
The Power of Biocatalysts for Highly Selective and Efficient Phosphorylation Reactions


1
Institute of Molecular and Industrial Biotechnology, Lodz University of Technology, 90-537 Lodz, Poland
2
Swiss Coordination Committee Biotechnology (SKB), 8021 Zurich, Switzerland
Catalysts 2022, 12(11), 1436; https://doi.org/10.3390/catal12111436
Submission received: 16 August 2022
/
Revised: 8 November 2022
/
Accepted: 11 November 2022
/
Published: 15 November 2022
(This article belongs to the Special Issue 10th Anniversary of Catalysts: Biocatalysis in Analysis and Synthesis—Past, Present and Future)
Abstract
:Reactions involving the transfer of phosphorus-containing groups are of key importance for maintaining life, from biological cells, tissues and organs to plants, animals, humans, ecosystems and the whole planet earth. The sustainable utilization of the nonrenewable element phosphorus is of key importance for a balanced phosphorus cycle. Significant advances have been achieved in highly selective and efficient biocatalytic phosphorylation reactions, fundamental and applied aspects of phosphorylation biocatalysts, novel phosphorylation biocatalysts, discovery methodologies and tools, analytical and synthetic applications, useful phosphoryl donors and systems for their regeneration, reaction engineering, product recovery and purification. Biocatalytic phosphorylation reactions with complete conversion therefore provide an excellent reaction platform for valuable analytical and synthetic applications.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
From the discovery of the element phosphorus and its chemistry to the current status of knowledge, many light as well as dark aspects have been emerging, connected to value and waste, to health and disease and to life and death. The element phosphorus is of central importance for all living organisms as well as for various human activities, from mining to agriculture, industry, science, the environment and society. Therefore, adequate attention must be paid to the global phosphorus cycle, which involves biochemical as well as geochemical reactions and pathways [1]. The resource-efficient use and recycling of phosphorus is essential at different scales on our planet as the biochemical flow of phosphorus has been identified as a planetary boundary at high risk [2]. As the phosphorus cycle is perturbed by human activities such as the increased utilization of phosphorus mineral resources and increasing abundance of phosphate in aqueous environments, work on closing the phosphorus loop by phosphorus recovery and recycling is important [3]. As phosphoric acid is produced as a key industrial intermediate for phosphate fertilizers on a very large scale by a sulfuric acid treatment of phosphate rock raw materials in a wet process, phosphoric acid is also of much interest for replacing white phosphorus as a precursor in manufacturing phosphorus-containing nonfertilizer chemicals [4]. Thereby, the amount of phosphogypsum waste generated in the wet process is four times larger than the phosphorus fertilizer and also contains toxic metals and radionuclides, which limit the resource efficiency and sustainability of this phosphorus value chain [5]. It is however not only at the global scale but also at the regional and local scale where a sustainable utilization of the nonrenewable element phosphorus is of key importance for a balanced phosphorus cycle, for example for the life and death of biological organisms in nature, for the maintenance of a resilient agriculture and for minimizing environmental pollution.
The long-standing scientific interest in many fundamental aspects of reactions involving the transfer of phosphorus-containing groups is therefore of key importance in obtaining new knowledge about reactions in phosphorus chemistry and their mechanisms, thermodynamics and kinetics. The unique kinetic and thermodynamic characteristics of phosphorus-containing molecules and ionized phosphate groups are of much interest in prebiotic phosphorylation [6], in the highly important role of phosphorus for the biosphere, life and nature on planet earth [7,8,9], as well as for present and future resource-efficient and sustainable phosphorylation reactions [10]. Spontaneous sugar phosphorylation has been discovered to occur in microdroplets at ambient pressure and temperature, and the ΔG of the phosphorylation reaction in microdroplets has been demonstrated to be negative and much lower than for the reaction in a bulk solution, thus making the phosphorylation reaction more favorable in microdroplets than in bulk solutions [11].
The thermodynamic challenge of a positive Gibbs free energy change (ΔG) for phosphorylation in bulk solutions can be overcome by different approaches depending on the type of phosphorylation reaction. The two main chemical approaches in stoichiometric phosphorylations, for example the conversion of alcohols to phosphate monoesters, require three reaction steps, including an oxidation step and protecting group removal when using trivalent P(III) or two reaction steps including a hydrolysis step when pentavalent P(V) is involved. The direct esterification of alcohols under mild conditions has been pioneered by Cramer using an activated phosphoric acid [12,13]. In the condensation of phosphoric acid with alcohols, the reaction can be favorably shifted towards the product by removing the water formed in the esterification azeotropically from the reaction [14]. The mixed anhydride acetyl phosphate, which was prepared by the activation of phosphoric acid by acetic anhydride, used a high energy phosphoryl donor in the monophosphorylation of alcohols [15]. Good isolated yields have been obtained by the use of tetrabutylammonium dihydrogenphosphate as a phosphate donor and trichloroacetonitrile as an esterification agent [16]. The need for robust and selective one-step phosphorylation reactions, which are sustainable, protecting-group free and versatile, has made the development of highly selective and efficient catalytic phosphorylation reactions an important goal. In the case of the direct phosphorylation of alcohols catalyzed by tetrabutylammonium hydrogensulfate, a favorable Gibbs free energy change can be achieved by the use of the high-energy phosphoryl donor potassium phosphoenolpyruvate [17].
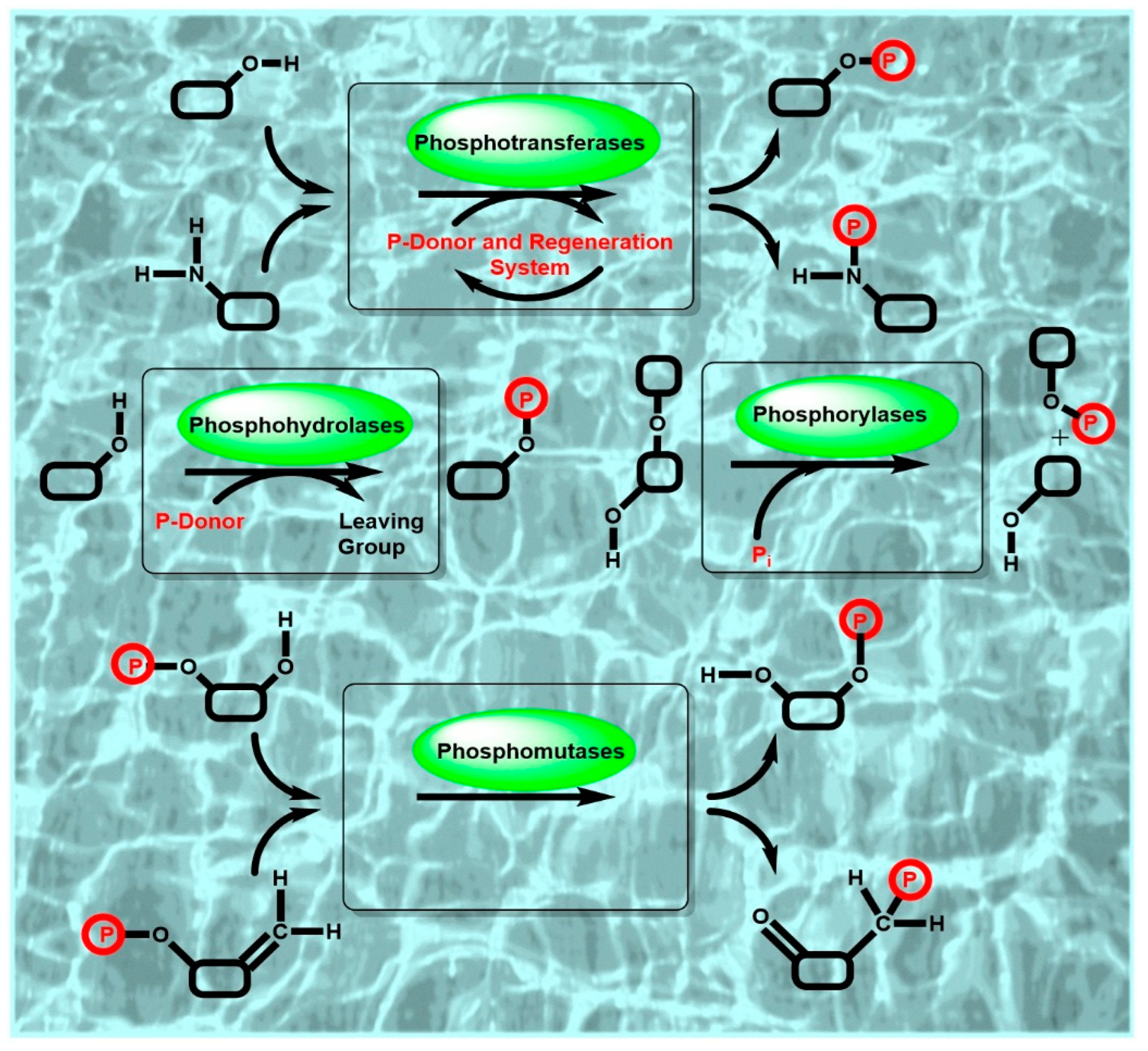
Due to the importance of highly selective and efficient catalytic phosphorylation reactions in the biosphere, phosphorylation biocatalysts, phosphoryl donors and biocatalytic systems are omnipresent in nature. Biocatalytic phosphorylations have therefore been of much interest for both in vitro applications such as biocatalytic syntheses as well for in vivo biotransformations, for example, biocatalytic prodrug activation to biologically active pharmaceuticals in the human body or biocatalytic antibiotic deactivation in drug-resistant microbes by the phosphorylation of antibiotics [10]. The discovery and characterization of phosphoryl-group transferring enzymes from nature (see Figure 1 for a schematic representation of key classes of phosphorylation biocatalysts and the involved reactions) continues therefore to be of much interest. In addition, computational and experimental enzyme engineering methodologies and tools have enabled the development of novel biocatalysts for specific reaction conditions, for catalyzing the phosphorylation of non-natural substrates, or for new-to-nature reactions.
The main goal of this review is to highlight the current state of the art and advances in highly selective biocatalytic phosphorylation reactions, fundamental and applied aspects of phosphorylation biocatalysts, novel phosphorylation biocatalysts, discovery methodologies and tools, analytical as well as synthetic applications, useful phosphoryl donors and systems for their regeneration, reaction engineering, product recovery and purification. The significant advances in all these areas towards improving the resource-efficiency of the overall bioprocess have made selective and efficient biocatalytic phosphorylation reactions an excellent reaction platform for valuable industrial applications.
2. Structures, Functions and Mechanisms of Phosphorylation Biocatalysts
The rational knowledge organization for a growing number of phosphorylation biocatalysts benefits from the unique classification of an enzymes according to its functions of catalyzing reactions from substrates to products, which was introduced more than 60 years ago and which is continuously developed further by the Enzyme Commission [18]. The description of enzymes by standardized nomenclature, classification and the four-digit Enzyme Commission number (EC number), which is well established for identifying an enzyme according to the reaction or transport catalyzed, is also very valuable for connecting information on phosphorylation biocatalysts, such as structures, functions and mechanisms.
Molecular information on phosphorylation biocatalysts can benefit greatly from resources accumulating structural data at a fast pace from sequencing work, from the first gene sequence to the present wealth of relevant sequences, from hypothetical or unreviewed annotations to well-characterized and experimentally validated genes. More than 2 billion nucleotide sequences [19] in the genetic sequence database GenBank in June 2022 and sequence data in the Universal Protein Resource Knowledgebase UniProtKB provide comprehensive sequence data of proteins [20], including phosphorylation biocatalysts. The UniProtKB Release 2022_02 contains in UniProtKB/TrEMBL more than 231 million entries, which are however largely predicted, automatically annotated and unreviewed, while 567,483 protein sequences have been manually annotated and reviewed in UniProtKB/SwissProt [20]. An impressive growth of nearly 10% per year can also be observed in the number of experimentally determined three-dimensional protein structures, from the first reported structure to the current number in the Protein Data Bank (PDB) [21]. The breakthrough of the artificial intelligence (AI) system Alphafold developed by Deepmind to predict protein structures with high accuracy [22,23] and the release of over 200 million protein structure predictions recently by the partnership of Deepmind with EMBL-EBI in AlphaFold Protein Structure Database provide great opportunities for accelerating research on phosphorylation biocatalysts.
In addition to the information resources on the sequences and structures of phosphorylation biocatalysts, easy access to the current status of knowledge about their functional properties, such as activities and kinetic parameters, substrate scope or stabilities, as well as other information about their biological context and occurrence in nature is of major importance. By focusing for 35 years on the continuous and ongoing extraction of data on classified enzymes from the literature, the Braunschweig Enzyme Database (BRENDA) has become the most comprehensive and globally utilized information resource on enzymes, which also enables fast access to existing knowledge about classified phosphorylation biocatalysts [24], which has been extracted retrospectively from publications. With the growing number of publications on enzymes and as the functional descriptions of biocatalysts in publications may vary, guidelines on standardizing how biocatalytic reactions should be reported [25] facilitate the exchange of enzyme function data. The Standards for Reporting Enzymology Data (STRENDA) database, which has been launched as an enzyme function database [26] incorporating the STRENDA guidelines, enables authors to check the completeness and validity of enzymology datasets before submission to a journal and supports the quality of the whole workflow from discovery to publication and information retrieval. The elucidation of relationships between the structure and function of phosphorylation biocatalysts is not only of fundamental interest but also highly valuable for the discovery and development of novel phosphorylation biocatalysts with desired function and performance.
2.1. Structures of Phosphorylation Biocatalysts
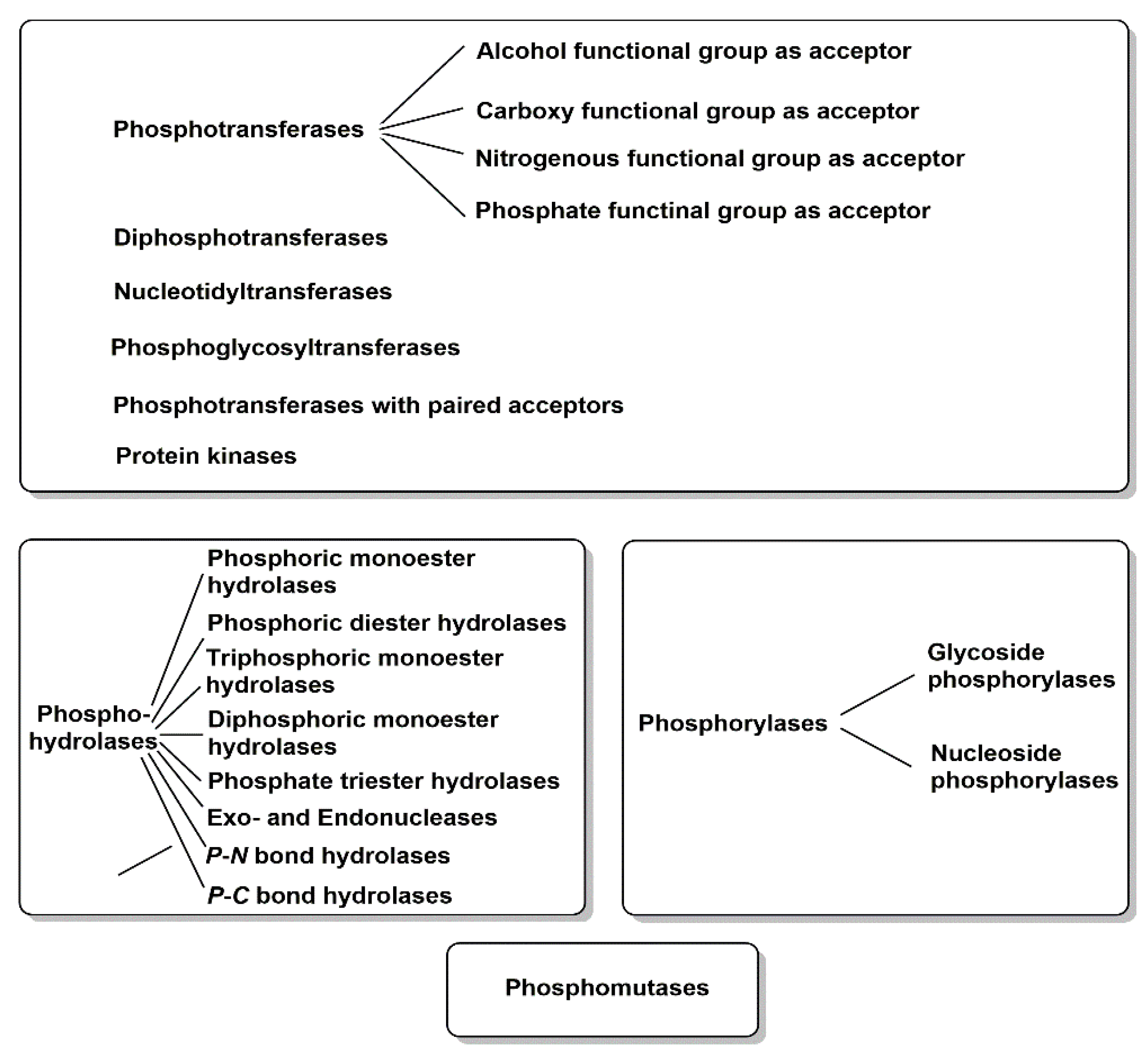
Biocatalysts which catalyze phosphoryl group transfer reactions are mainly classified in the class of transferases, which have an Enzyme Commission (EC) four-digit number starting with two, but there are also various phosphorylation biocatalysts from the hydrolases of EC class 3 and the isomerases from EC class 5. Therefore, phosphotransferases and phosphorylases of EC class 1, phosphohydrolases of EC class 3 and phosphomutases of EC class 5 form the scope of this review. Beyond the scope of this review are biocatalysts which, in addition to phosphorylation, catalyze other reactions, such as D-glyceraldehyde 3-phosphate dehydrogenase from the oxidoreductases of EC class 1 catalyzing the oxidative phosphorylation of D-glyceraldehyde 3-phosphate to D-1,3-diphosphosphate glycerate, or carbamoyl phosphate synthases, which utilize ammonia or hydrolyze L-glutamine, from the ligases of EC class 6 catalyzing carbon–nitrogen bond formation in addition to phosphorylation.
2.1.1. Phosphotransferase Structures
The largest number of phosphorylation biocatalyst structures has been reported for phosphotransferases in EC class 2.7, with 36,705 reviewed protein sequences listed under EC class 2.7 in UniProtKB/SwissProt and 5,559,483 unreviewed protein sequences listed under EC class 2.7 in UniProtKB/TrEMBL [20]. The growing number of phosphotransferase/kinases has been classified according to sequences and different criteria such as structural folds, catalyzed reaction types, evolutionary relationships or biological organisms into protein families, which have also been assembled into fold groups according to similar structural folds [27,28,29,30]. A total of 213,201 experimentally determined three-dimensional structures of phosphotransferases, among which 1401 structures were obtained with a resolution of 1.5 A or better, have been deposited in PDB [21]. The first three-dimensional kinase structures, which were obtained more than 4 decades ago by x-ray diffraction, involved the metabolic enzymes hexokinase B from Saccharomyces cerevisiae [31], pyruvate kinase from cat muscle [32] and phosphoglycerate kinase from Saccharomyces cerevisiae [33]. In the meantime, the number of kinase structures with small molecules as substrates has been continuously growing, for example, for carbohydrate kinases, lipid kinases, nucleoside kinases, nucleoside monophosphate and nucleoside diphosphate kinases, hydroxyacid kinases and amino acid kinases [21]. More than 17,000 kinase sequences were classified based on the similarity of their sequences into 30 families, whereby 98% of all the sequences are represented by 19 families and fall into seven general structural folds [27]. These fold groups with known three-dimensional structure include the Rossmann fold, ferredoxin fold, ribonuclease H fold and TIM β/α-barrel, which are some of the most widespread folds [27]. Numerous structures of human and viral nucleoside kinases, nucleoside monophosphate kinases and nucleoside diphosphate kinases have been determined and are highly relevant for the activation cascade of nucleoside analogs to their active triphosphate forms at high concentrations and at the desired site [34]. Structures determined in different environments may differ, as described for example for the small integral membrane protein diacylglycerol kinase DgkA, which catalyzes the ATP-dependent phosphorylation of diacylglycerol to phosphatidic acid, where the domain swapping observed by NMR in the solution structure [35] was not observed by X-ray in the crystal structure [36] or by magic angle spinning solid-state NMR in phospholipid bilayers [37]. The analysis of protein kinase structures continues to attract major interest since the first report, describing the structure of a catalytic subunit of a protein kinase depending on cyclic adenosine monophosphate (cAMP), more than three decades ago [38], because of their fundamental importance in the post-translation modification of proteins, signaling and drug discovery [39,40,41]. The discovery of ribonucleic acid kinase ArkI and the determination of its structure opens a window into a whole new range of kinases and is of great interest in the post-transcriptional modification of tRNA [42].
