Fe-ZSM-5 catalysts of low Fe content: Part II. Assessing the function of different Fe sites by spectroscopic in situ studies. J. Catal. 2006, 239, 173–186. [Google Scholar] [CrossRef]
- Bordiga, S.; Buzzoni, R.; Geobaldo, F.; Lamberti, C.; Giamello, E.; Zecchina, A.; Leofanti, G.; Petrini, G.; Tozzola, G.; Vlaic, G. Structure and Reactivity of Framework and Extraframework Iron in Fe-Silicalite as Investigated by Spectroscopic and Physicochemical Methods. J. Catal. 1996, 158, 486–501. [Google Scholar] [CrossRef]
- Setyawati, I.A.; Rettig, S.J.; Orvig, C. Cationic iron(III) complex with a hexadentate N2, N′2′, O2-aminopyridylphenolate ligand. Can. J. Chem. 1999, 77, 2033–2038. [Google Scholar] [CrossRef]
- Rtimi, S.; Baghriche, O.; Sanjines, R.; Pulgarin, C.; Bensimon, M.; Kiwi, J. TiON and TiON-Ag sputtered surfaces leading to bacterial inactivation under indoor actinic light. J. Photochem. Photobiol. A Chem. 2013, 256, 52–63. [Google Scholar] [CrossRef] [Green Version]
- Mejía, M.I.; Restrepo, G.; Marín, J.M.; Sanjines, R.; Pulgarín, C.; Mielczarski, E.; Mielczarski, J.; Kiwi, J. Magnetron-Sputtered Ag Surfaces. New Evidence for the Nature of the Ag Ions Intervening in Bacterial Inactivation. ACS Appl. Mater. Interfaces 2010, 2, 230–235. [Google Scholar] [CrossRef]
- Lopez-Salido, I.; Lim, D.C.; Kim, Y.D. Ag nanoparticles on highly ordered pyrolytic graphite (HOPG) surfaces studied using STM and XPS. Surf. Sci. 2005, 588, 6–18. [Google Scholar] [CrossRef]
- Lim, D.C.; Lopez-Salido, I.; Kim, Y.D. Size selectivity for CO-oxidation of Ag nanoparticles on highly ordered pyrolytic graphite (HOPG). Surf. Sci. 2005, 598, 96–103. [Google Scholar] [CrossRef]
- Suzuki, Y.; Miyanaga, T.; Hoshino, H.; Matsumoto, N.; Ainai, T. In-Situ XAFS Study of Ag Clusters in Zeolite 4A. Phys. Scr. 2005, 2005, 765. [Google Scholar] [CrossRef]
- Borgna, A.; Stagg, S.M.; Resasco, D.E. Interference Phenomena in the EXAFS Spectra of Pt−Sn Bimetallic Catalysts. J. Phys. Chem. B 1998, 102, 5077–5081. [Google Scholar] [CrossRef]
- Niggli, P. XII. Die Kristallstruktur einiger Oxyde I. Z. Für Krist.-Cryst. Mater. 1922, 57, 253–299. [Google Scholar] [CrossRef] [Green Version]
- Boix, A.V.; Aspromonte, S.G.; Miro, E.E. Deactivation studies of the SCR of NOx with hydrocarbons on Co-mordenite monolithic catalysts. Appl. Catal. A Gen. 2008, 341, 26–34. [Google Scholar] [CrossRef]
- Dutta, P.K.; Rao, K.M.; Park, J.Y. Correlation of Raman Spectra of Zeolites with Framework Architecture. J. Phys. Chem. 1991, 95, 6654–6656. [Google Scholar] [CrossRef]
- Dutta, P.K.; Puri, M. Synthesis and Structure of Zeolite ZSM-5: A Raman Spectroscopic Study. J. Phys. Chem. 1987, 91, 4329–4333. [Google Scholar] [CrossRef]