2.1.2. Phosphohydrolase Structures
Phosphohydrolases or phosphatases in EC class 3.1, which in nature catalyze the hydrolytic reaction direction, can also be used for catalyzing the reverse reaction of phosphorylation. A large number of phosphatase sequences are known and have been deposited in UniProtKB/SwissProt and UniProt/TrEMBL: 4399 reviewed and 669,414 unreviewed protein sequences of the phosphoric-monoester hydrolases of enzyme class EC 3.1.3, 1043 reviewed and 247,424 unreviewed protein sequences of the phosphoric-diester hydrolases of enzyme class EC 3.1.4 and 34 reviewed and 7396 unreviewed protein sequences of the diphosphoric-monoester hydrolases of enzyme class EC 3.1.7 [20]. Numerous three-dimensional phosphatase structures have been determined experimentally and deposited in PDB: 2639 3D structures of the phosphoric-monoester hydrolases of enzyme class EC 3.1.3, 936 3D structures of the phosphoric-diester hydrolases of enzyme class EC 3.1.4 and 16 3D structures of the diphosphoric-monoester hydrolases of enzyme class EC 3.1.7 [21].
A wealth of structural information is now available for phosphatases, and the structural classification of proteins (SCOP) database [43] now lists 13 folds, 23 superfamilies and 48 families for phosphatases, enabling new insights about the role of the protein scaffold in the transfer of phosphoryl groups [44]. The example of the crystal structures determined for the enzyme encoded by the gene Rv2131c from Mycobacterium tuberculosis, which was originally assumed to be a bifunctional enzyme inositol monophosphatase/fructose-1,6-bisphosphatase and then discovered to be a 3′-phosphoadenosine-5′-phosphatase and showing 31% identity of its amino acid sequence with the regulator protein CysQ of sulfate assimilation in E. coli [45], illustrates the value of paying attention to molecular details and the experimental verification of annotations [46]. The CysQ structures have been determined in a ligand-free form; in the substrate-bound form, which contains phosphoadenosine-phosphate and is lithium-inhibited; and the product-bound form, in which AMP, phosphate and 3 Mg2+ ions are bound [46].
As the phosphotransferase versus phosphohydrolase balance of most phosphatases is unfavorable for phosphorylation reactions, the identification of structural determinants for shifting this balance towards high phosphotransferase activity and low hydrolases activity is desirable. The structures of nonspecific acid phosphatases have attracted much interest in order to understand the factors enabling their use in phosphorylation reactions with inexpensive phosphoryl donors for a broad range of substrates. The structure of the nonspecific acid phosphatase from Escherichia blattae [47] was important for the development of a phosphatase with enhanced phosphoryl group transfer activity over its inherent phosphatase activity. Thereby, the discovery that in the homologous nonspecific acid phosphatase from Morganella morganii the productivity of the 5′-phosphorylation of inosine was increased when the value for the Michaelis–Menten constant Km for inosine was reduced, was important for guiding the structure-based engineering of the phosphatase for the inosine 5′-phosphorylation [48]. The replacement of Gly74 by Aspartic acid and Ile153 by Threonine in the wild-type enzyme of Escherichia blattae practically led to no tertiary structure change in the G74D/I153T mutant, but it reduced the Km value for inosine [49]. Further mutations around the binding site of inosine have resulted in the S72F/G74D/I153T mutant enzyme, which showed Vmax to be 2.7-fold higher and Km to be 5.4-fold lower than the wild-type enzyme [49,50]. The same phosphotransferase level as in Morganella morganii was reached for the acid phosphatase by increasing the number of amino acid substitutions to 11 by site-directed mutagenesis, elucidating amino acid positions relevant for phosphotransferase activity [50].
The crystal structure of the acid phosphatase from Pseudomonas aeruginosa, containing in the asymmetric unit 3 identical units, each consisting of 10 α-helices, showed His132 acting as the key acid−base catalyst for phosphorylation reaction [51]. The introduction of charged residues near the active site has demonstrated an increase in the phosphorylation/hydrolysis ratio, for example with the optimal Asp135 → Arg135 mutation showing a 2.9-fold increase in the 2-phosphorylation of L-ascorbic acid [51].
2.1.3. Phosphorylase Structures
Phosphorylases in EC class 2.4, which catalyze the reversible phosphorolytic breakdown of an O- or an N-glycosidic bond using inorganic phosphate to generate a specific glycosyl phosphate, are listed with 1191 reviewed protein sequences in UniProtKB/SwissProt and with 282,371 unreviewed protein sequences in UniProtKB/TrEMBL [20]. Prominent phosphorylase biocatalysts are the glycoside phosphorylases, which have already been known for more than eight decades [52], and the nucleoside phosphorylases, which are involved in the reversible phosphorolysis of purine and pyrimidine nucleosides [53]. A total of 1319 experimentally determined three-dimensional structures of phosphorylases, among which 57 structures have been obtained with a resolution of 1.5 A or better, have been deposited in PDB [21].
The glycoside phosphorylases are mainly classified in the glycoside hydrolase family (GH family) of the carbohydrate-active enzymes database (CAZy database) [54] and more than one X-ray structure for each family of disaccharide phosphorylases, which are popular for phosphorolysis reactions, have been determined [55].
Recently, the crystal structures of a new class of nucleoside phosphorylases were determined, which showed a conserved dimeric Cupin fold with a high hydrophobic dimer interface [56], while the classical two families of nucleoside phosphorylases had a different structure. The nucleoside phosphorylase-I (NP-I) family enzymes are trimeric or hexameric and share a common α/β-subunit fold, while the nucleoside phosphorylase-II (NP-II) family enzymes have dimeric structures.
2.1.4. Phosphomutase Structures
Among the intramolecular transferases of EC class 5.4, the phosphotransferases or phosphomutases of EC class 5.4.2 are of much interest: 1933 reviewed and 156,197 unreviewed protein sequences of the phosphomutases can be found in UniProtKB/SwissProt and UniProtKB/TrEMBL, while 244 experimentally determined 3D structures of the phosphomutases of EC class 5.4.2, among which 107 structures have been obtained with a resolution of 1.5 A or better, have been deposited in PDB [21].
Phosphopentomutase from Bacillus cereus, the crystal structure of which was the first structure published of a procaryotic phosphopentomutase, has been shown to fold into a core domain organized around an alkaline phosphatase fold and a cap domain, with the active site housed by an electropositive cleft at the interface between the core and cap domains [57]. Phosphomannomutase HAD5 from Plasmodium falciparum, which is essential for this malaria-causing parasite and also has phosphoglucomutase activity, has recently been shown to have a similar structure to the human enzyme with two domains, although significant sequence variations have been found in an active site loop [58].
2.2. Functions of Phosphorylation Biocatalysts
The importance of powerful biocatalysts for the highly selective covalent introduction of polar and charged phosphoryl-groups to a large variety of small and large substrate molecules is reflected by the great functional diversity of phosphorylation biocatalysts. An overview of the major functions of the phosphoryl-group transferring biocatalysts of EC classes 1, 3 and 5 is summarized in Figure 2.
2.2.1. Phosphotransferase Functions
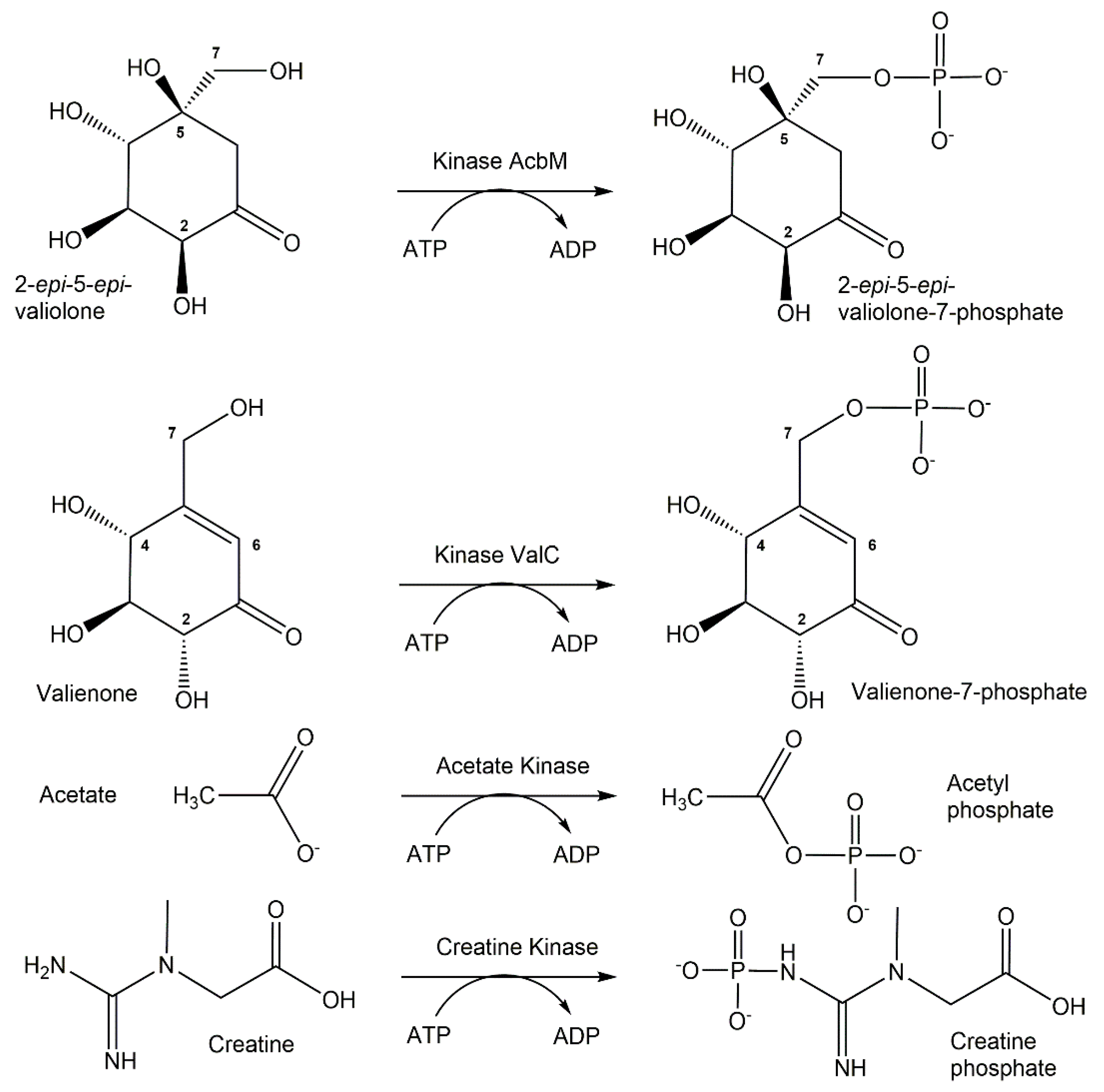
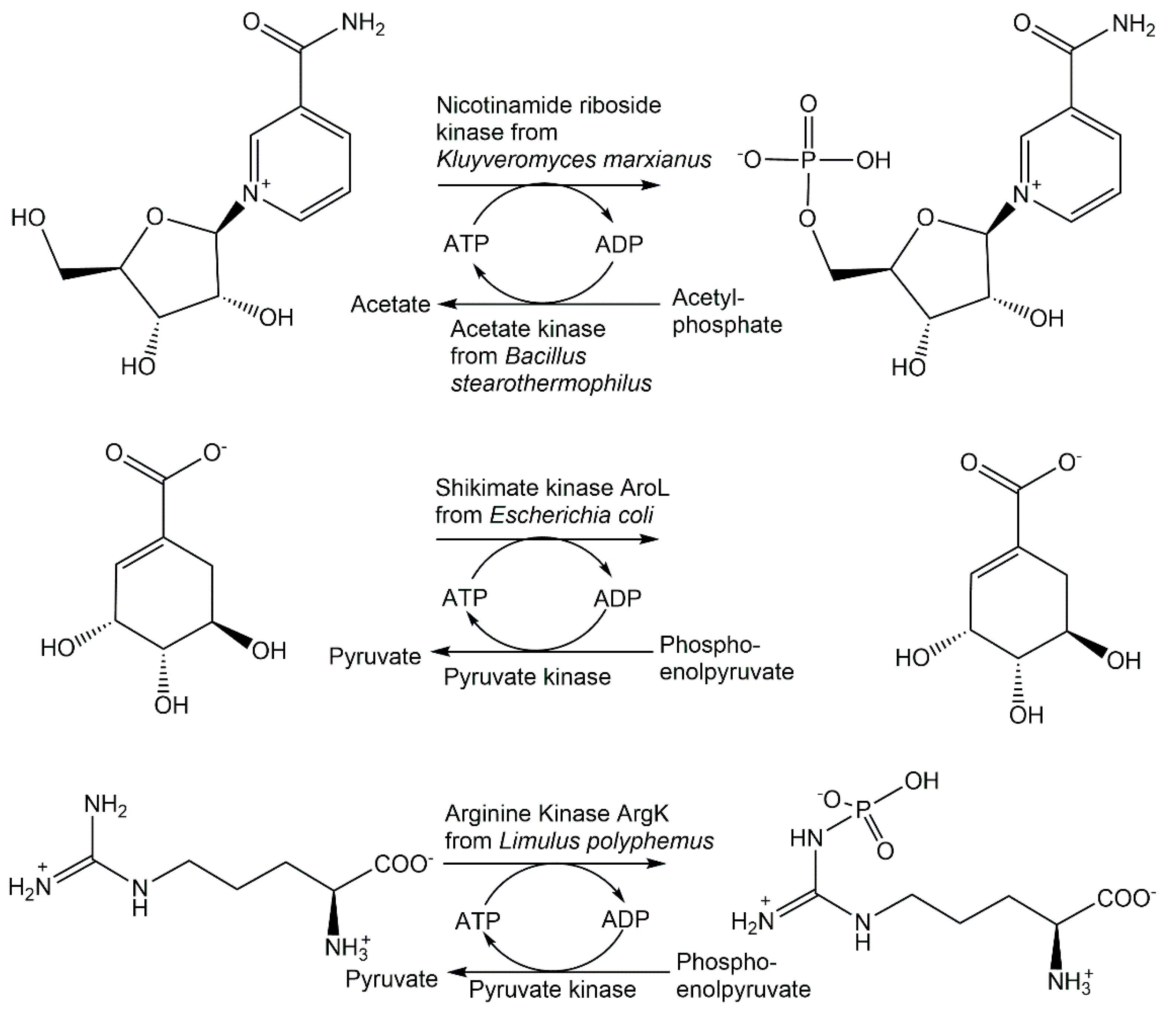
The large functional diversity of phosphotransferases, illustrated also by the many different phosphorylation reactions and acceptor substrates ranging from metabolites and other small molecular weight compounds to large biomolecules such as proteins, enables the highly selective biocatalytic transfer of a defined number and the molecular nature of a phosphoryl-group-containing donor to one specific functional group of an acceptor molecule [59,60]. Thereby, the highly selective formation of new covalent bonds of phosphorus to oxygen, nitrogen, sulfur and carbon is very attractive, as a large number of phosphotransferases exist in nature and no protecting groups for donors and acceptors are needed. The exquisite selectivity of phosphotransferases in catalyzing the phosphorylation of a particular hydroxy group is illustrated in Figure 3 by the two ATP-dependent kinases AcbM and ValC. The kinase AcbM plays an important role in the biosynthetic pathway to the seven-carbon cyclitol unit of the antidiabetic drug acarbose in the Actinoplanes sp. and catalyzes the formation of 2-epi-5-epi-valiolone-7-phosphate [61]. For the phosphorylation of the closely related substrates valienone and validone to valienone-7-phosphate and validone-7-phosphate, respectively, the different kinase ValC is required, which has a critical function in the biosynthesis of the antifungal compound validamycin A [62]. Short chain fatty acid kinases are involved in central metabolic pathways catalyzing the reversible ATP-dependent phosphorylation of carboxylic acids leading to the formation of acyl-phosphates, such as acetate kinase (see Figure 3) catalyzing the phosphorylation of acetate to acetyl-phosphate [63,64]. Phosphagen kinases, such as creatine kinases (see Figure 3) catalyzing the ATP-dependent reversible N-phosphorylation of creatine, are key enzymes in catalyzing the formation of high energy phosphorus nitrogen bonds [65,66].
The global analysis, identification and cataloguing of the functional diversity of all protein kinases in the genome of an organism, which was first introduced for the protein kinase complement of the human genome and was named kinome by Manning et al. [39], has been greatly expanded to animal, plant and microbial organisms, thus further extending the functional diversity. Although tremendous knowledge about protein kinase functions concerning the phosphorylations of serine, threonine and tyrosine residues in proteins exists, much less is known about the noncanonical phosphorylations of histidine, lysine, arginine, cysteine, aspartate and glutamate residues [67]. Evidence is emerging that protein kinase functions can also be exerted by metabolic enzymes, such as hexokinase, pyruvate kinase M2 and phosphoglycerate kinase 1 [68]. Despite the long history of metabolic kinases having small molecules as substrates, the functional diversity of kinases unrelated to protein kinases is understudied and is therefore of much interest [69,70].
2.2.2. Phosphohydrolase Functions
A broad functional diversity of EC class 3 enzymes is known to catalyze the hydrolysis of phosphorus–oxygen bonds in phosphoric-monoesters, phosphoric-diesters, diphosphoric-monoesters, triphosphoric acid monoesters, phosphoric acid triesters, RNA, DNA, phosphorus-containing anhydrides and the hydrolysis of phosphorus–nitrogen and phosphorus–carbon bonds.
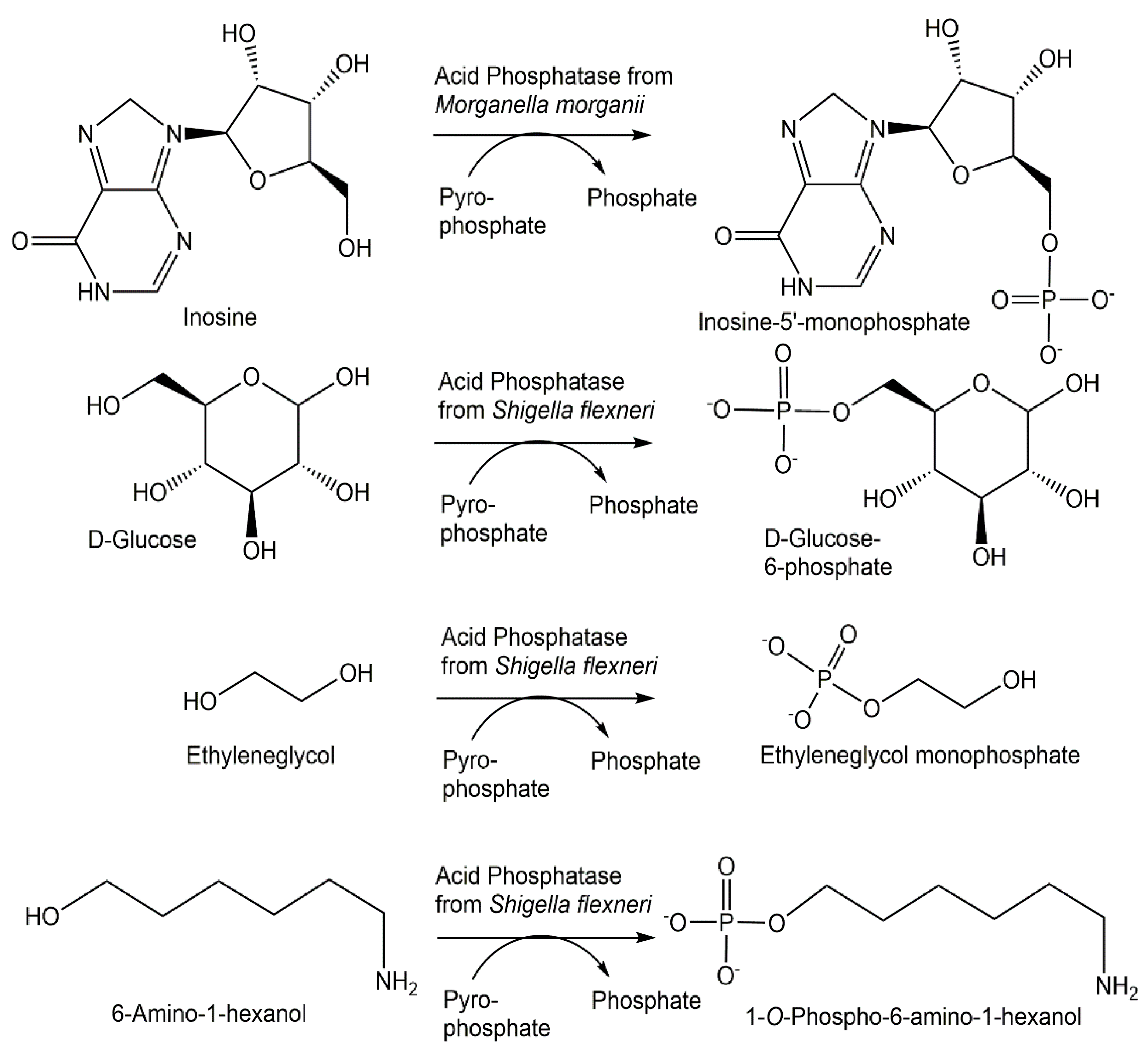
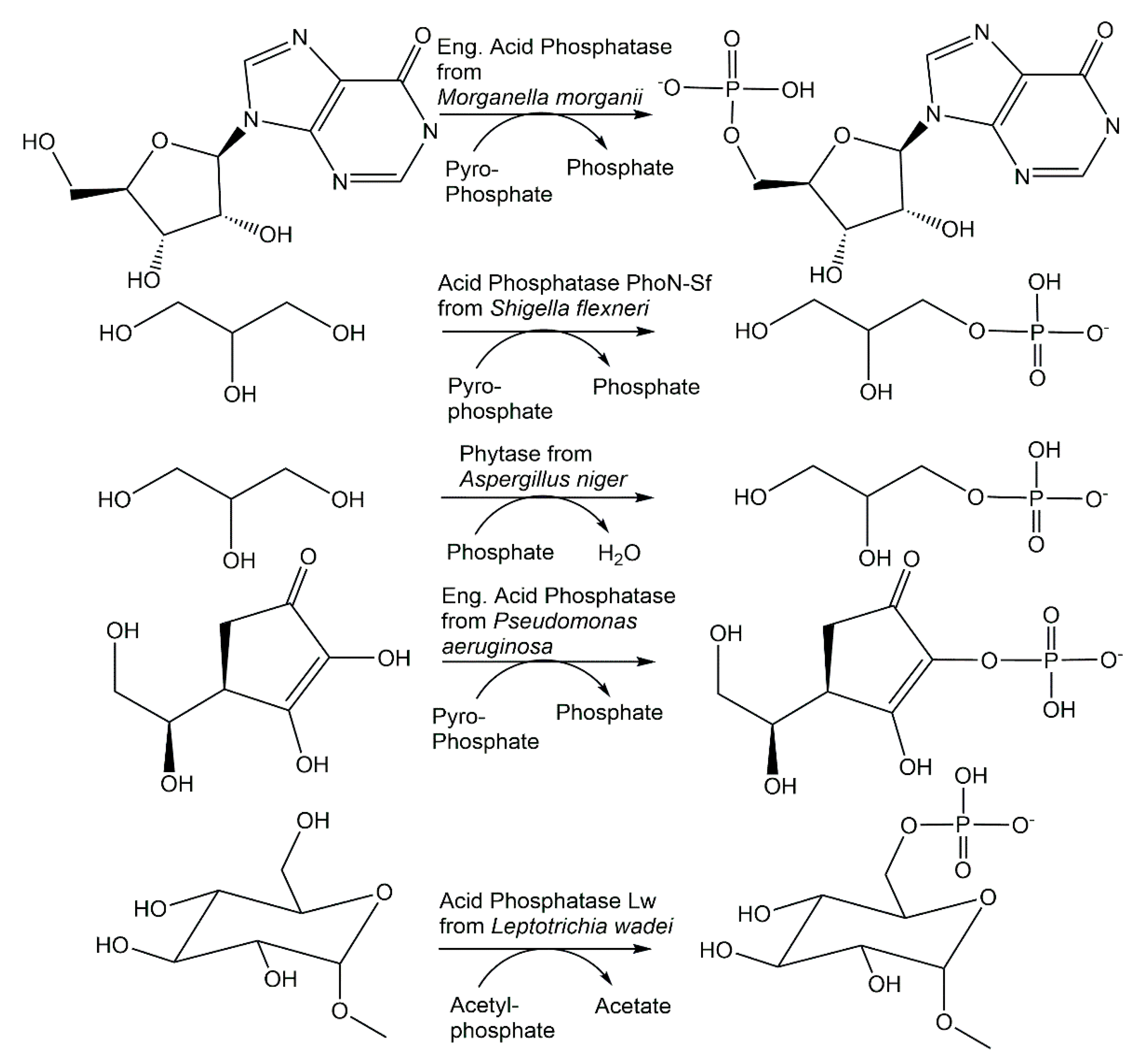
The ability of a number of phosphatases to catalyze not only hydrolysis reactions but also phosphorylation reactions has been investigated for more than seven decades [71]. Although hydrolytic activities can counteract phosphorylation reactions by reducing the yield of phosphorylated products, the differential hydrolysis of enantiomers to chiral precursors of antiviral compounds by phosphotriesterase variants [72], increasing the transferase/hydrolase ratio of phosphatases by protein engineering and the combined use of suitable phosphatase, inexpensive phosphoryl donors and reaction engineering offer new opportunities for broadly applicable and scalable phosphorylation reactions. Acid phosphatase from Morganella morganii has been found to also catalyze the 5′-phosphorylation of nucleosides (see Figure 4) using not only the energy-rich carbamoyl phosphate and acetylphosphate, but also pyrophosphate as an inexpensive phosphoryl donor [73]. While the closely related acid phosphatase from Providencia stuartii exhibited phosphorylation activity similar to the M. morganii enzyme, the acid phosphatases from Enterobacter aerogenes, Escherichia blattae and Klebsiella planticola showed lower phosphorylation activities [74]. Nonspecific acid phosphatases from Shigella flexneri and Salmonella enterica, which have been shown to be selective for phosphorylate D-glucose to D-glucose-6-phosphate, without the formation of D-glucose-1-phosphate, using pyrophosphate [75], have also been demonstrated (see Figure 4), together with other acid phosphatases such as PiACP from Prevotella intermedia, to catalyze the selective phosphorylation of monoalcohols and diols [76].
The transferase/hydrolase ratio is also of much fundamental interest in the regulation of the energy production and control of glycolysis by the bifunctional 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase isoenzymes (PFKFBs), with the PFKFB3, which is overexpressed in many cancer types, having the highest kinase/phosphatase ratio [77].
2.2.3. Phosphorylase Functions
Retaining and inverting glycoside phosphorylases catalyze the reversible formation of glycosyl 1-phosphates with the anomeric configuration corresponding to both the enzyme type and the configuration of the substrate, for which a rather limited number of disaccharides, oligosaccharides and polysaccharides is known, although 544 glycoside phosphorylase entries from nine GH families and six GT families are listed in the CAZy database [54]. The reversible formation of the same α-D-ribose 1-phosphate and a purine base or a pyrimidine base from the corresponding purine or pyrimidine nucleoside is catalyzed by nucleoside phosphorylases of family NP-I or NP-II [53]. Improving the stability and broadening the substrate scope of nucleoside phosphorylases is of much interest for both the phosphorolysis as well as the synthesis direction [78,79,80].
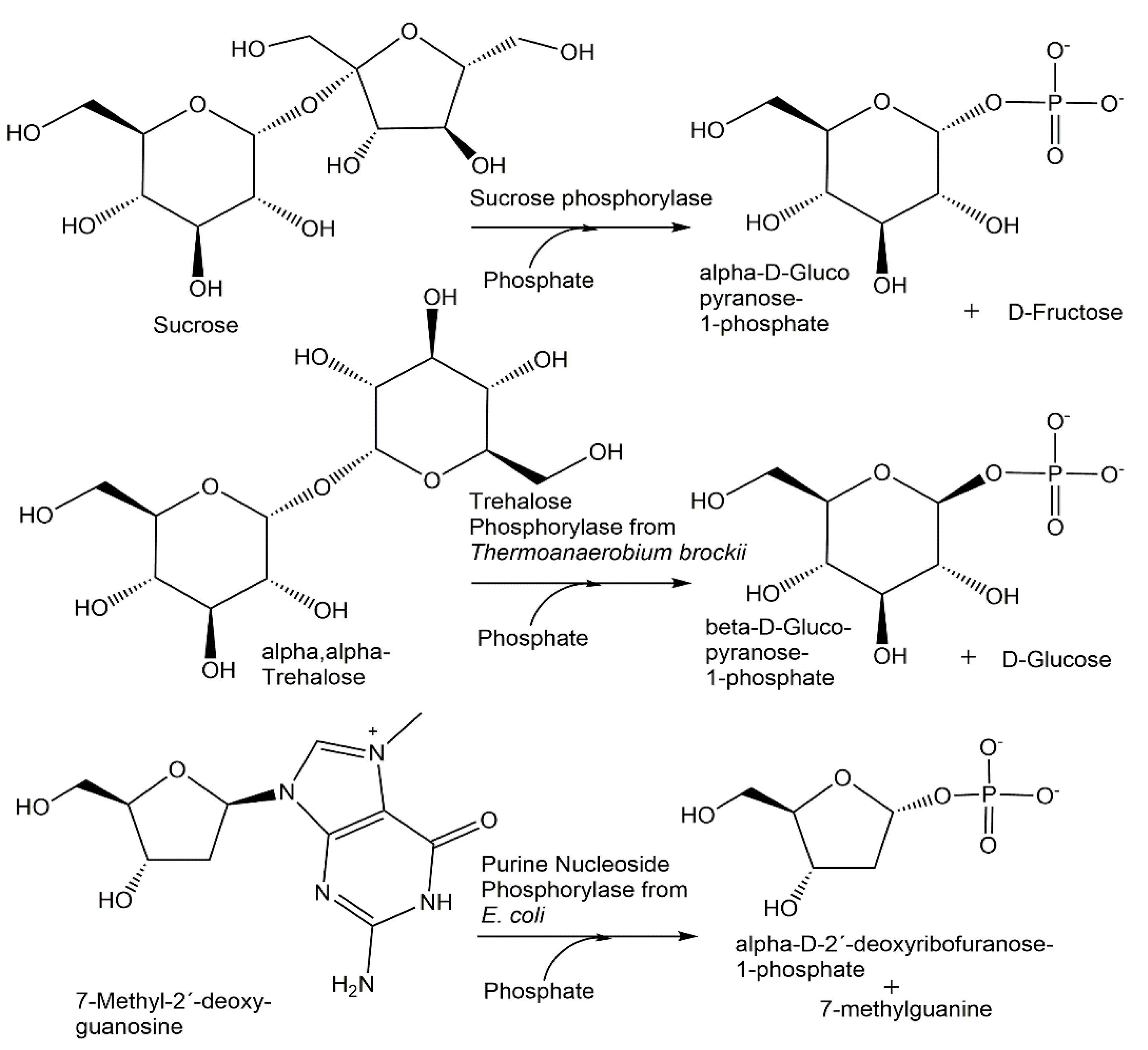
Sucrose phosphorylase catalyzes the reversible phosphorolysis of sucrose using phosphate, whereby α-D-glucopyranose-1-phosphate is formed (see Figure 5) and D-fructose is obtained as a byproduct [81], while trehalose phosphorylase leads to the anomeric β-D-glucopyranose-1-phosphate using α,α-trehalose and phosphate as substrates [82]. A nearly quantitative phosphorolysis of 7-methyl- 2′-deoxyguanosine to α-D-2′-deoxyribofuranose-1-phosphate can be obtained when purine nucleoside phosphorylase from E. coli is used [83].
2.2.4. Phosphomutase Functions
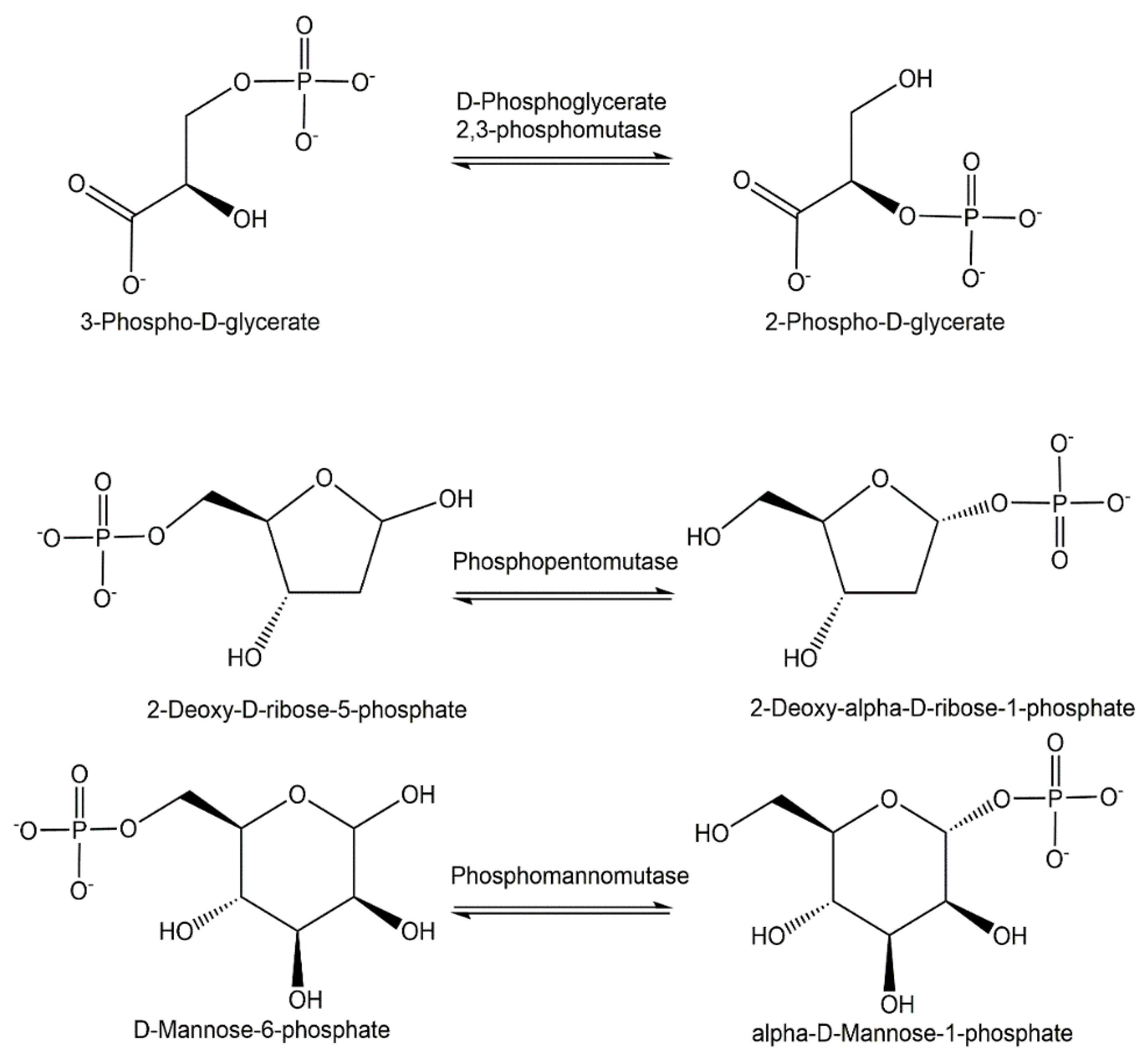
Phosphotransferases catalyzing the reversible intramolecular transfer of phosphoryl groups, also called phosphomutases, represent a metabolically important group of enzymes, not only for creating new phosphorus–oxygen bonds, but also for forming phosphorus–carbon bonds, while the phosphorus–oxygen bond of the starting substrate is cleaved. The reversible conversion of glyceric acids, pentoses and hexoses which are phosphorylated can thereby be achieved by the use of phosphoglycerate mutases, phosphopentomutases and phosphohexomutases with high selectivity. The reversible isomerization of 3-phospho-D-glycerate and 2-phospho-D-glycerate can be catalyzed (see Figure 6) by cofactor-independent as well as 2,3-bisphosphoglycerate-dependent phosphoglycerate mutases [84]. The phosphopentomutase function in catalyzing the reversible intramolecular phosphoryl group transfer is of much physiological and preparative interest, as shown for example in the isomerization of 2-deoxy-D-ribose-5-phosphate (see Figure 6) and 2-deoxy-alpha-D-ribose-1-phosphate [85]. Figure 6 also shows the enzymatic interconversion of D-mannose-6-phosphate and alpha-D-mannose-1-phosphate, which is catalyzed by phosphomannomutase and is essential in the activation of D-mannose and glycoconjugate biosynthesis in eukaryotes [86].
While D-glucose 6-phosphate interconversion catalyzed by α-D-phospho-glucomutases leads to α-D-glucose 1-phosphate [87], β-D-glucose 1-phosphate is obtained when its interconversion is catalyzed by β-D-phosphoglucomutases [88] in the thermodynamically unfavorable rearrangement of phosphoenolpyruvate to 3-phosphonopyruvate catalyzed by phosphoenolpyruvate mutase [89].
2.3. Mechanisms of Phosphorylation Biocatalysts
The mechanisms by which biocatalysts achieve the enormous rate accelerations by several orders of magnitude of the phosphoryl-group transfer reactions in comparison with the slow reaction rates without biocatalysts are of fundamental interest and continue to attract much attention [90,91,92]. Various distinct phosphoryl group transfer mechanisms have been elucidated in numerous enzyme families [93].
2.3.1. Phosphotransferases
The large 1012–1014-fold rate enhancements produced by hexokinase, homoserine kinase and N-acetylgalactosamine kinase has been demonstrated in a thermodynamic analysis to be due to two effects of these representative kinases compared with nonenzymatic phosphoryl group transfer: a more favorable entropy of activation and major reductions in the enthalpy of activation [94]. The catalytic mechanism by which adenylate kinase achieves this rate acceleration, which has been investigated in much detail by a combination of various techniques such as NMR and crystallography, involves the activation of phosphoryl transfer and lid opening, both by two orders of magnitude, by placing the charged cofactor Mg2+ in the active site organized before [95]. Based on structural and biochemical analyses of the ADP-phosphorylating class I polyphosphate kinase 2 from Francisella tularensis and the AMP- and ADP-phosphorylating class III polyphosphate kinase 2 from Meiothermus ruber, a mechanism of action has been proposed [96]. A key feature of the proposed mechanism is the in-line nucleophilic attack of the nucleotide on polyphosphate, which is activated by the active-site Lewis acidic Mg2+ upon the binding of polyphosphate [96].
2.3.2. Phosphohydrolases
The investigation of acid phosphatase α-D-glucose 1-phosphate phosphatase from Escherichia coli provided evidence for a 104-fold phosphatase efficiency advantage of a histidine compared to an aspartate nucleophile in position 18 and an additional 100-fold phosphatase efficiency advantage by the cooperative interaction of the catalytic nucleophile His18 with general acid catalytic groups, which is lost when His18 is replaced with Asp18 [97].
2.3.3. Phosphorylases
Structural and kinetic studies of hexameric purine nucleoside phosphorylase have provided detailed insights into the mechanism of purine nucleoside and phosphate binding, subunit conformations in open and closed forms, phosphate-induced conformational change, the sequence of nucleoside binding and subunit cooperation for effective catalysis [98].
2.3.4. Phosphomutases
Detailed NMR and X-ray investigations of the β-phosphoglucomutase-catalyzed isomerization of β-D-glucose 1-phosphate and D-glucose 6-phosphate via β-D-glucose 1,6-bisphosphate have revealed the importance of a conserved transition state organization by an invariant protein conformation, and priority for phosphate positioning over hexose in the substrate recognition [99]. The unusual mechanism of the phospho-enolpyruvate mutase-catalyzed intramolecular phosphoryl group transfer, which requires the cleavage of a low-energy oxygen–phosphorus bond and the formation of a high-energy carbon–phosphorus bond, is currently understood as a pericyclic rearrangement, which is retaining the configuration and placing the incoming nucleophile not strictly in line with the leaving group [100].