- Król, M.; Mozgawa, W.; Barczyk, K.; Bajda, T.; Kozanecki, M. Changes in the vibrational spectra of zeolites due to sorption of heavy metal cations. J. Appl. Spectrosc. 2013, 80, 644–650. [Google Scholar] [CrossRef]
- Jin, S.; Feng, Z.; Fan, F. UV Raman Spectroscopic Characterization of Catalysts and Catalytic Active Sites. Catal. Lett. 2015, 145, 468–481. [Google Scholar] [CrossRef]
- Yu, Y.; Xiong, G.; Li, C.; Xiao, F.-S. Characterization of Iron Atoms in the Framework of MFI-Type Zeolites by UV Resonance Raman Spectroscopy. J. Catal. 2000, 194, 487–490. [Google Scholar] [CrossRef]
- Sun, K.; Fan, F.; Xia, H.; Feng, Z.; Li, W.-X.; Li, C. Framework Fe Ions in Fe-ZSM-5 Zeolite Studied by UV Resonance Raman Spectroscopy and Density Functional Theory Calculations. J. Phys. Chem. C 2008, 112, 16036–16041. [Google Scholar] [CrossRef]
- Ju, X.; Tian, F.; Wang, Y.; Fan, F.; Feng, Z.; Li, C. A novel synthetic strategy of Fe-ZSM-35 with pure framework Fe species and its formation mechanism. Inorg. Chem. Front. 2018, 5, 2031–2037. [Google Scholar] [CrossRef]
- Li, Y.; Feng, Z.; Xin, H.; Fan, F.; Zhang, J.; Magusin, P.C.M.M.; Hensen, E.J.M.; van Santen, R.A.; Yang, Q.; Li, C. Effect of Aluminum on the Nature of the Iron Species in Fe-SBA-15. J. Phys. Chem. B 2006, 110, 26114–26121. [Google Scholar] [CrossRef]
- Chlebda, D.K.; Stachurska, P.; Jedrzejczyk, R.J.; Kuterasinski, Ł.; Dziedzicka, A.; Górecka, S.; Chmielarz, L.; Łojewska, J.; Sitarz, M.; Jodłowski, P.J. DeNOx Abatement over Sonically Prepared Iron-Substituted Y, USY and MFI Zeolite Catalysts in Lean Exhaust Gas Conditions. Nanomaterials 2018, 8, 21. [Google Scholar] [CrossRef] [Green Version]
- Bremard, C.; Le Maire, M. Low-frequency Raman spectra of dehydrated faujasite zeolites. J. Phys. Chem. 1993, 97, 9695–9702. [Google Scholar] [CrossRef]
- Brandenberger, S.; Kröcher, O.; Tissler, A.; Althoff, R. The State of the Art in Selective Catalytic Reduction of NOx by Ammonia Using Metal-Exchanged Zeolite Catalysts. Catal. Rev. 2008, 50, 492–531. [Google Scholar] [CrossRef]
- Ravel, B.; Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 2005, 12, 537–541. [Google Scholar] [CrossRef] [Green Version]
- Newville, M. IFEFFIT: Interactive XAFS analysis and FEFF fitting. J. Synchrotron Radiat. 2001, 8, 322–324. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | SBET, m2∙g−1 | Vtotal, cm3∙g−1 | Vmicro, cm3∙g−1 | Pore Diameter, Å |
|---|---|---|---|---|
| NaMOR | 338 | 0.19 | 0.16 | 22.8 |
| AgMOR | 322 | 0.19 | 0.14 | 23.2 |
| FeMOR | 398 | 0.23 | 0.17 | 23.3 |
| AgFeMOR | 343 | 0.20 | 0.15 | 23.7 |
| FeAgMOR | 336 | 0.20 | 0.14 | 24.1 |
| Sample | Atomic % | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Si | Al | O | Ag | Fe | Na | Si/Al | EIEM | ||
| EIEM-Fe2+ | EIEM–Fe3+ | ||||||||