3. Discovery Methodologies and Tools for Phosphorylation Biocatalysts
The range of methodologies and tools for the discovery of novel biocatalysts and function–sequence relationships has been very much advanced from classical protein purification from a biological organism and its subsequent biochemical characterization of the purified protein, which also require the availability or synthesis of suitable enzyme substrates for both the activity-guided purification as well as for measuring enzyme kinetics. A number of methodologies, ranging from bioinformatics and genome mining to metagenomics, are available for finding phosphorylation biocatalysts with known functions that are based on sequence similarity. Important tools for the discovery of phosphorylation biocatalysts with novel functions are meaningful analytical assays of the enzyme-catalyzed substrate to product conversion. Robust and sensitive enzyme activity assays with a high information content, which enable the precise identification of the phosphorylation site in the substrate, continue to be a key prerequisite for the discovery of novel enzyme functions, or the experimental proof or disproof of automatically annotated protein sequences of phosphorylation biocatalysts. There are however many different approaches to utilizing enzyme activity assays for accelerating the assignment of the enzyme functions of the catalyzing substrate to product conversions to protein sequences and structures. Multiplexing selected enzyme activity assays in a single reaction vessel has been valuable for facilitating single protein kinase assays by using a kinase activity assay for kinome profiling in a single reaction, whereby 90 different peptide phosphorylation rates are obtained by mass spectrometry [101], requiring, however, stable isotope-labeled phosphorylated peptides as internal standards. The miniaturization and parallelization of enzyme activity assays have the advantages of saving costs, materials and time, but require higher sensitivity. This can be provided by highly informative and sensitive mass spectrometry methodologies, which have enabled the profiling of in vitro enzymatic activities of the 1,275 Escherichia coli proteins, which are functionally uncharacterized, to discover 241 enzymes which are potentially novel enzymes, of which 12 have been validated experimentally. This activity-based metabolite profiling (ABMP) of an overexpressed or purified protein is based on measuring by nontargeted mass spectrometry metabolites which are accumulated or depleted after the incubation of the protein of interest in a metabolome extract [102]. The identification in central enzymes of Escherichia coli of 34 new phosphorylation sites, which are functional in the regulation of enzyme activity, is of much interest for discovering novel protein kinases or for finding a relaxed specificity of the known serine/threonine or tyrosine kinases of Escherichia coli [103]. Chemical probes which are designed to specifically measure all proteins with the same characteristic activity in one class of phosphorylation biocatalysts can be used in activity-based protein profiling (ABPP), for example, for profiling a specific class of protein kinases, lipid kinases or other metabolic kinases [104].
A number of broadly applicable methodologies for the assignment of a novel or known enzyme function to a domain of unknown function (DUF) are provided by genomic enzymology [105], by the ligand-oriented screening for solute-binding transport proteins, by the analysis tools of sequence similarity networks (SSN) and genome neighborhood networks (GNN), enabling the identification of DUF1537 as a novel kinase family in acid sugar catabolic pathways and the discovery of four DUF1537 family members as novel C4-sugar kinases which depend on ATP [106].
The great methodological advances for the discovery and development of novel biocatalysts which have been provided by directed evolution, protein engineering and computational approaches to protein design have enabled the optimization of biocatalyst properties such as activities, selectivities or stabilities, and their adaptation to reaction conditions [107,108]. The power and potential of these tools and methodologies has attracted much interest as they provide the opportunity to develop numerous phosphorylation biocatalysts since the directed evolution of thymidine kinase for the phosphorylation of zidovudine [109], and their use in tackling challenging new-to-nature phosphorus chemistry is very interesting [110].
4. Novel Phosphorylation Biocatalysts
Based on the progress with biocatalysts for the formation of new O-P linkages, for example, in the synthesis of phosphorylated sugars or nucleotides, novel phosphorylation biocatalysts are highly desirable for efficient, highly selective and sustainable phosphorylation reactions and because biocatalytic X-P bond formation has been anticipated to lead to the faster development of nucleoside and nucleotide therapeutics in drug discovery and development [111].
4.1. Novel Phosphotransferases
Since the first demonstration 55 years ago that an enzyme preparation of an antibiotic-resistant E. coli strain could phosphorylate kanamycin and related antibiotics [112], the antibiotic resistome and assignments of enzyme functions to antibiotic resistance genes have attracted much interest [113,114]. Numerous novel phosphorylation biocatalysts have been discovered in microorganisms to inactivate antibiotics, such as aminoglycoside and macrolide antibiotics, for defense or self-protection by catalyzing their O-phosphorylation. A new antibiotic-resistance kinase AmiN from Bacillus subtilis has been discovered which inactivates amicoumacin by catalyzing the phosphorylation of the 2-hydroxy group of amicoumacin and was misannotated as a homoserine kinase [115]. From the recently discovered rifamycin inactivating phosphotransferase family, an unusual antibiotic resistance kinase, rifampicin phosphotransferase from Listeria monocytogenes, has been described to depend on ATP in phosphorylating the 21-hydroxy group of rifampicin, with the concomittant formation of AMP and inorganic phosphate [116]. The phosphotransferase Cpr17, which was discovered by cloning and characterizing the biosynthetic gene cluster of the capuramycin-type nucleoside antibiotic A-102395, was demonstrated to provide self-resistance and to prefer GTP as the phosphoryl donor [117].
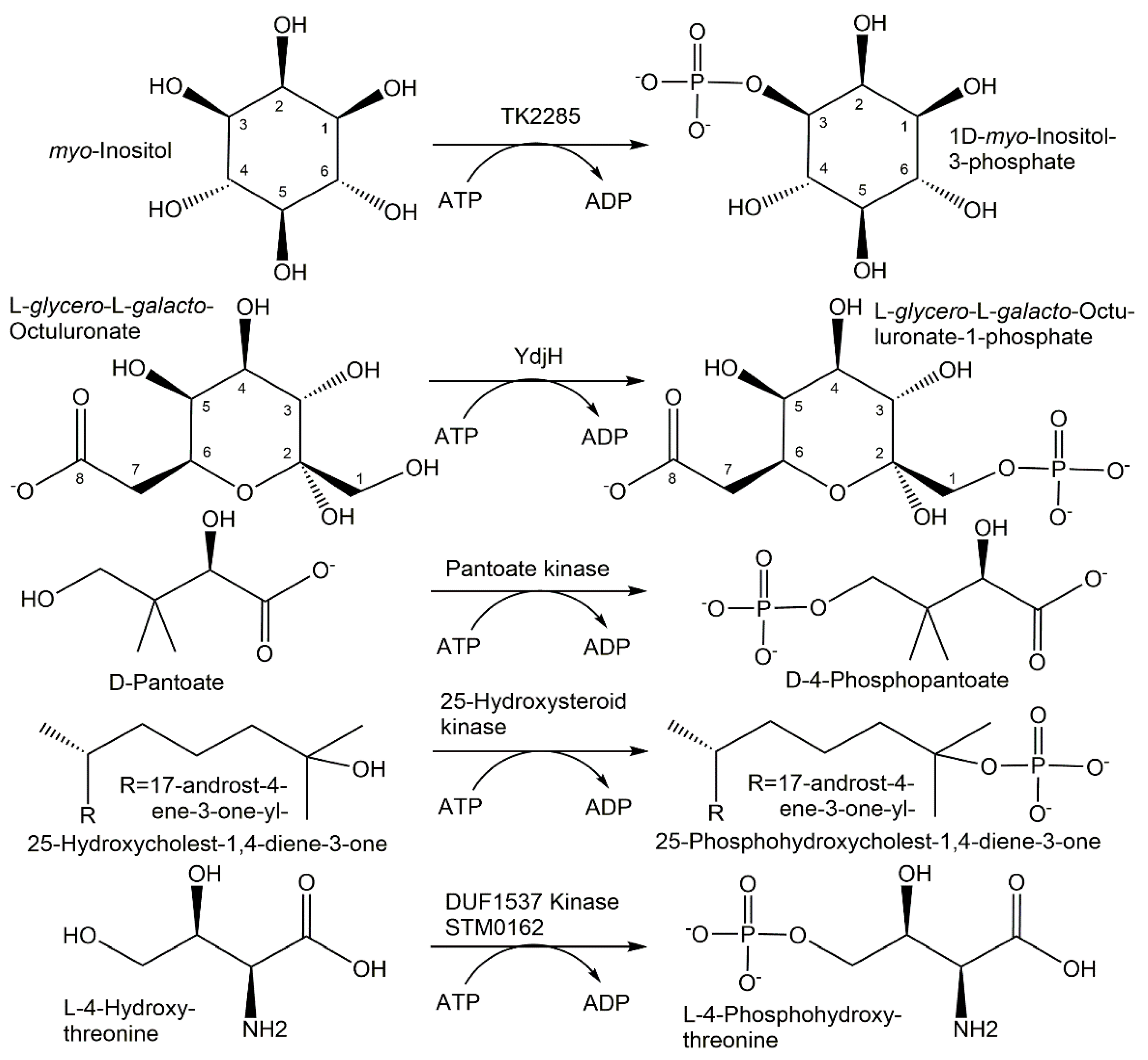
Enzymes for the selective phosphorylation of hydroxycarboxylic acids are of much interest, and the recently discovered ATP-dependent pantoate kinase from Thermococcus kodakaraensis, which catalyzes the 4-phosphorylation of D-pantoate to D-4-phosphopantoate (see Figure 7), is not regulated by CoA and has been demonstrated to accept the nucleotides CTP, GTP and UTP [118].
A novel flavonoid phosphotransferase from Bacillus subtilis, which has been discovered to catalyze the regioselective ATP-dependent phosphorylation of a broad range of flavonoids to the corresponding flavonoid monophosphates and the generation of AMP and phosphate, enables the efficient and sustainable conversion of poorly water-soluble natural flavones, isoflavones, flavonols, flavanones and flavonolignans into their monophosphosphorylated forms with improved water solubilities [119].
A highly active 25-hydroxysteroid kinase has been found to be involved in the microbial degradation of the side chain of cholesterol and sitosterol in Sterolibacterium denitrificans and catalyzed the selective and fully reversible phosphorylation of the tertiary hydroxy-group at the C25 of steroid alcohols such as the cholesterol metabolites 25-hydroxy-cholest-4-en-3-one and 25-hydroxy-cholest-1,4-diene-3-one (see Figure 7), the sitosterol metabolite 25-hydroxy-sitost-1,4-diene-3-one or 25-hydroxy vitamin D3 [120].
Numerous novel carbohydrate kinases have been discovered in microbial pathways of carbohydrate utilization, by expressing and characterizing DUFs, or by directed evolution. When directed evolution was used to broaden the substrate scope of galactokinase GalK from Escherichia coli, it was found that the Y371H variant, containing only a single amino acid exchange from tyrosine in the wild-type enzyme to histidine in the variant, already showed a wider substrate scope and accepted seven additional D-sugars, L-altrose and L-glucose as substrates [121]. The TK2285 protein from Thermococcus kodakaraensis has been discovered to function as an ATP-dependent myo-inositol kinase (see Figure 7) catalyzing the phosphorylation of the 3-hydroxy group of myo-inositol to 1D-myo-inositol 3-phosphate [122]. The same enzyme function could also be achieved by an engineered pyrophosphate-dependent myo-inositol 1-kinase by changing residues to recognize myo-inositol [123]. A highly active galactokinase from Leminorella grimontii has been found to catalyze the 1-phosphorylation of a number of galactose analogues [124]. From screening a series of wild-type enzymes, seven galacto- and six N-acetylhexosamine kinases, new enzyme phosphorylation activities towards fluorinated monosaccharides and four novel N-acetylhexosamine kinases have been discovered [125]. The novel sugar kinase YdhJ is the first kinase accepting C8-monosaccharides as substrates and has been discovered in the course of the functional characterization of the ydj gene cluster [126]. The 1-hydroxy group of a number of 2-keto-monosaccharides has been shown to be phosphorylated by YdhJ, and L-glycero-L-galacto-octuluronate was the best substrate (see Figure 7) being converted to L-glycero-L-galacto-octuluronate-1-phosphate [126]. The functional characterization of Cj1415, involved in the biosynthetic pathway to the O-methyl phosphoramidate modified capsular polysaccharide from Campylobacter jejuni, has led to its identification as cytidine diphosphoramidate kinase catalyzing 3′-phosphorylation [127].
The substrate scope of the ATP-dependent 5′-phosphorylation of all four 2-deoxynucleosides and analogues catalyzed by deoxynucleoside kinase from Drosophila melanogaster has been broadened to also include an unnatural nucleoside of an artificially expanded genetic information system by the Q81E variant [128].
The area of amino acid kinases has experienced a very interesting series of recent discoveries. Novel serine kinases catalyzing the O-phosphorylation of free serine have been discovered and were found to be dependent on ADP in Thermococcus kodakaraensis [129], while the serine kinase from Staphylothermus marinus was found to be ATP dependent [130]. An interesting novel amino acid kinase has been discovered as a DUF1537 family member STM0162, which catalyzes not only the phosphorylation of D-threonate, but also the phosphorylation of the toxic intermediate L-4-hydroxythreonine to the essential metabolite L-4-phosphohydroxythreonine (see Figure 7), providing thereby a recycling and detoxification path [131].
The protein Cj1418 has been discovered to be a unique ATP-dependent L-glutamine kinase catalyzing the N-phosphorylation of L-glutamine at its amide nitrogen, which is required in the already-mentioned biosynthetic pathway to the O-methyl phosphoramidate modification of the Campylobacter jejuni capsular polysaccharide [132,133].
4.2. Novel Phosphohydrolases
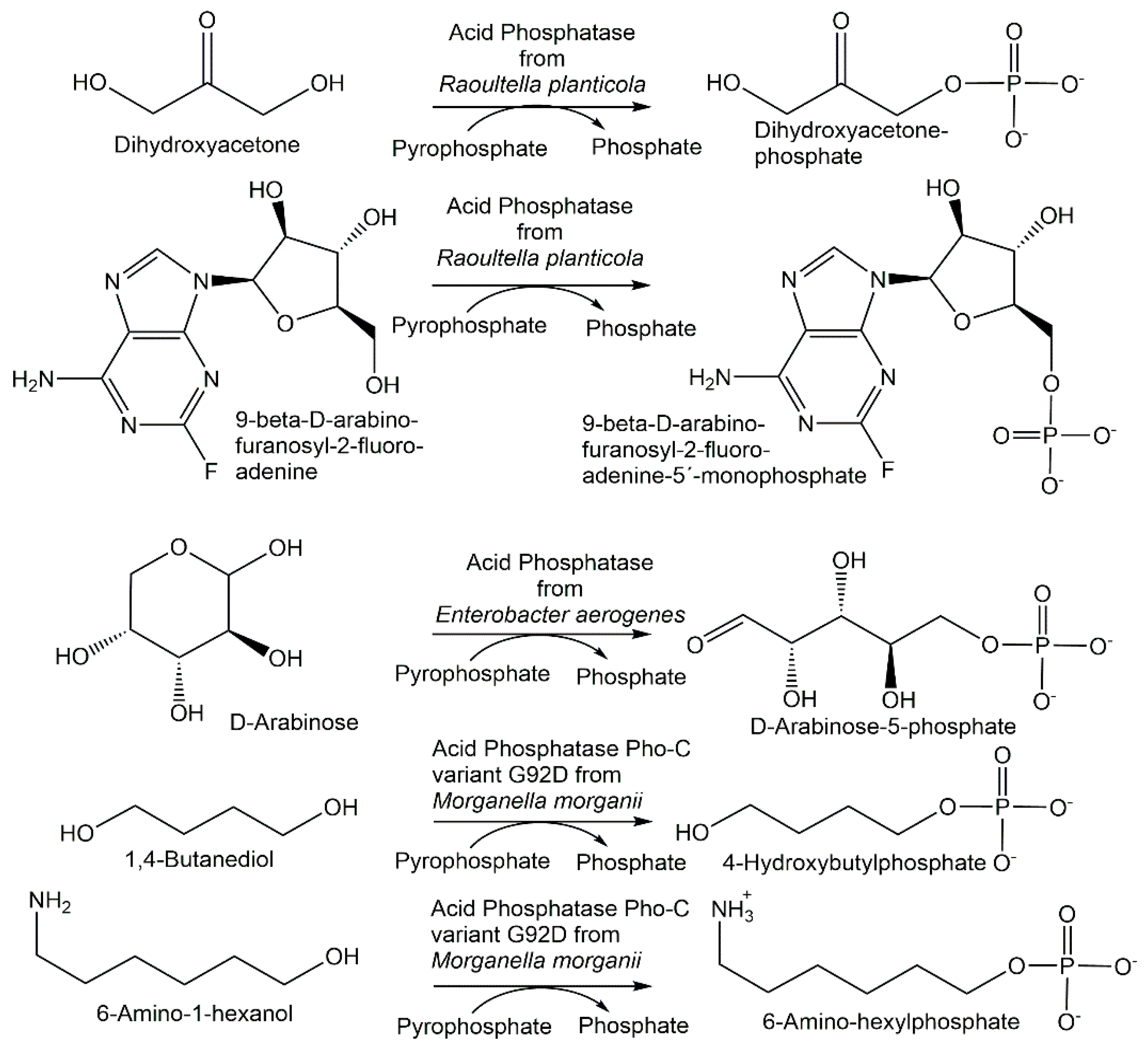
Novel phosphohydrolases which can be used in the reverse direction with inexpensive phosphoryl donors to catalyze phosphorylations have been found from microbial sources as well as by enzyme engineering. Nonspecific acid phosphatases from wild-type and genetically modified strains of Enterobacter aerogenes and Raoultella planticola have been found to also catalyze the phosphorylation of nucleosides and sugars (see Figure 8) using pyrophosphate as the phosphoryl donor [134].
The I171T-G92D mutant of the M. morganii acid phosphatase PhoC enabled the decrease in the Km value for inosine in the phosphorylation reaction using pyrophosphate (see Figure 4) by two thirds of that of the wild-type enzyme, to a value well below the inosine solubility under the phosphorylation reaction conditions [135]. The double mutant I171T-G92D and the single mutants G92D, I171T, G92A and G92N of the M. morganii acid phosphatase PhoC and a nonspecific acid phosphatase variant from E. blattae have been investigated with respect to the phosphorylation of primary alcohols using pyrophosphate [135]. Beneficial mutations, for example G92D and I171T-G92D, have led to an increased affinity for alcohol substrates such as 1,4-butanediol, glycerol, ethylene glycol and 6-amino-1-hexanol (see Figure 8), as well as the decreased hydrolysis of phosphorylated products and the extension of the optimum pH towards a neutral pH [136]. The covalent immobilization of the nonspecific acid phosphatases PhoN from Shigella flexneri and Salmonella enterica ser. typhimurium LT2 enabled a better phosphorylation of alcohol substrates in an aqueous medium using pyrophosphate as the phosphoryl donor [137].
4.3. Novel Phosphorylases
The search for novel glycoside phosphorylases has attracted much interest for extending the rather limited functional diversity of known glycoside phosphorylases.
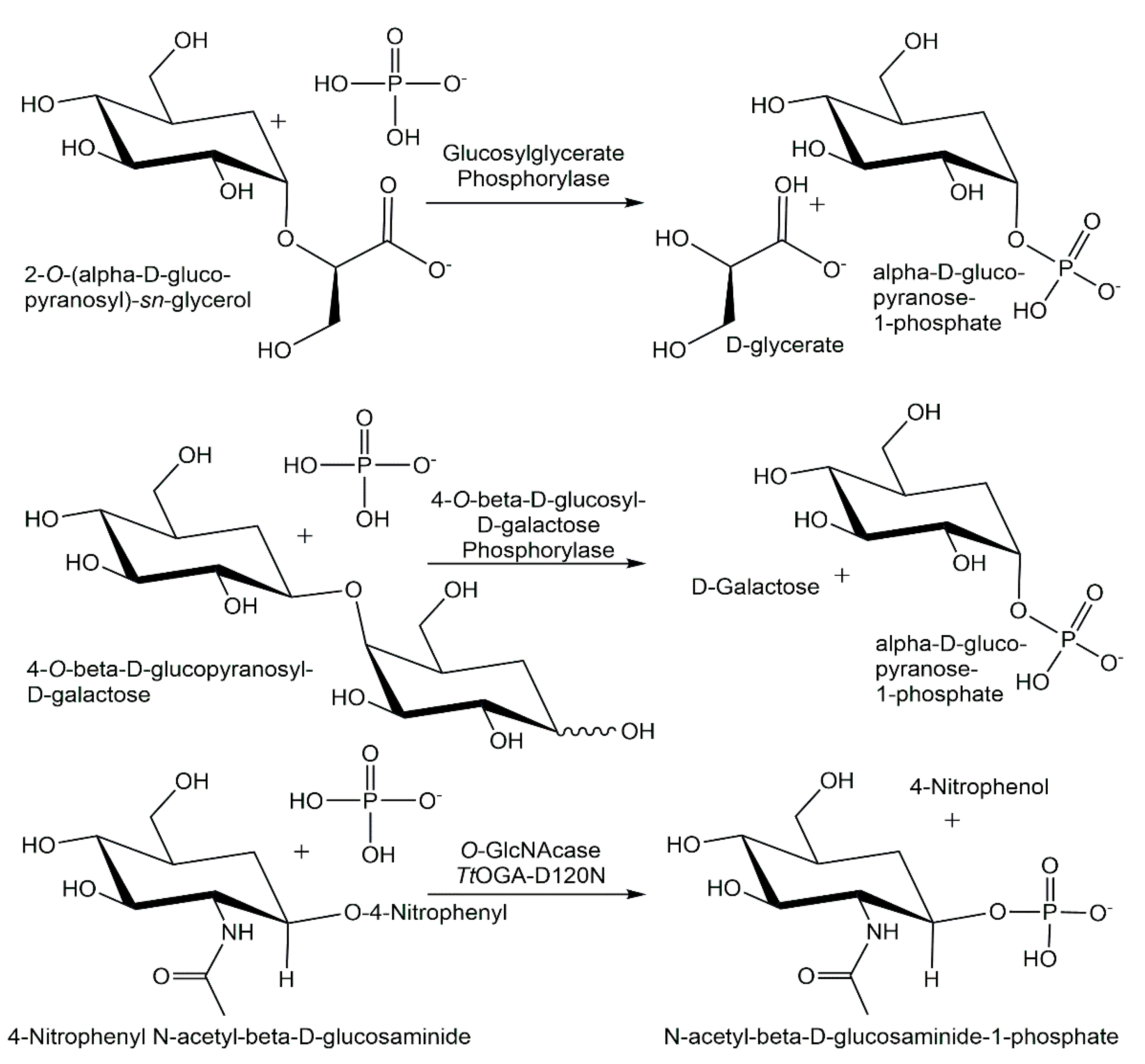
A high-throughput assay has been used to screen a metagenomic source library for glycoside phosphorylases, whereby seven new glycoside phosphorylases from the CAZy family GH94, among them two cellobiose phosphorylases, two cellodextrin phosphorylases and two laminaribiose phosphorylases, and a β-1,3-oligoglucan phosphorylase from the CAZy family GH149 [138]. Expressing and characterizing what was thought to be a sucrose phosphorylase from Meiothermus silvanus, residing in an unexplored branch of glycoside hydrolase family GH13, enabled the discovery of a glucosylglycerate phosphorylase (see Figure 9) [139]. By identifying and experimentally screening unknown clusters of the glycoside hydrolase family GH94 sequence space, using a combination of phylogenetic analysis and SSN, it was possible to discover a new 4-O-β-D-glucosyl-D-galactose phosphorylase (see Figure 9) from Paenibacillus polymyxa [140]. Efficient glucosaminide phosphorylases (see Figure 9), which have been engineered from GH84 O-GlcNacases by a single point mutation, have been shown to be 10-fold more active than their naturally occurring counterparts [141]. Widening the substrate spectrum is also of much interest for nucleoside phosphorylases, which has been achieved by engineering novel variants of purine nucleoside phosphorylases [142] and by the wider substrate spectrum of thermophilic nucleoside phosphorylases [143].
4.4. Novel Phosphomutases
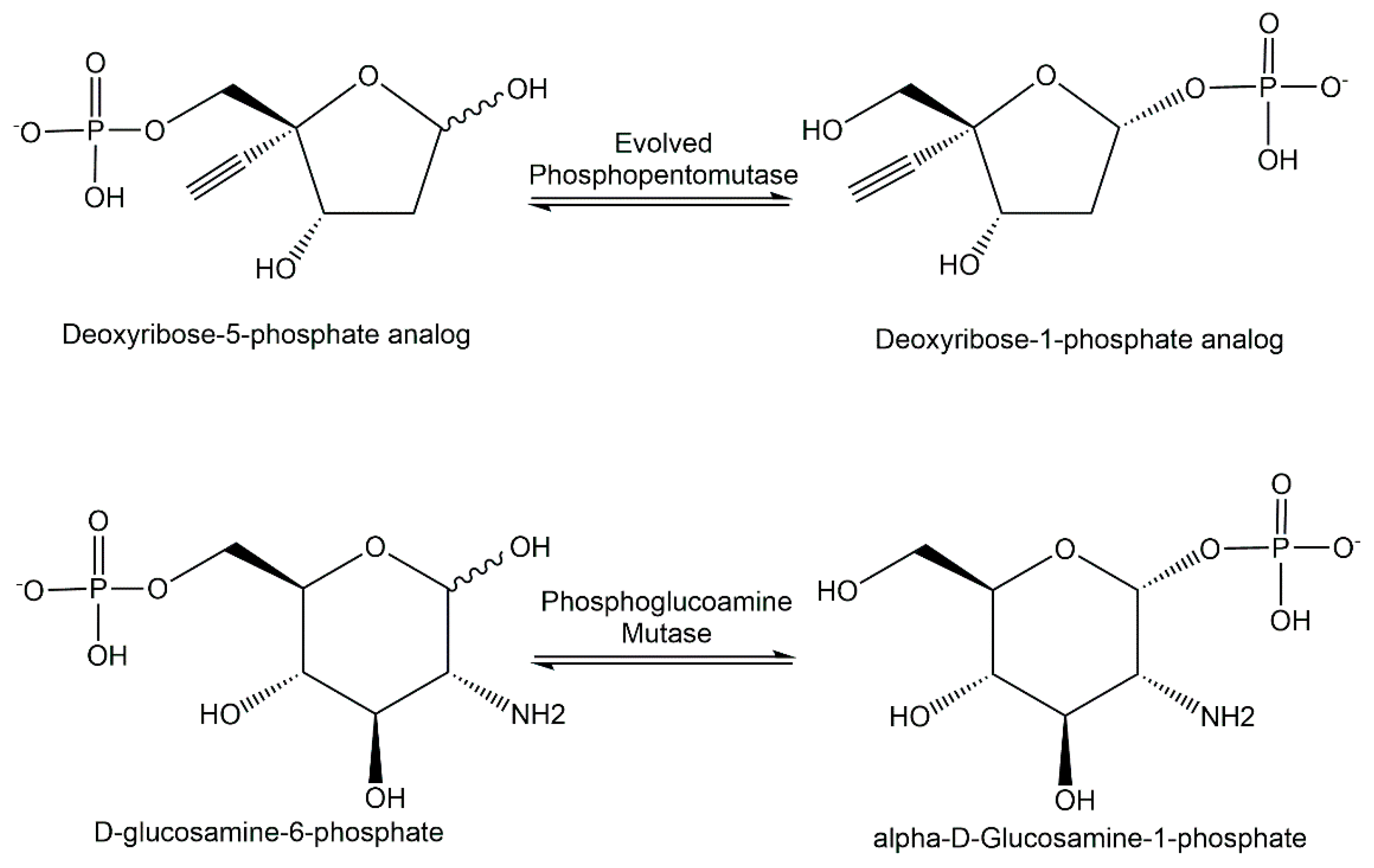
The discovery of novel enzymes catalyzing intramolecular phosphoryl-group transfer has been especially interesting in the area of the phosphopentomutases, for example, engineered phosphopentomutase variant enzymes (see Figure 10) catalyzing the isomerization of a deoxyribose-5-phosphate analog to the corresponding deoxyribose-1-phosphate analog [144]. The recombinant expression of the ST0242 protein from Sulfolobus tokadaii has led to the discovery of its phosphoglucosamine-mutase and phosphogalactosamine-mutase activity [145].
5. Analytical Applications of Phosphorylation Biocatalysts
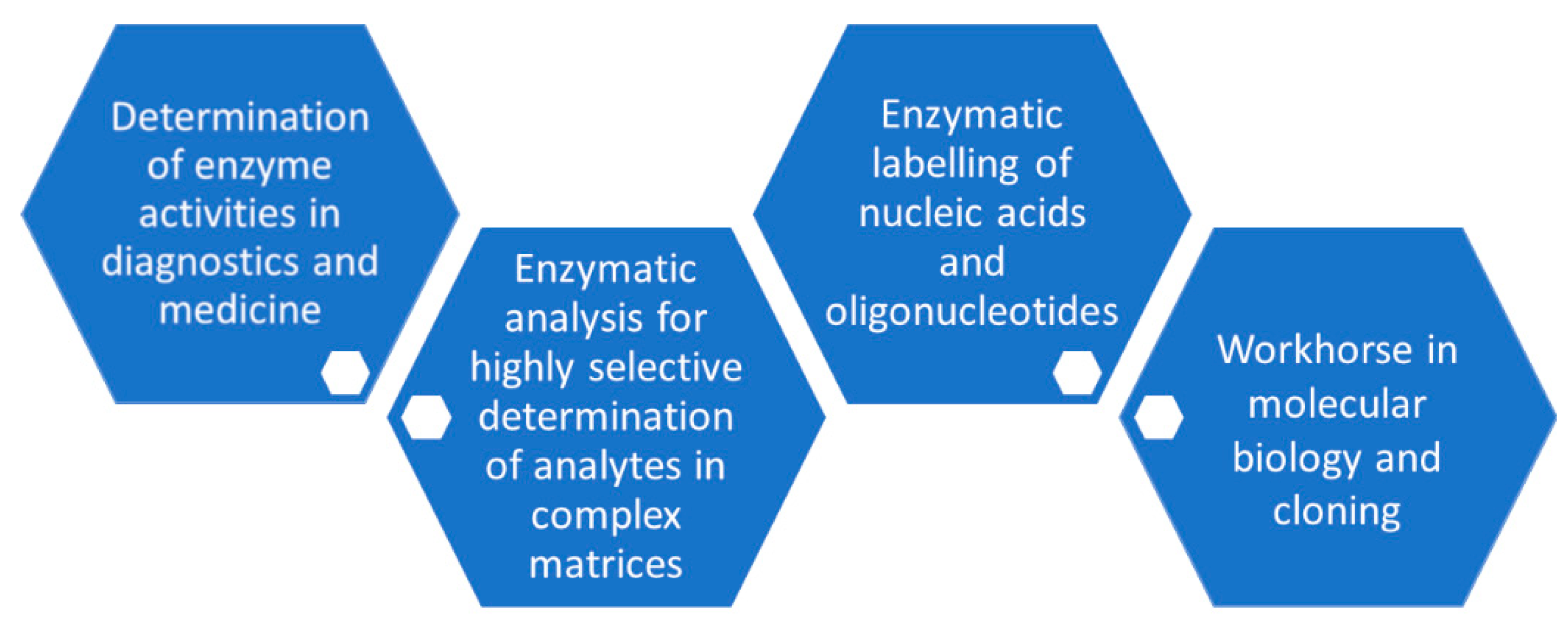
The manufacturing of suitable and highly active phosphorylation biocatalysts which have been made available as fit-for-purpose enzymes and with negligible levels of undesired contaminating enzyme activities continue to be important prerequisites for analytical applications (see Figure 11), as already shown with the development of methods for enzymatic analysis several decades ago [146,147], for example, in the applications of glycerol kinase for the determination of glycerol or creatine kinase for the analysis of creatine. Significant improvements in the individual steps of the overall approach to enzymatic analysis, by efficient enzyme production using recombinant technologies, analytical method optimization, international standardization, miniaturization and automation have facilitated analytical applications and have led to its routine use in diagnostics. Building on the principles of enzymatic analysis established long ago and the power of phosphorylation biocatalysts, the highly selective determination of analytes in complex matrices can be achieved without time-consuming prior sample preparation.
Analytical applications connected with phosphorylation biocatalysts are also important with respect to the selective measurement of the corresponding enzyme activities, for example, in medicine as diagnostic tools for human health and disease and in the development of enzyme inhibitors by determining their characteristics for inhibiting a desired phosphorylation biocatalyst activity.
5.1. Analytical Applications of Phosphotransferases
The basic principle of the highly selective phosphorylation of glycerol to L-glycerol-3-phosphate catalyzed by ATP-dependent glycerol kinase continues to be a key reaction, which is then coupled to indicator reactions in modern quantitative assays of glycerol. The enzymatic determination of glycerol can be applied in the food, beverages and cosmetics process analysis and control in microbial fermentation and cell culture media, for example, in process analysis, or additionally following an enzymatic hydrolysis reaction for obtaining glycerol which is then also determined by applying glycerol kinase and an indicator reaction in triglycerides diagnostics in medicine. Phosphorylation biocatalysts are applied not only for the enzymatic analysis of substrates, but also for the determination of enzyme activities, and kinases are themselves target enzymes for biomedical analysis. An important application is the activity assay of creatine kinase and its isoenzymes for disease and emergency diagnosis, such as acute myocardial infarction and skeletal muscle diseases. The recommendation of the International Federation of Clinical Chemistry (IFCC) for the measurement of creatine kinase activity includes the utilization of hexokinase [148].
Phosphorylation biocatalysts have also found broad applications in the analysis of nucleic acids and oligonucleotides as well as for the analytical scale preparation of selectively phosphorylated nucleic acids. Recombinant T4 polynucleotide kinase has become a workhorse for molecular biology [149] and is used in molecular cloning for the 5′-phosphorylation of DNA or RNA. T4 polynucleotide kinase is also used for labeling nucleic acids and oligonucleotides at the 5′ end for generating analytical reagents for detection, size marker in electrophoresis or hybridization probes, which can be used in techniques for locating and binding the nucleic acids of a complementary sequence, such as Southern Blotting, Northern Blotting and in situ hybridization [150]. The acceptance of ATP-biotin as a substrate by T4 polynucleotide kinase is of much interest for the kinase-catalyzed biotinylation of single-stranded DNA [151].
5.2. Analytical Applications of Phosphohydrolases
Increased alkaline phosphatase activity in serum, if not caused by bone growth or pregnancy, occurs in bone and/or liver disease and therefore alkaline phosphatase is a target enzyme in clinical chemistry, with IFCC reference procedures available for measuring catalytic concentrations of alkaline phosphatase [152]. Alkaline phosphatase has also been widely used as a reporter enzyme in enzyme immunoassays due to its advantages, such as enzyme properties, easy preparation of enzyme–antibody conjugates and signal response for various detection types, making it a privileged label for enzyme immunoassays [153].
5.3. Analytical Applications of Phosphorylases
The substrate specificity of cellobiose phosphorylase has been attractive for the colorimetric quantification of cellobiose in the presence of other saccharides, whereby linear calibration curves have been obtained [154]. Maltose phosphorylase has been applied for the selective detection of phosphate without interference from other anions by using the immobilized enzyme in a biosensor with conductometric detection [155]. The activity of the glycogen phosphorylase isoenzyme BB is of much clinical interest in tissue hypoxia and ischemic myocardial damage [156].
5.4. Analytical Applications of Phosphomutases
As phosphoglycerate mutase 1 is the rate-limiting glycolytic enzyme in tumor cells, leukocytes and heart tissue, the analysis of its activity and its regulation is of much biomedical interest [157], for example, as an effector of the mammalian target of the rapamycin signaling pathway and as prognostic biomarker of non-small cell lung cancer [158].
6. Synthetic Applications of Phosphorylation Biocatalysts
Simple and broadly applicable process designs for synthetic phosphorylation reactions are attractive for reducing complexity when the required process targets with regard to selectivity and conversion can be achieved while meeting safety, health, environment and sustainability goals [159]. Synthetic applications of phosphorylation biocatalysts as well as the number and types of products have grown (see Figure 12), thus facilitating the further development of processes.
As the applications, boundary conditions and approaches differ across the specific reactions catalyzed by the key classes of phosphorylation biocatalysts, the approaches towards the synthetic applications of phosphohydrolases, phosphotransferases, phosphorylases and phosphomutases are described individually within separate subsections. A separate subsection summarizes the most recent approaches in applying phosphorylation biocatalysts in cascade reactions.
6.1. Synthetic Applications of Phosphohydrolases
The application of simple and inexpensive phosphoryl donors such as pyrophosphate with phosphatases for catalyzing phosphorylation reactions for the synthesis of phosphorylated products (see Figure 13) requires, however, measures to counteract or prevent the favored phosphatase-catalyzed hydrolysis of the newly synthesized phosphorylated products.
Reaction and enzyme engineering of the phosphatase-catalyzed 5′-phosphorylation of nucleosides using pyrophosphate as a phosphoryl donor has enabled the production of 5′-nucleotides at an industrial large scale, such as the production of inosine-5′-phosphate from inosine [160,161]. Bacterial nonspecific acid phosphatases have been exploited in the phosphorylation of a large range of monoalcohols and diols [162,163,164,165,166,167]. High product concentrations could be achieved in the phosphorylation of glycerol (see Figure 13), where 167 g per liter of racemic glycerol-1-phosphate was obtained when acid phosphatase PhoN-Sf from Shigella flexneri and pyrophosphate was used, and 104 g per liter of racemic glycerol-1-phosphate with the simple application of phytase from Aspergillus niger and monophosphate [167]. Even higher product concentrations of greater than 400 g per liter could be achieved in the 6-phosphorylation of maltotriose and methyl-α-D-glucopyranoside (see Figure 13) [166]. The phosphorylation efficiency of acid phosphatase from Pseudomonas aeruginosa towards 2-phosphorylation of L-ascorbic acid has been improved by protein engineering, and L-ascorbic acid-2-phosphate has been obtained with 48.6% conversion at a product concentration of 61.5 g per liter [168].
6.2. Synthetic Applications of Phosphotransferases
Another biocatalytic 5′-phosphorylation of the β-D-ribofuranoside substructure has been achieved in the synthesis of β-nicotinamide mononucleotide from the NAD+ precursor nicotinamide riboside using nicotinamide riboside kinase from Kluyveromyces marxianus and the system acetyl phosphate/acetate kinase from Bacillus stearothermophilus for ATP regeneration (see Figure 14), whereby a product concentration of 93.5 g per liter and a productivity of 281 g per liter and per day could be achieved for NMN [169]. The synthesis of L-glyceraldehyde-3-phosphate has been performed by the enantioselective phosphorylation of glyceraldehyde catalyzed by glycerol kinase, whereby only the L-glyceraldehyde is taken as a substrate [170,171]. The dihydroxyacetone kinase-catalyzed phosphorylation has been shown to be enantioselective for the D-glyceraldehyde, which has enabled the synthesis of D-glyceraldehyde-3-phosphate [172].
Recombinant hydroxyacid kinases have been very valuable for selectively phosphorylating hydroxycarboxylic acids containing two or more hydroxy functional groups. Mevalonate-5-kinase from Thermococcus kodakaraensis has been applied in the synthesis of (R)-5-phosphomevalonate in >98% purity and a 65% isolated yield [173]. Glycerate-2-kinase from Thermotoga maritima was applied to manufacture D-glycerate-2-phosphate in excellent purity and with a 72% isolated yield [174]. The highly active shikimate kinase AroL from Escherichia coli illustrates the power of phosphorylation biocatalysts by its application in a highly efficient one-step synthesis of shikimate-3-phosphate (see Figure 14), which is superior to other approaches by avoiding lengthy synthetic routes, low yields, hazardous reagents or side product formation [175].
Carbohydrate kinases provide great advantages for the protecting group free and selective phosphorylation of a specific hydroxy functional group. Enantiocomplementary D- and L-xylulokinases have been applied for phosphorylating D- and L-xylulose with the complete conversion to the D- and L-enantiomers of xylulose-5-phosphate, respectively [176]. A facile one-step enzymatic synthesis of D-tagatose-1,6-phosphate using a D-tagatose-6-phosphate kinase-catalyzed 1-phosphorylation of D-tagatose-6-phosphate with ATP regeneration has been demonstrated to be highly efficient and scalable [177]. The selectivity of a series of carbohydrate kinases has been used for the synthesis of eight phosphorylated ketopentoses, where the high substrate selectivity towards the ketopentoses but not to the ketoses D-xylose and L-arabinose enabled the control of the coupling of the reversible isomerization/epimerization reactions with the subsequent phosphorylation i one pot [178,179].
The highly selective biocatalytic O-phosphorylation of psilocyn to psilocybin catalyzed by recombinant ATP-dependent Psilocybe cubensis 4-hydroxytryptamine kinase PsiK has been demonstrated at a gram scale without ATP regeneration, with an isolated yield of 88.5% [180]. Avoiding low-yield phosphorylation chemistry using protecting groups and their removal by hydrogenolysis depending on heavy metals, and optimization opportunities by bioprocess development, such as reducing ATP cofactor amounts by ATP regeneration, downstream processing and product recovery, illustrate the power of phosphorylation biocatalysts in natural product synthesis.
The application of the recombinant arginine kinase ArgK from Limulus polyphemus enabled a straightforward and highly efficient one-step synthesis of Nω-phospho-L-arginine by the highly selective enzymatic N-phosphorylation of L-arginine at the ω-nitrogen (see Figure 14), thus avoiding lengthy routes with the introduction and removal of protecting groups [181].