| NaMOR | 48.9 | 7.5 | 33.9 | - | - | 9.7 | 6.5 | 1.29 | |
| AgMOR | 38.1 | 5.8 | 49.9 | 4.6 | - | 1.6 | 6.5 | 1.07 | |
| FeMOR | 44.0 | 6.4 | 45.4 | - | 0.8 | 3.4 | 6.8 | 0.78 | 0.91 |
| FeAgMOR | 37.2 | 5.4 | 51.9 | 3.6 | 0.4 | 1.5 | 6.9 | 1.09 | 1.17 |
| AgFeMOR | 39.7 | 5.9 | 49.7 | 3.1 | 0.9 | 0.7 | 6.7 | 0.95 | 1.10 |
| Samples | Ag ions | Ag-Support Interaction | Ag0 | Agn Clusters, Ø < 2 nm |
|---|---|---|---|---|
| 366.2 eV | 367.4–368 eV | 368.0–368.2 eV | ≥369 eV | |
| AgMOR | - | 367.07-(12%) | - | 368.79-(88%) |
| AgFeMOR | 366.56-(20%) | 368.52-(80%) | ||
| FeAgMOR | - | 367.65-(29%) | - | 369.14-(71%) |
| Sample | Scattering Path | Coordination Number | Interatomic Distance, Å | Debye Factor, Å2 | R-Factor, % |
|---|---|---|---|---|---|
| Ref. Ag foil | Ag-Ag | 12 | 2.89 | - | - |
| Ref. Ag2O | Ag-O | 4 | 2.04 [44] | - | - |
| Ag-Ag | 8 | 3.34 | - | ||
| AgMOR | Ag-O | 1.4 | 1.91 | 0.0001 | 3.1 |
| 1.4 | 2.05 | ||||
| 1.4 | 2.12 | ||||
| 1.4 | 2.24 | ||||
| Ag-(Si or Al) | 1.3 | 2.82 | 0.0020 | ||
| 1.3 | 2.96 | ||||
| 1.3 | 3.02 | ||||
| 1.3 | 3.15 | ||||
| Ag-Ag | 0 | - | - | ||
| AgFeMOR | Ag-O | 1 | 1.90 | 0.0001 | 2.3 |
| 1 | 2.04 | ||||
| 1 | 2.10 | ||||
| 1 | 2.22 | ||||
| Ag-(Si or Al) | 1 | 2.92 | 0.0028 | ||
| 1 | 2.92 | ||||
| 1 | 3.12 | ||||
| 1 | 3.21 | ||||
| Ag-Ag | 1.6 | 2.82 | 0.0059 | ||
| FeAgMOR | Ag-O | 1 | 1.90 | 0.0009 | 1.3 |
| 1 | 2.04 | ||||
| 1 | 2.11 | ||||
| 1 | 2.22 | ||||
| Ag-(Si or Al) | 1 | 2.90 | 0.0030 | ||
| 1 | 2.90 | ||||
| 1 | 3.02 | ||||
| 1 | 3.16 | ||||
| Ag-Ag | 0.5 | 2.76 | 0.0043 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-López, P.; Kotolevich, Y.; Antúnez-García, J.; Chávez-Rivas, F.; Khramov, E.; Berlier, G.; Moreno-Ruiz, L.; Zubavichus, Y.; Petranovskii, V.; Fuentes-Moyado, S.; et al. Influence of Components Deposition Order on Silver Species Formation in Bimetallic Ag-Fe System Supported on Mordenite. Catalysts 2022, 12, 1453. https://doi.org/10.3390/catal12111453
Sánchez-López P, Kotolevich Y, Antúnez-García J, Chávez-Rivas F, Khramov E, Berlier G, Moreno-Ruiz L, Zubavichus Y, Petranovskii V, Fuentes-Moyado S, et al. Influence of Components Deposition Order on Silver Species Formation in Bimetallic Ag-Fe System Supported on Mordenite. Catalysts. 2022; 12(11):1453. https://doi.org/10.3390/catal12111453
Chicago/Turabian StyleSánchez-López, Perla, Yulia Kotolevich, Joel Antúnez-García, Fernando Chávez-Rivas, Evgeny Khramov, Gloria Berlier, Luis Moreno-Ruiz, Yan Zubavichus, Vitalii Petranovskii, Sergio Fuentes-Moyado, and et al. 2022. "Influence of Components Deposition Order on Silver Species Formation in Bimetallic Ag-Fe System Supported on Mordenite" Catalysts 12, no. 11: 1453. https://doi.org/10.3390/catal12111453