6.3. Synthetic Applications of Phosphorylases
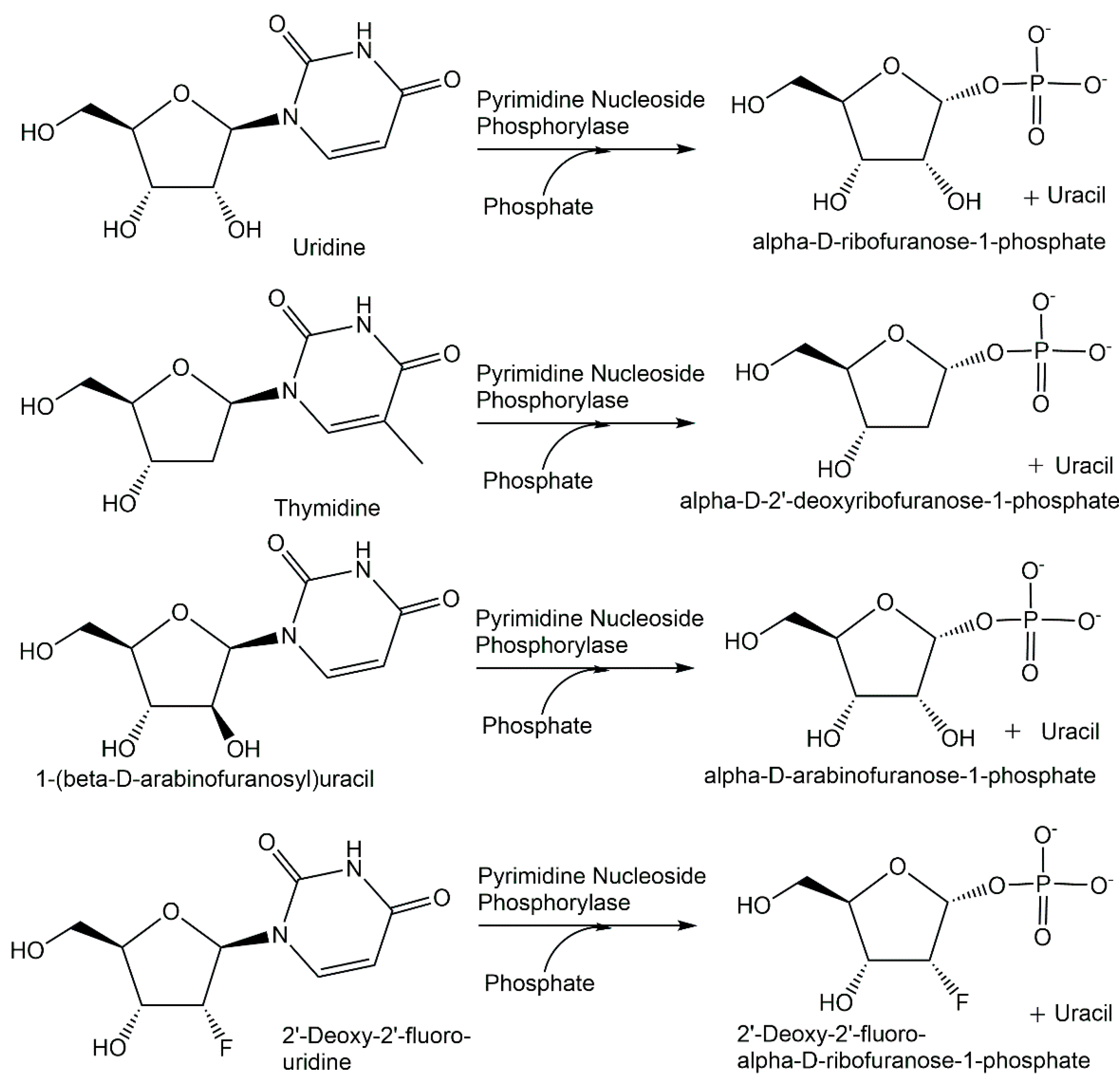
Sucrose phosphorylase from Leuconostoc mesenteroides has been covalently immobilized on Eupergit C for the continuous production in a packed bed reactor of α-D-glucose-1-phosphate from sucrose and phosphate (see Figure 5) at 0.6 M concentration each, whereby the reactor could be operated with a 91% conversion which remained stable up to a reaction time of 650 h [81]. When sucrose phosphorylase from Bifidobacterium adolescentis immobilized on Sepabeads for the continuous production at 60 °C of α-D-glucose-1-phosphate from sucrose and phosphate, a space–time yield of 179 g per liter and per hour was achieved [182]. A-D-glucose-1-phosphate has also been obtained in good yield from starch and phosphate at 0.7 M concentration each by using α-glucan phosphorylase from Thermus caldophilus [183]. Whole cells of E. coli expressing trehalose phosphorylase have been applied for the production of β-D-glucose-1-phosphate from trehalose and phosphate (see Figure 5) at 60 °C, with a 26% conversion [82]. The use of nucleoside phosphorylases for the synthesis of natural and modified α-D-pentofuranose-1-phosphates is of much interest for facilitated access to these key building blocks and metabolites. Highly efficient syntheses of α-D-ribose-1-phosphate and 2-deoxy-α-D-ribose-1-phosphate from 7-methylguanosine and 7-methly-2′-deoxyguanosine, respectively, and phosphate, have been achieved in a 74–96% yield after isolation and purification by enzymatic phosphorolysis (see Figure 5) using purine nucleoside phosphorylase [83].
Thermostable pyrimidine nucleoside phosphorylases have been applied in the facile and rapid enzymatic synthesis of a series of natural and modified α-D-pentofuranose-1-phosphates (see Figure 15) in gram quantities with a purity of greater than 95% from the corresponding nucleosides and phosphate [184]. After optimizing enzymatic phosphorolysis procedures and reaction conditions for the conversion of uridine and phosphate to α-D-ribofuranose-1-phosphate and uracil, and for the conversion of thymidine and phosphate to α-D-2′-deoxyribofuranose-1-phosphate and uracil, the protocols have been applied for the pyrimidine nucleoside phosphorylase-catalyzed synthesis of α-D-arabinofuranose-1-phosphate, 2′-deoxy-2′-fluoro-α-D-ribofuranose-1-phosphate and 2′-deoxy-2′-fluoro-α-D-arabinofuranose-1-phosphate [184].
6.4. Synthetic Applications of Phosphomutases
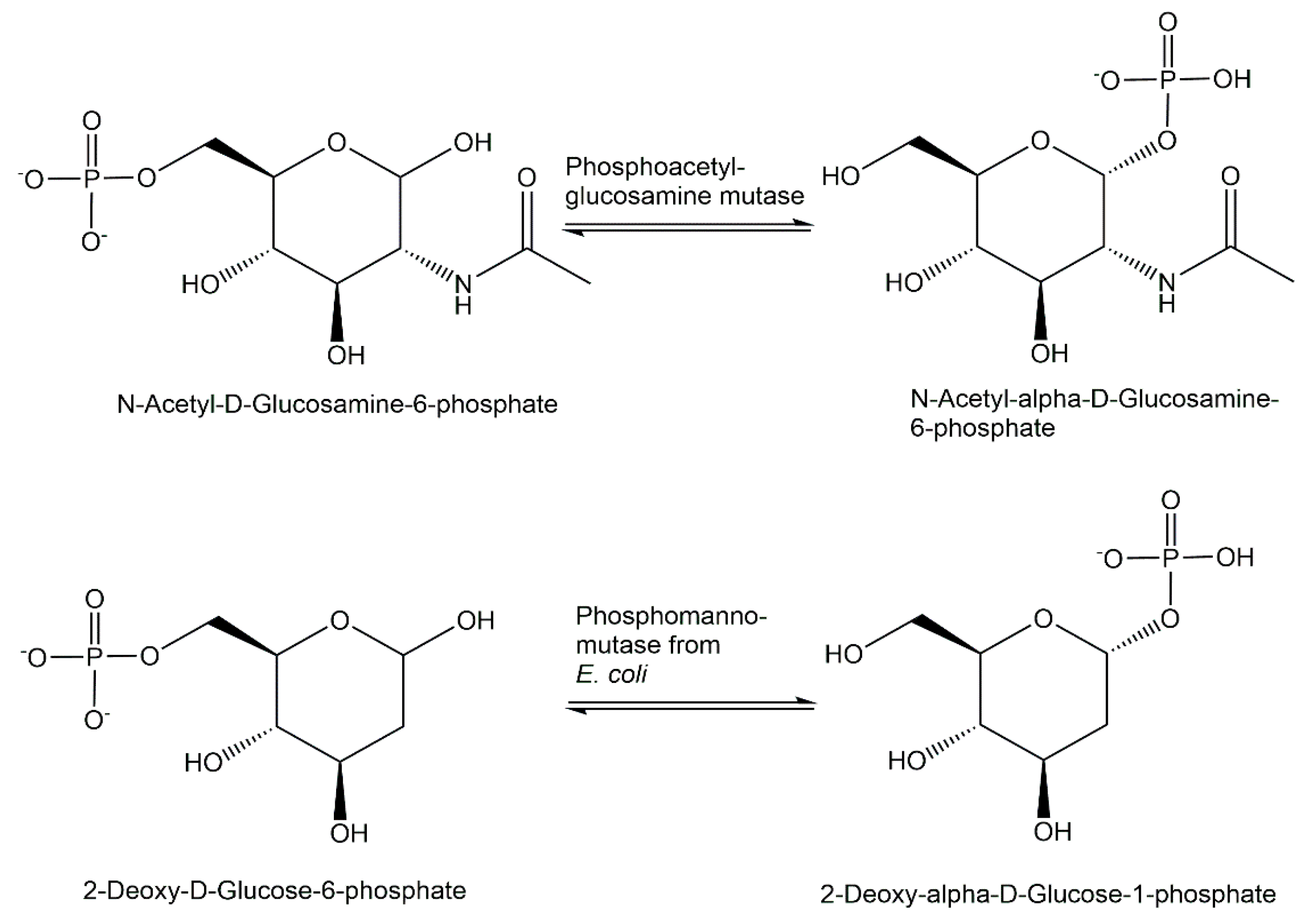
The equilibrium thermodynamics of intramolecular isomerization reactions catalyzed by phosphomutases needs to be overcome in order to avoid challenging product recovery and purification. Coupling the phosphomutase-catalyzed reaction step with a subsequent reaction provides an elegant strategy, also used by nature, to shift the thermodynamic equilibrium towards the product side. An early application has been the use of crude preparations of phosphoacetylglucosamine mutase from Neurospora crassa for catalyzing the conversion of labeled N-acetylglucosamine-6-phosphate to labeled N-acetylglucosamine-1-phosphate coupled with the subsequent reaction catalyzed by uridinetriphosphate: N-acetylglucosamine 1-phosphate phosphotransferase for the synthesis of labeled uridinediphosphate-N-acetylglucosamine [185]. The recombinant expression of phosphoacetylglucosamine mutase Agm1 from Saccharomyces cerevisiae has been advantageous for the conversion of N-acetylglucosamine-6-phosphate to N-acetylglucosamine-1-phosphate (see Figure 16) and subsequently to uridinediphosphate-N-acetylglucosamine as part of a de novo pathway to legionaminic acid [186].
A phosphopentomutase from a Bacillus sphaericus strain, which was found by screening for an acetaldehyde- and phosphorylated-compound-tolerant enzyme, was applied in its recombinant version with the corresponding gene cloned and expressed in E. coli, for catalyzing the isomerization of 2-deoxyribose-5-phosphate to 2-deoxyribose-1-phosphate (see Figure 6), towards the enzymatic synthesis of 2′-deoxynucleoside [187]. Phosphomannomutase from E. coli has been applied to catalyze the conversion of 2-deoxy-D-glucose-6-phosphate to 2-deoxy-D-glucose-1-phosphate (see Figure 16) as part of the enzymatic synthesis of deoxythymidinediphosphate-2-deoxy-α-glucose [188].
6.5. Phosphorylation Biocatalysts in Cascades
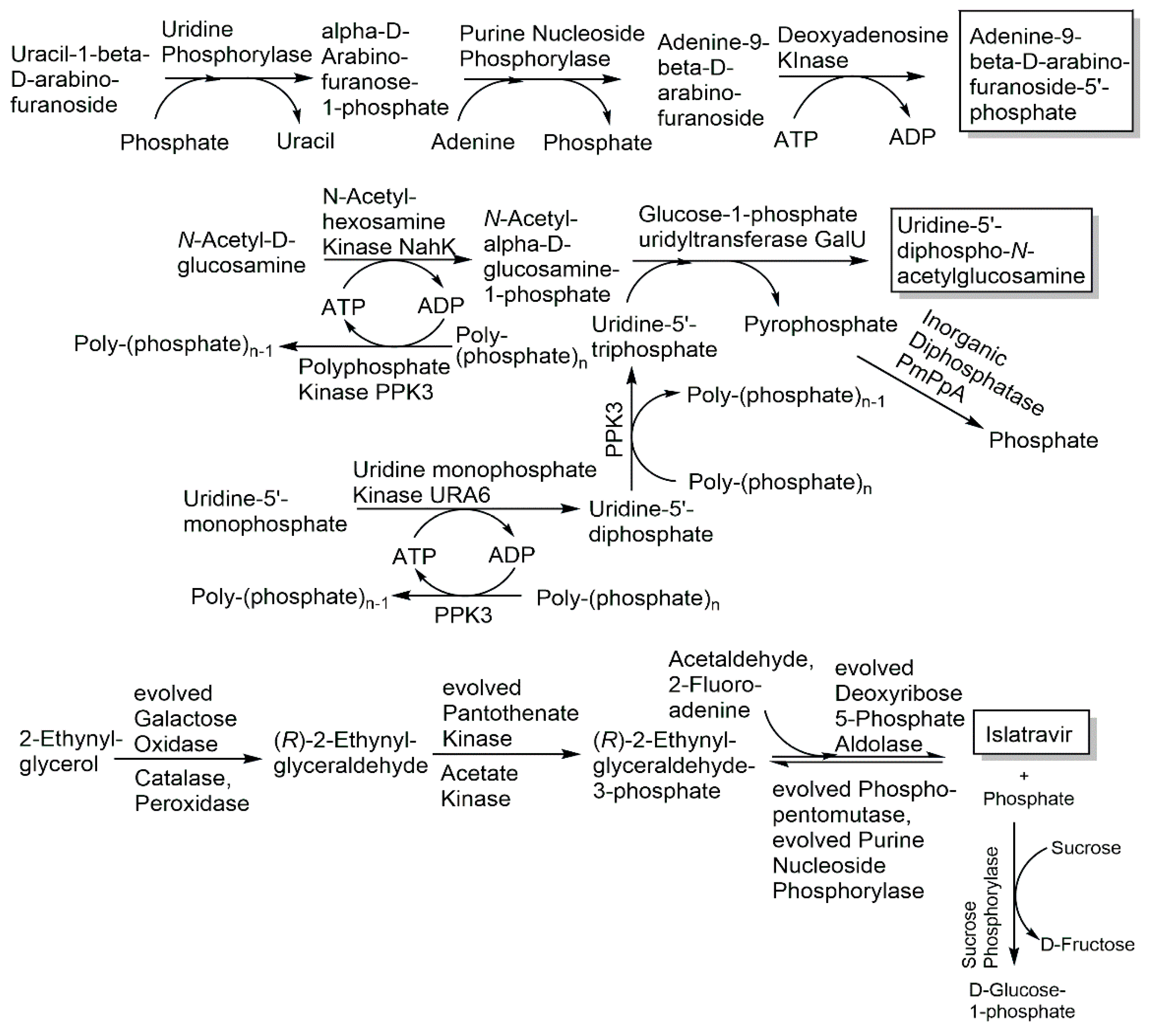
The five recombinant enzymes N–acetylhexosamine kinase NahK, glucose-1-phosphate uridyltransferase GalU, uridine monophosphate kinase URA6, polyphosphate kinase PPK3 and inorganic diphosphatase PmPpA have been applied in a cascade reaction in one pot for the production of uridine-5′-diphospho-N-acetylglucosamine (see Figure 17) from uridine-5′-monophosphate, N-acetylglucosamine and polyphosphate with a space–time yield of about 0.8 g per liter per hour [189]. The three enzymes uridine phosphorylase from Clostridium perfringens, purine nucleoside phosphorylase from Aeromonas hydrophila and deoxyadenosine kinase from Dictyostelium discoideum have been used for the synthesis of the antiviral arabinosyladenine-5′-monophosphate (see Figure 17) with 90% purity and 55% yield [190]. Kinases have also been successfully applied in synthetic reaction cascades for preparing key phosphorylated intermediates, for example dihydroxyacetone kinase in synthesizing phosphorylated D- and L-monosaccharides [191,192]. The most recent developments of various optimized phosphorylation biocatalysts in highly efficient and selective routes to antivirals are impressive examples of the fast move of biocatalytic phosphorylations into industrial processes of the pharmaceutical industry. Five phosphorylation biocatalysts, the evolved pantothenate kinase, phosphopentomutase, purine nucleoside phosphorylase and the two auxiliary enzymes acetate kinase from Thermotoga maritima and sucrose phosphorylase from Alloscardovia omnicolens, have been key for the success of the multistep enzymatic manufacturing process to the antiviral islatravir (see Figure 17) against HIV [193]. The advantages of biocatalytic cascade reactions for complexity reduction, faster delivery and higher yield are also important in the synthesis of stable isotope-labeled compounds, such as the enzymatic one-pot synthesis of 13C- and 15N-labeled ATP and GTP with up to 66% isolated yields using various kinases [194], due to the costs of the stable isotope-labeled starting materials. When radioactive isotopes are involved, the minimization of radioactive waste and the elimination of handling radioactive intermediates are further advantages, such as in the one-pot biocatalytic cascade reaction to the 14C-labeled antiHIV nucleoside islatravir from 2-14C-acetaldehyde, which gave a five-fold improvement in the overall radiochemical yield compared with the chemical route [195]. Evolving S-methyl-5-thioribose kinase activity for the 1-phosphorylation of the 5-isobutyryl-D-ribose with an α:β diastereomer ratio of >99:1, using propionyl-phosphate as phosphoryl donor, and engineering the uridine phosphorylase activity for the synthesis of 5′-isobutyryluridine from 5′-isobutyryl-D-ribose-1-phosphate and uracil have been key, together with other auxiliary enzymes, in a novel short reaction cascade to the antiviral molnupiravir, which decreased the 10 reaction steps of the initial chemical route from D-ribose and uracil to 3 steps, while increasing the overall yield from less than 10% to 69% [196].
The chemically challenging diastereoselective synthesis of P-chiral nucleotide 1-thiotriphosphates has been achieved by a combination of the enzyme optimization of adenylate kinase, guanylate kinase and the phosphoryl donor regenerating acetate kinase by several rounds of directed evolution with reaction engineering [197].
6.6. Phosphoryl Donors and Systems for their Regeneration
Easily accessible, safe and inexpensive inorganic phosphates are attractive phosphoryl donors for a variety of biocatalytic phosphorylation reactions. Therefore, phosphorylation biocatalysts, which can accept inorganic phosphoryl donors, such as phosphorylases accepting phosphate, phosphatases accepting pyrophosphate or kinases accepting polyphosphates, are of much interest for phosphorylations at an industrial large scale. The full utilization of the phosphoryl donor is realized with simple phosphate as a donor in phosphorylase-catalyzed processes. The use of pyrophosphate for the phosphorylation of inosine using an engineered acid phosphatase (see Figure 4) has enabled an industrial large-scale production process for inosine-5-monophosphate with an 88% yield and a product concentration of greater than 100 g per liter [73,74]. As pyrophosphate can be easily prepared from phosphate, the phosphate byproduct formed in the phosphorylation reaction can also be recycled to pyrophosphate. Polyphosphate has not only received much attention as an easily accessible, inexpensive and stable phosphoryl donor for industrial phosphorylations but is also of fundamental interest [198,199], and early work on metaphosphate as a phosphoryl donor has led to the discovery of an NAD kinase [200] accepting metaphosphate, but not pyrophosphate or polyphosphate.
A great number of phosphorylation biocatalysts are however not dependent on inorganic phosphoryl donors but require high-energy organic compounds as phosphoryl donors [59], among which ATP is in wide use in natural and synthetic phosphorylation reactions. Therefore, systems for the synthesis and regeneration of ATP are of much interest, not only for phosphorylation biocatalysts with respect to possible enzyme inhibition, downstream processing aspects or costs of the phosphorylation process, but also in the wider context of ATP-dependent biocatalysts [201,202]. Regeneration systems utilizing sacrificial high energy phosphorylating agents together with the corresponding enzymes to catalyze the conversion of ADP to ATP, such as phosphoenolpyruvate and pyruvate kinase, or acetyl phosphate and acetate kinase, have been in wide use since the pioneering work of Whitesides and coworkers [203,204,205]. The further development of new ATP regeneration systems using easily accessible polyphosphates and polyphosphate kinases [206], high energy agents such as propionylphosphate [196] and phosphorus recycling enzymes in cascade reactions such as pyruvate oxidase [196,207], provides great opportunities for complexity reduction. A new electrochemical system using electricity instead of high energy phosphates or stoichiometric oxidants has been applied for ATP regeneration at the 20 g scale, whereby current and enzymatic processes need to be balanced [208].
6.7. Phosphorylation Reaction Engineering
The in-depth knowledge and characterization of the phosphorylation biocatalyst and the dependence of its properties, such as its activity, stability, activation and inhibition, on parameters such as temperature, pH, buffer or ionic strength for the reaction in focus is essential for achieving an optimized performance of a kinetically and thermodynamically feasible phosphorylation reaction. Kinetic as well as thermodynamic parameters and the determination of the Michaelis constant KM, kcat or inhibitor constants, the equilibrium constant of the reaction and complexation constants, for example of ATP with a divalent cation such as Mg2+, are of major importance not only for optimizing a single enzymatic reaction but also for a reaction cascade. The properties and stabilities of reaction components can be decisive for reaching optimal reaction engineering parameters, for example in the product half life of the glycerol kinase-catalyzed phosphorylation of L-glyceraldehyde to L-glyceraldehyde-3-phosphate [209] and the effect of the Mg/ATP-ratio on glycerol kinase [210].
6.8. Product Recovery and Purification
What is already being taken into account in the reaction design is how the phosphorylation can be driven to complete conversion and how a phosphorylated product is isolated from the reaction mixture is also essential for later operations. Facile product recovery and purification can be a significant factor for the economy and sustainability of an overall phosphorylation process. Therefore, making use of the most suitable and effective methodologies [211] and developing innovative new approaches for recovering and purifying the highly charged products from aqueous media is of major importance.
7. Opportunities and Outlook
The planetary boundary of the phosphorus biochemical flow at high risk and the essential and unique status of phosphorus bonds for many central features of life require resource-efficient use and the re-use of the nonrenewable phosphorus in order to close the phosphorus cycle. The characteristic and prevailing form of phosphorus occurs in nature in a stable oxidation state of five, which is what scholars have proposed chemists should use too [212], and in chemical bonds to oxygen in phosphates, with an impressive balance of stability and reactivity as pH-dependent ionized species.
The opportunities for discovering and utilizing the power of phosphorylation biocatalysts look exciting towards highly selective and efficient phosphorylation reactions, towards understanding fundamental and applied aspects and towards sustainable industrial chemistry [213]. Due to its benefits for selectivity, safety, health, the environment and sustainability, the use of phosphorylation biocatalysts enabling complete conversion to the phosphorylated product in one reaction step is a highly attractive and powerful synthetic strategy for replacing lengthy phosphorylation methodologies, which require protected phosphoryl group donors as well as the introduction of suitable protecting groups into the starting compound. The predominant use by nature of the phosphorus oxidation state +5 and of the phosphate group, which can be introduced from inorganic or organic phosphoryl donors and can occur in different ionization states carrying multiple negative charges, provides inspirations for making further valuable use of phosphorylation biocatalysts in various directions. Cost-efficient industrial large-scale processes can be envisaged by developing phosphorylation biocatalysts accepting inorganic phosphoryl donors. The preferential use of phosphate as a good leaving group in various biochemical reactions, such as carbon–carbon bond formation, decarboxylation, substitution and elimination reactions [7], is an opportunity to develop new reaction cascades involving phosphorylation biocatalysts and subsequent reactions, thereby replacing leaving groups traditionally used in synthetic chemistry, such as halides, tosylates or triflates. Finally, the biocatalytic introduction of the negatively charged phosphate group into poorly water-soluble compounds is of interest for improving the solubilities in aqueous media and for the retention of compounds within biological cells as negatively charged compounds after their poorly water-soluble precursors have passed the cell membranes and have undergone intracellular phosphorylation.
Therefore, the further exploration of the capabilities and power of natural and engineered phosphorylation biocatalysts from different angles and perspectives will be very beneficial, not only for highly selective and efficient phosphorylation reactions, but also for biocatalytic routes involving phosphate as a leaving group and for systems biocatalysis approaches.
Funding
This research received no external funding.
Conflicts of Interest
The author declares no conflict of interest.
References
- Ruttenberg, K.C. The Global Phosphorus Cycle. Treatise Geochem. 2003, 8, 585–645. [Google Scholar] [CrossRef]
- Steffen, W.; Richardson, K.; Rockström, J.; Cornell, S.E.; Fetzer, I.; Bennett, E.M.; Biggs, R.; Carpenter, S.R.; De Vries, W.; De Wit, C.A.; et al. Planetary boundaries: Guiding human development on a changing planet. Science 2015, 347, 1259855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jupp, A.R.; Beijer, S.; Narain, G.C.; Schipper, W.; Slootweg, J.C. Phosphorus recovery and recycling–closing the loop. Chem. Soc. Rev. 2021, 50, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Geeson, M.B.; Cummins, C.C. Phosphoric acid as a precursor to chemicals traditionally synthesized from white phosphorus. Science 2018, 359, 1383–1385. [Google Scholar] [CrossRef] [Green Version]
- Willey, N.; Timbs, P. Radioactivity in Future Phosphogypsum: New predictions based on estimates of ‘Peak P’ and rock phosphate resources. J. Environ. Radioact. 2022, 244–245, 106828. [Google Scholar] [CrossRef]
- Pasek, M.A. Thermodynamics of Prebiotic Phosphorylation. Chem. Rev. 2020, 120, 4690–4706. [Google Scholar] [CrossRef]
- Westheimer, F.H. Why Nature Chose Phosphates. Science 1987, 235, 1173–1178. [Google Scholar] [CrossRef]
- Bowler, M.W.; Cliff, M.J.; Waltho, J.P.; Blackburn, G.M. Why did Nature select phosphate for its dominant roles in biology? New J. Chem. 2010, 34, 784–794. [Google Scholar] [CrossRef]
- Kamerlin, S.C.; Sharma, P.K.; Prasad, R.B.; Warshel, A. Why Nature Really Chose Phosphate. Q. Rev. Biophys. 2013, 246, 1–132. [Google Scholar] [CrossRef]
- Wohlgemuth, R. Key advances in biocatalytic phosphorylations in the last two decades: Biocatalytic syntheses in vitro and biotransformations in vivo (in humans). Biotechnol. J. 2021, 16, 2000090. [Google Scholar] [CrossRef]
- Nam, I.; Lee, J.K.; Nam, H.G.; Zare, R.N. Abiotic production of sugar phosphates and uridine ribonucleoside in aqueous micro-droplets. PNAS 2017, 114, 12396–12400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cramer, F. Neuere Methoden der präparativen organischen Chemie III 2. Darstellung von Estern, Amiden und Anhydriden der Phosphorsäure. Angew. Chem. 1960, 72, 236–249. [Google Scholar] [CrossRef]
- Cramer, F.; Weimann, G. Imidoester, VII. Trichloracetonitril, ein Reagenz zur selektiven Veresterung von Phosphorsäuren. Chem. Ber. 1961, 94, 996–1007. [Google Scholar] [CrossRef]
- Sakakura, A.; Katsukawa, M.; Ishihara, K. Selective synthesis of phosphate monoesters by dehydrative condensation of phosphoric acid and alcohols promoted by nucleophilic bases. Org. Lett. 2005, 7, 1999–2002. [Google Scholar] [CrossRef] [PubMed]
- Dueymes, C.; Pirat, C.; Pascal, R. Facile synthesis of simple mono-alkyl phosphates from phosphoric acid and alcohols. Tetrahedron Lett. 2008, 49, 5300–5301. [Google Scholar] [CrossRef]
- Lira, L.M.; Vasilev, D.; Pilli, R.A.; Wessjohann, L.A. One-pot synthesis of organophosphate monoesters from alcohols. Tetrahedron Lett. 2013, 54, 1690–1692. [Google Scholar] [CrossRef] [Green Version]
- Domon, K.; Puripat, M.; Fujiyoshi, K.; Hatanaka, M.; Kawashima, S.A.; Yamatsugu, K.; Kanai, K. Catalytic Chemoselective O-Phosphorylation of Alcohols. ACS Cent. Sci. 2020, 6, 283–292. [Google Scholar] [CrossRef] [Green Version]
- McDonald, A.G.; Tipton, K.F. Enzyme nomenclature and classification: The state of the art. FEBS J. 2022. [Google Scholar] [CrossRef]
- Sayers, E.W.; Cavanaugh, M.; Clark, K.; Pruitt, K.D.; Schoch, C.L.; Sherry, S.T.; Karsch-Mizrachi, I. GenBank. Nucleic Acids Res. 2021, 49, D92–D96. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Di Costanzo, L.; Duarte, J.M.; et al. RCSB Protein Data Bank: Powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2021, 49, D437–D451. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Chang, A.; Jeske, L.; Ulbrich, S.; Hofmann, J.; Koblitz, J.; Schomburg, I.; Neumann-Schaal, M.; Jahn, D.; Schomburg, D. BRENDA, the ELIXIR core data resource in 2021: New developments and updates. Nucleic Acids Res. 2021, 49, D498–D508. [Google Scholar] [CrossRef] [PubMed]
- Gardossi, L.; Poulsen, P.B.; Ballesteros, A.; Hult, K.; Švedas, V.K.; Vasić-Rački, Đ.; Carrea, G.; Magnusson, A.; Schmid, A.; Wohlgemuth, R.; et al. Guidelines for reporting of biocatalytic reactions. Trends Biotechnol. 2010, 28, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Swainston, N.; Baici, A.; Bakker, B.M.; Cornish-Bowden, A.; Fitzpatrick, P.F.; Halling, P.; Leyh, T.S.; O’Donovan, C.; Raushel, F.M.; Reschel, U.; et al. STRENDA DB: Enabling the validation and sharing of enzyme kinetics data. FEBS J. 2018, 285, 2193–2204. [Google Scholar] [CrossRef]
- Cheek, S.; Zhang, H.; Grishin, N.V. Sequence and structure classification of kinases. J. Mol. Biol. 2002, 320, 855–881. [Google Scholar] [CrossRef] [Green Version]
- Cheek, S.; Ginalski, K.; Zhang, H.; Grishin, N.V. A comprehensive update of the sequence and structure classification of kinases. BMC Struct. Biol. 2005, 5, 6. [Google Scholar] [CrossRef] [Green Version]
- Blum, M.; Chang, H.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 2021, 49, D344–D354. [Google Scholar] [CrossRef]
- Kannan, N.; Taylor, S.S.; Zhai, Y.; Venter, J.C.; Manning, G. Structural and Functional Diversity of the Microbial Kinome. PloS Biology 2007, 5, e17. [Google Scholar] [CrossRef]
- Anderson, C.M.; Stenkamp, R.E.; Steitz, T.A. Sequencing a protein by X-ray crystallography: II. Refinement of yeast hexokinase B Co-ordinates and sequence at 2.1 Å resolution. J. Mol. Biol. 1978, 123, 15–33. [Google Scholar] [CrossRef]
- Stuart, D.I.; Levine, M.; Muirhead, H.; Stammers, D.K. Crystal structure of cat muscle pyruvate kinase at a resolution of 2.6 Å. J. Mol. Biol. 1979, 134, 109–142. [Google Scholar] [CrossRef]
- Watson, H.C.; Walker, N.P.; Shaw, P.J.; Bryant, T.N.; Wendell, P.L.; Fothergill, L.A.; Perkins, R.E.; Conroy, S.C.; Dobson, M.J.; Tuite, M.F. Sequence and structure of yeast phosphoglycerate kinase. EMBO J. 1982, 1, 1635–1640. [Google Scholar] [CrossRef] [PubMed]
- Deville-Bonne, D.; El Amri, C.; Meyer, P.; Chen, Y.; Agrofoglio, L.A.; Janin, J. Human and viral nucleoside/nucleotide kinases involved in antiviral drug activation: Structural and catalytic properties. Antivir. Res. 2010, 86, 101–120. [Google Scholar] [CrossRef] [PubMed]
- Van Horn, W.D.; Kim, H.-J.; Ellis, C.D.; Hadziselimovic, A.; Sulistijo, E.S.; Karra, M.D.; Tian, C.; Sönnichsen, F.D.; Sanders, C.R. Solution NMR Structure of Membrane-Integral Diacylglycerol Kinase. Science 2009, 324, 1726–1729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Lyons, J.A.; Pye, V.E.; Vogeley, L.; Araga, D.; Kenyon, C.P.; Shah, S.T.A.; Doherty, C.; Aherne, M.; Caffrey, M. Crystal structure of the integral membrane diacylglycerol kinase. Nature 2013, 497, 521–524. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Shen, Y.; Chen, Y.; Zhang, Z.; Ma, S.; Wan, Q.; Tong, Q.; Glaubitz, C.; Liu, M.; Yang, J. Structure of membrane diacylglycerol kinase in lipid bilayers. Commun. Biol. 2021, 4, 282. [Google Scholar] [CrossRef]
- Knighton, D.R.; Zheng, J.; Ten Eyck, L.F.; Ashford, V.A.; Xuong, N.H.; Taylor, S.S. and Sowadski, J.M. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science 1991, 253, 407–414. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [Green Version]
- Arter, C.; Trask, L.; Ward, S.; Yeoh, S. and Bayliss, R. Structural features of the protein kinase domain and targeted binding by small molecule inhibitors. J. Biol. Chem. 2022, 298, 102247. [Google Scholar] [CrossRef]
- Modi, Y.; Dunbrack Jr, R.L. Kincore: A web resource for structural classification of protein kinases and their inhibitors. Nucleic Acids Res. 2022, 50, D654–D664. [Google Scholar] [CrossRef] [PubMed]
- Ohira, T.; Minowa, K.; Sugiyama, K.; Yamashita, S.; Sakaguchi, Y.; Miyauchi, K.; Noguchi, R.; Kaneko, A.; Orita, I.; Fukui, T.; et al. Reversible RNA phosphorylation stabilizes tRNA for cellular thermotolerance. Nature 2022, 605, 372–379. [Google Scholar] [CrossRef] [PubMed]
- SCOP 2 Database. Available online: http://scop.mrc-lmb.cam.ac.uk/ (accessed on 15 August 2022).
- Allen, K.N.; Dunaway-Mariano, D. Catalytic scaffolds for phosphoryl group transfer. Curr. Opin. Struct. Biol. 2016, 41, 172–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatzios, S.K.; Iavarone, A.T.; Bertozzi, C.R. Rv2131c from Mycobacterium tuberculosis Is a CysQ 3′-Phosphoadenosine-5′-phosphatase. Biochemistry 2008, 47, 5823–5831. [Google Scholar] [CrossRef] [Green Version]
- Erickson, A.I.; Sarsam, R.D.; Fisher, A.J. Crystal Structures of Mycobacterium tuberculosis CysQ, with Substrate and Products Bound. Biochemistry 2015, 54, 6830–6841. [Google Scholar] [CrossRef]
- Ishikawa, K.; Mihara, Y.; Gondoh, K.; Suzuki, E.; Asano, Y. X-ray structures of a novel acid phosphatase from Escherichia blattae and its complex with the transition-state analog molybdate. EMBO J. 2000, 19, 2412–2423. [Google Scholar] [CrossRef] [Green Version]
- Asano, Y. Overview of screening for new microbial biocatalysts and their uses in organic synthesis – selection and optimization of biocatalysts. J. Biotechnol. 2002, 94, 65–72. [Google Scholar] [CrossRef]
- Ishikawa, K.; Mihara, Y.; Shimba, N.; Ohtsu, N.; Kawasaki, H.; Suzuki, E.-i.; Asano, Y. Enhancement of nucleoside phosphorylation activity in an acid phosphatase. Protein Eng. 2002, 15, 539–543. [Google Scholar] [CrossRef] [Green Version]
- Mihara, Y.; Ishikawa, K.; Suzuki, E.-i.; Asano, Y. Improving the Pyrophosphate-inosine Phosphotransferase Activity of Escherichia blattae Acid Phosphatase by Sequential Site-directed Mutagenesis. Biosci. Biotechnol. Biochem. 2004, 68, 1046–1050. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Yan, S.; Hou, X.; Song, W.; Wang, L.; Wu, T.; Qi, M.; Wu, J.; Rao, Y.; Wang, B.; et al. Local Electric Field Modulated Reactivity of Pseudomonas aeruginosa Acid Phosphatase for Enhancing Phosphorylation of L-Ascorbic Acid. ACS Catal. 2021, 11, 13397–13407. [Google Scholar] [CrossRef]
- Puchart, V. Glycoside phosphorylases: Structure, catalytic properties and biotechnological potential. Biotechnol. Adv. 2015, 33, 261–276. [Google Scholar] [CrossRef] [PubMed]
- Pugmire, M.J.; Ealick, S.E. Structural analyses reveal two distinct families of nucleoside phosphorylases. Biochem. J. 2002, 361, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Drula, E.; Garron, M.-L.; Dogan, S.; Lombard, V.; Henrissat, B.; Terrapon, N. The carbohydrate-active enzyme database: Func-tions and literature. Nucleic Acids Res. 2022, 50, D571–D577. [Google Scholar] [CrossRef]
- Sun, S.; You, C. Disaccharide phosphorylases: Structure, catalytic mechanisms and directed evolution. Synth. Syst. Biotechnol. 2021, 6, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Li, X.; Guo, W.; Wu, B. Crystal structures of a new class of pyrimidine/purine nucleoside phosphorylase revealed a Cupin fold. Proteins 2022, 90, 1233–1241. [Google Scholar] [CrossRef]
- Panosian, T.D.; Nannemann, D.P.; Watkins, G.R.; Phelan, V.V.; McDonald, W.H.; Wadzinski, B.E.; Bachmann, B.O.; Iverson, T.M. Bacillus cereus phosphopentomutase is an alkaline phosphatase family member that exhibits an altered entry point into the catalytic cycle. J. Biol. Chem. 2011, 286, 8043–8054. [Google Scholar] [CrossRef] [Green Version]
- Frasse, P.M.; Miller, J.J.; Polino, A.J.; Soleimani, E.; Zhu, J.S.; Jakeman, D.L.; Jez, J.M.; Goldberg, D.E.; John, A.R.O. 2022. Enzymatic and structural characterization of HAD5, an essential phosphomannomutase of malaria-causing parasites. J. Biol. Chem. 2022, 298, 101550. [Google Scholar] [CrossRef]
- Gauss, D.; Schönenberger, B.; Molla, G.S.; Kinfu, B.M.; Chow, J.; Liese, A.; Streit, W.R.; Wohlgemuth, R. Biocatalytic phosphorylation of metabolites. In Applied Biocatalysis–From Fundamental Science to Industrial Applications; Hilterhaus, L., Liese, A., Kettling, U., Antranikian, G., Eds.; Wiley-VCH: Weinheim, Germany, 2016; pp. 147–177. [Google Scholar]
- Wohlgemuth, R.; Liese, A.; Streit, W. Biocatalytic phosphorylations of metabolites: Past, present, and future. Trends Biotechnol. 2017, 35, 452–465. [Google Scholar] [CrossRef]
- Tsunoda, T.; Samadi, A.; Burade, S.; Mahmud, T. Complete biosynthetic pathway to the antidiabetic drug acarbose. Nat. Commun. 2022, 13, 3455. [Google Scholar] [CrossRef]
- Minagawa, K.; Zhang, Y.; Ito, T.; Bai, L.; Deng, Z.; Mahmud, T. ValC, a New Type of C7-Cyclitol Kinase Involved in the Biosynthesis of the Antifungal Agent Validamycin, A. ChemBioChem 2007, 8, 632–641. [Google Scholar] [CrossRef]
- Smith, C.-I.; Martin, S.R.; and Smith, K.S. Acetate kinase: Not just a bacterial enzyme. Trends Microbiol. 2006, 14, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Bachochin, M.J.; Van Allen, M.; Barber, R.D. Characterization of a Rhodobacter sphaeroides primary fatty acid kinase. Arch. Microbiol. 2021, 203, 861–864. [Google Scholar] [CrossRef] [PubMed]
- Wallimann, T.; Tokarska-Schlattner, M.; Schlattner, U. The creatine kinase system and pleiotropic effects of creatine. Amino Acids 2011, 40, 1271–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.-S.; Wang, Y.; Li, C.; Chen, Z.; Yan, Y.-B.; Zhou, H.-M. Dissecting the key residues crucial for the species-specific thermostability of muscle-type creatine kinase. Int. J. Biol. Macromol. 2010, 47, 366–370. [Google Scholar] [CrossRef]
- Hunter, T. A journey from phosphotyrosine to phosphohistidine and beyond. Mol. Cell 2022, 82, 2190–2220. [Google Scholar] [CrossRef]
- Lu, Z.; Hunter, T. Metabolic kinases moonlighting as protein kinases. Trends Biochem. Sci. 2018, 43, 301–310. [Google Scholar] [CrossRef]
- Berginski, M.E.; Moret, N.; Liu, C.; Goldfarb, D.; Sorger, P.K.; Gomez, S.M. The Dark Kinase Knowledgebase: An online compendium of knowledge and experimental results of understudied kinases. Nucleic Acids Res. 2021, 49, D529–D535. [Google Scholar] [CrossRef]
- Moret, N.; Liu, C.; Gyori, B.M.; Bachman, J.A.; Steppi, A.; Hug, C.; Taujale, R.; Huang, L.C.; Berginski, M.E.; Gomez, S.M.; et al. A resource for exploring the understudied human kinome for research and therapeutic opportunities. BioRxiv 2021. Available online: https://www.biorxiv.org/content/10.1101/2020.04.02.022277v3 (accessed on 15 August 2022).
- Morton, R.K. The Phosphotransferase Activity of Phosphatases. Biochem. J. 1958, 70, 139–155. [Google Scholar] [CrossRef] [Green Version]
- Holden, H.M.; Raushel, F.M. From the Three-Dimensional Structure of Phosphotriesterase. Biochemistry 2021, 60, 3413–3415. [Google Scholar] [CrossRef]
- Asano, Y.; Mihara, Y.; Yamada, H. A novel selective nucleoside phosphorylation enzyme from Morganella morganii. J. Biosci. Bioeng. 1999, 87, 732–738. [Google Scholar] [CrossRef]
- Mihara, Y.; Utagawa, T.; Yamada, H.; Asano, Y. Acid phosphatase/phosphotransferases from enteric bacteria. J. Biosci. Bioeng. 2001, 92, 50–54. [Google Scholar] [CrossRef]
- Tanaka, N.; Hasan, Z.; Hartog, A.F.; van Herk, T.; Wever, R. Phosphorylation and dephosphorylation of polyhydroxy compounds by class A bacterial acid phosphatases. Org. Biomol. Chem. 2003, 1, 2833–2839. [Google Scholar] [CrossRef] [PubMed]
- Tasnádi, G.; Lukesch, M.; Zechner, M.; Jud, W.; Hall, M.; Ditrich, K.; Baldenius, K.; Hartog, A.F.; Wever, R.; Faber, K. Exploiting acid phosphatases in the synthesis of phosphorylated monoalcohols and diols. Eur. J. Org. Chem. 2016, 1, 45–50. [Google Scholar] [CrossRef] [Green Version]
- Ros, S.; Schulze, A. Balancing glycolytic flux: The role of 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatases in cancer metabolism. Cancer Metab. 2013, 1, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nannemann, D.P.; Kaufmann, K.W.; Meiler, J.; Bachmann, B.O. Design and directed evolution of a dideoxy purine nucleoside phosphorylase. Protein Eng. Des. Sel. 2010, 23, 607–616. [Google Scholar] [CrossRef] [Green Version]
- Birmingham, W.R.; Starbird, C.A.; Panosian, T.D.; Nannemann, D.P.; Iverson, T.M.; Bachmann, B.O. Bioretrosynthetic construction of a didanosine biosynthetic pathway. Nat. Chem. Biol. 2014, 10, 392–399. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Szeker, K.; Janocha, B.; Böhme, T.; Albrecht, D.; Mikhailopulo, I.A.; Neubauer, P. Recombinant purine nucleoside phosphorylases from thermophiles: Preparation, properties and activity towards purine and pyrimidine nucleosides. FEBS J. 2013, 280, 1475–1490. [Google Scholar] [CrossRef]
- Goedl, C.; Schwarz, A.; Minani, A.; Nidetzky, B. Recombinant sucrose phosphorylase: Characterization, kinetic studies of transglucosylation, and application of immobilized enzyme for production of alpha-D-glucose 1-phosphate. J. Biotechnol. 2007, 129, 77–86. [Google Scholar] [CrossRef]
- Van der Borght, J.; Desmet, T.; Soetaert, W. Enzymatic production of β-d-glucose-1-phosphate from trehalose. Biotechnol. J. 2010, 5, 986–993. [Google Scholar] [CrossRef] [Green Version]
- Kulikova, I.V.; Drenichev, M.S.; Solyev, P.N.; Alexeev, C.S.; Mikhailov, C.N. Enzymatic Synthesis of 2-Deoxyribose 1-phosphate and Ribose 1-phosphate and Subsequent Preparation of Nucleosides. Eur. J. Org. Chem. 2019, 6999–7004. [Google Scholar] [CrossRef]
- Fothergill-Gilmore, L.A.; Watson, H.C. The phosphoglycerate mutases. Adv. Enzymol. Relat. Areas Mol. Biol 1989, 62, 227–313. [Google Scholar] [CrossRef]
- Tozzi, M.G.; Camici, M.; Mascia, L.; Sgarrella, F.; Ipata, P.L. Pentose phosphates in nucleoside interconversion and catabolism. FEBS.J. 2006, 273, 1089–1101. [Google Scholar] [CrossRef] [PubMed]
- Kedzierski, L.; Malby, R.L.; Smith, B.J.; Perugini, M.A.; Hodder, A.N.; Ilg, T.; Colman, P.M.; Handman, E. Structure of Leishmania mexicana phosphomannomutase highlights similarities with human isoforms. J. Mol. Biol. 2006, 363, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Stiers, K.M.; Muenks, A.G.; Beamer, L.J. Biology, mechanism, and structure of enzymes in the α-D-phosphohexomutase superfamily. Adv. Protein Chem. Struct. Biol. 2017, 109, 265–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, J.; Finci, L.; Zhang, C.; Lahiri, S.; Zhang, G.; Peisach, E.; Allen, K.N.; Dunaway-Mariano, D. Analysis of the structural determinants underlying discrimination between substrate and solvent in β-phosphoglucomutase catalysis. Biochemistry 2009, 48, 1984–1995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Lu, Z.; Han, Y.; Jia, Y.; Howard, A.; Dunaway-Mariano, D.; Herzberg, O. Conformational flexibility of PEP mutase. Biochemistry 2004, 43, 4447–4453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knowles, J.R. Enzyme-Catalyzed Phosphoryl Transfer Reactions. Annu. Rev. Biochem. 1980, 49, 877–919. [Google Scholar] [CrossRef]
- Cleland, W.W.; Hengge, A.C. Enzymatic Mechanisms of Phosphate and Sulfate Transfer. Chem. Rev. 2006, 106, 3252–3278. [Google Scholar] [CrossRef]
- Lassila, J.K.; Zalatan, J.G.; Herschlag, D. Biological Phosphoryl-Transfer Reactions: Understanding Mechanism and Catalysis. Ann. Rev. Biochem. 2011, 80, 669–702. [Google Scholar] [CrossRef] [Green Version]
- Allen, K.N.; Dunaway-Mariano, D. Phosphoryl group transfer: Evolution of a catalytic scaffold. Trends Biochem. Sci. 2004, 29, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Stockbridge, R.B.; Wolfenden, R. The intrinsic reactivity of ATP and the catalytic proficiencies of kinases acting on glucose, N-acetylgalactosamine, and homoserine: A thermodynamic analysis. J. Biol. Chem. 2009, 284, 22747–22757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerns, S.J.; Agafonov, R.V.; Cho, Y.-J.; Pontiggia, F.; Otten, R.; Pachov, D.V.; Kutter, S.; Phung, L.A.; Murphy, P.N.; Thai, V.; et al. The energy landscape of adenylate kinase during catalysis. Nat. Struct. Mol. Biol. 2015, 22, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Parnell, A.E.; Mordhorst, S.; Kemper, F.; Giurrandino, M.; Prince, J.P.; Schwarzer, N.J.; Hofer, A.; Wohlwend, D.; Jessen, H.J.; Gerhardt, S.; et al. Substrate recognition and mechanism revealed by ligand-bound polyphosphate kinase 2 structures. Proc. Natl. Acad. Sci. USA 2018, 115, 3350–3355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeiffer, M.; Crean, R.M.; Moreira, C.; Parracino, A.; Oberdorfer, G.; Brecker, L.; Hammerschmidt, F.; Lynn Kamerlin, S.C.; Nidetzky, B. Essential Functional Interplay of the Catalytic Groups in Acid Phosphatase. ACS Catal. 2022, 12, 3357–3370. [Google Scholar] [CrossRef]
- Štefanić, Z.; Narczyk, M.; Mikleušević, G.; Kazazić, S.; Bzowska, A.; Luić, M. Crystallographic snapshots of ligand binding to hexameric purine nucleoside phosphorylase and kinetic studies give insight into the mechanism of catalysis. Sci. Rep. 2018, 8, 15427. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Bhattasali, D.; Pellegrini, E.; Forget, S.M.; Baxter, N.J.; Cliff, M.J.; Bowler, M.W.; Jakeman, D.L.; Blackburn, G.M.; Waltho, J.P. α-Fluorophosphonates reveal how a phosphomutase conserves transition state conformation over hexose recognition in its two-step reaction. Proc. Natl. Acad. Sci. USA 2014, 111, 12384–12389. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Figueroa, J.S.; Palmer, D.R.; Horsman, G.P. Phosphoenolpyruvate mutase-catalyzed C-P bond formation: Mechanistic ambiguities and opportunities. ChemBioChem 2022, 23, e202200285. [Google Scholar] [CrossRef]
- Kubota, K.; Anjum, R.; Yu, Y.; Kunz, R.C.; Andersen, J.N.; Kraus, M.; Keilhack, H.; Nagashima, K.; Krauss, S.; Paweletz, C.; et al. Sensitive multiplexed analysis of kinase activities and activity-based kinase identification. Nat. Biotechnol. 2009, 27, 933–940. [Google Scholar] [CrossRef] [Green Version]
- Sévin, D.C.; Fuhrer, T.; Zamboni, N.; Sauer, U. Nontargeted in vitro metabolomics for high-throughput identification of novel enzymes in Escherichia coli. Nat. Methods 2017, 14, 187–194. [Google Scholar] [CrossRef]
- Schastnaya, E.; Raguz Nakic, Z.; Gruber, C.H.; Doubleday, P.F.; Krishnan, A.; Johns, N.I.; Park, J.; Wang, H.H.; Sauer, U. Extensive regulation of enzyme activity by phosphorylation in Escherichia coli. Nat. Commun. 2021, 12, 5650. [Google Scholar] [CrossRef] [PubMed]
- Cravatt, B.F.; Wright, A.T.; Kozarich, J.W. Activity-based protein profiling: From enzyme chemistry to proteomic chemistry. Annu. Rev. Biochem. 2008, 77, 383–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zallot, R.; Oberg, N.; Gerlt, J.A. Discovery of new enzymatic functions and metabolic pathways using genomic enzymology web tools. Curr. Opin. Biotechnol. 2021, 69, 77–90. [Google Scholar] [CrossRef]
- Zhang, X.; Carter, M.S.; Vetting, M.W.; San Francisco, B.; Zhao, S.; Al-Obaidi, N.F.; Solbiati, J.O.; Thiaville, J.J.; de Crécy-Lagard, V.; Jacobson, M.P.; et al. Assignment of function to a domain of unknown function: DUF1537 is a new kinase family in catabolic pathways for acid sugars. Proc. Natl. Acad. Sci. USA 2016, 113, E4161–E4169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, F.H. Directed evolution: Bringing new chemistry to life. Angew. Chem. Int. Ed. 2018, 57, 4143–4148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina, R.S.; Rix, G.; Mengiste, A.A.; Álvarez, B.; Seo, D.; Chen, H.; Hurtado, J.E.; Zhang, Q.; García-García, J.D.; Heins, Z.J.; et al. In vivo hypermutation and continuous evolution. Nat. Rev. Methods Prim. 2022, 2, 1–22. [Google Scholar] [CrossRef]
- Christians, F.C.; Scapozza, L.; Crameri, A.; Folkers, G.; Stemmer, W.P. Directed evolution of thymidine kinase for AZT phosphorylation using DNA family shuffling. Nat. Biotechnol. 1999, 17, 259–264. [Google Scholar] [CrossRef]
- Miller, D.C.; Athavale, S.V.; Arnold, F.H. Combining chemistry and protein engineering for new-to-nature biocatalysis. Nat. Synth. 2022, 1, 18–23. [Google Scholar] [CrossRef]
- Fryszkowska, A.; Devine, P.N. Biocatalysis in drug discovery and development. Curr. Opin. Chem. Biol. 2020, 55, 151–160. [Google Scholar] [CrossRef]
- Umezawa, H.; Okanishi, M.; Kondo, S.; Hamana, K.; Utahara, R.; Maeda, K.; Mitsuhashi, S. Phosphorylative inactivation of aminoglycosidic antibiotics by Escherichia coli carrying R factor. Science 1967, 157, 1559–1561. [Google Scholar] [CrossRef]
- Crofts, T.S.; Gasparrini, A.J.; Dantas, G. Next-generation approaches to understand and combat the antibiotic resistome. Nat. Rev. Microbiol. 2017, 15, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Surette, M.D.; Spanogiannopoulos, P.; Wright, G.D. The enzymes of the rifamycin antibiotic resistome. Acc. Chem. Res. 2021, 54, 2065–2075. [Google Scholar] [CrossRef] [PubMed]
- Terekhov, S.S.; Mokrushina, Y.A.; Nazarov, A.S.; Zlobin, A.; Zalevsky, A.; Bourenkov, G.; Golovin, A.; Belogurov Jr, A.; Osterman, I.A.; Kulikova, A.A.; et al. A kinase bioscavenger provides antibiotic resistance by extremely tight substrate binding. Sci. Adv. 2020, 6, eaaz9861. [Google Scholar] [CrossRef] [PubMed]
- Stogios, P.J.; Cox, G.; Spanogiannopoulos, P.; Pillon, M.C.; Waglechner, N.; Skarina, T.; Koteva, K.; Guarné, A.; Savchenko, A.; Wright, G.D. Rifampin phosphotransferase is an unusual antibiotic resistance kinase. Nat. Commun. 2016, 7, 11343. [Google Scholar] [CrossRef] [Green Version]
- Cai, W.; Goswami, A.; Yang, Z.; Liu, X.; Green, K.D.; Barnard-Britson, S.; Baba, S.; Funabashi, M.; Nonaka, K.; Sunkara, M.; et al. The Biosynthesis of Capuramycin-type Antibiotics. J. Biol. Chem. 2015, 290, 13710–13724. [Google Scholar] [CrossRef] [Green Version]
- Kita, A.; Kishimoto, A.; Shimosaka, T.; Tomita, H.; Yokooji, Y.; Imanaka, T.; Atomi, H.; Miki, K. Crystal structure of pantoate kinase from Thermococcus kodakarensis. Proteins 2020, 88, 718–724. [Google Scholar] [CrossRef]
- Hsu, C.; Tsai, H.Y.; Chang, C.F.; Yang, C.C.; Su, N.W. Discovery of a novel phosphotransferase from Bacillus subtilis that phos-phorylates a broad spectrum of flavonoids. Food Chem. 2022, 400, 134001. [Google Scholar] [CrossRef]
- Jacoby, C.; Goerke, M.; Bezold, D.; Jessen, H.; Boll, M. A fully reversible 25-hydroxy steroid kinase involved in oxygen-inde-pendent cholesterol side-chain oxidation. J. Biol. Chem. 2021, 297, 101105. [Google Scholar] [CrossRef]
- Hoffmeister, D.; Yang, J.; Liu, L.; Thorson, J.S. Creation of the first anomeric D/L-sugar kinase by means of directed evolution. Proc. Natl. Acad. Sci. USA 2003, 100, 13184–13189. [Google Scholar] [CrossRef]
- Nagata, R.; Fujihashi, M.; Sato, T.; Atomi, H.; Miki, K. Crystal Structure and Product Analysis of an Archaeal myo-Inositol Kinase Reveal Substrate Recognition Mode and 3-OH Phosphorylation. Biochemistry 2015, 54, 3494–3503. [Google Scholar] [CrossRef]
- Tashiro, R.; Sato, T.; Atomi, H.; Miki, K.; Fujihashi, M. Altering the phosphorylation position of pyrophosphate-dependent myo-inositol-1-kinase based on its crystal structure. ACS Chem. Biol. 2021, 16, 794–799. [Google Scholar] [CrossRef]
- Huang, H.; Parmeggiani, F.; Pallister, E.; Huang, C.-J.; Liu, F.-F.; Li, Q.; Birmingham, W.R.; Both, P.; Thomas, B.; Liu, L.; et al. Characterisation of a Bacterial Galactokinase with High Activity and Broad Substrate Tolerance for Chemoenzymatic Synthesis of 6-Aminogalactose-1-Phosphate and Analogues. ChemBioChem 2018, 19, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Keenan, T.; Parmeggiani, F.; Malassis, J.; Fontenelle, C.Q.; Vendeville, J.B.; Offen, W.; Both, P.; Huang, K.; Marchesi, A.; Heyam, A.; et al. Profiling substrate promiscuity of wild-type sugar kinases for multi-fluorinated monosaccharides. Cell Chem. Biol. 2020, 27, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Huddleston, J.P.; Raushel, F.M. Functional Characterization of YdjH, a Sugar Kinase of Unknown Specificity in Escherichia coli K12. Biochemistry 2019, 58, 3354–3364. [Google Scholar] [CrossRef] [PubMed]
- Taylor, Z.W.; Raushel, F.M. Cytidine Diphosphoramidate Kinase: An Enzyme Required for the Biosynthesis of the O-Methyl Phosphoramidate Modification in the Capsular Polysaccharides of Campylobacter jejuni. Biochemistry 2018, 57, 2238–2244. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, M.F.; Winiger, C.B.; Shaw, R.W.; Kim, M.-J.; Kim, M.-S.; Daugherty, A.B.; Chen, F.P.; Moses, J.D.; Lutz, S.; Benner, S.A. A Single Deoxynucleoside Kinase Variant from Drosophila melanogaster Synthesizes Monophosphates of Nucleosides That Are Components of an Expanded Genetic System. ACS Synth. Biol. 2017, 6, 388–394. [Google Scholar] [CrossRef]
- Makino, Y.; Sato, T.; Kawamura, H.; Hachisuka, S.I.; Takeno, R.; Imanaka, T.; Atomi, H. An archaeal ADP-dependent serine kinase involved in cysteine biosynthesis and serine metabolism. Nat. Commun. 2016, 7, 13446. [Google Scholar] [CrossRef] [Green Version]
- Mori, Y.; Kawamura, H.; Sato, T.; Fujita, T.; Nagata, R.; Fujihashi, M.; Miki, K.; Atomi, H. Identification and Enzymatic Analysis of an Archaeal ATP-Dependent Serine Kinase from the Hyperthermophilic Archaeon Staphylothermus marinus. J. Bact. 2021, 203, e00025-21. [Google Scholar] [CrossRef]
- Thiaville, J.J.; Flood, J.; Yurgel, S.; Prunetti, L.; Elbadawi-Sidhu, M.; Hutinet, G.; Forouhar, F.; Zhang, X.; Ganesan, V.; Reddy, P.; et al. Members of a Novel Kinase Family (DUF1537) Can Recycle Toxic Intermediates into an Essential Metabolite. ACS Chem. Biol. 2016, 11, 2304–2311. [Google Scholar] [CrossRef]
- Taylor, Z.W.; Brown, H.A.; Narindoshvili, T.; Wenzel, C.Q.; Szymanski, C.M.; Holden, H.M.; Raushel, F.M. Discovery of a glutamine kinase required for the biosynthesis of the O-methyl phosphoramidate modifications found in the capsular poly-saccharides of Campylobacter jejuni. J. Am. Chem. Soc. 2017, 139, 9463–9466. [Google Scholar] [CrossRef]
- Taylor, Z.W.; Chamberlain, A.R.; Raushel, F.M. Substrate specificity and chemical mechanism for the reaction catalyzed by glutamine kinase. Biochemistry 2018, 57, 5447–5455. [Google Scholar] [CrossRef] [PubMed]
- Médici, R.; Garaycoechea, J.I.; Valino, A.I.; Pereira, C.A.; Lewkowicz, E.S.; Iribarren, A.M. A comparative study on phosphotransferase activity of acid phosphatases from Raoultella planticola and Enterobacter aerogenes on nucleosides, sugars and related compounds. Appl. Microbiol. Biotechnol. 2014, 98, 3013–3022. [Google Scholar] [CrossRef] [PubMed]
- Mihara, Y.; Utagawa, T.; Yamada, H.; Asano, Y. Phosphorylation of Nucleosides by the Mutated Acid Phosphatase from Morganella morganii. Appl. Environ. Microbiol. 2000, 66, 2811–2816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tasnádi, G.; Zechner, M.; Hall, M.; Baldenius, K.; Ditrich, K.; Faber, K. Investigation of acid phosphatase variants for the synthesis of phosphate monoesters. Biotechnol. Bioeng. 2017, 114, 2187–2195. [Google Scholar] [CrossRef]
- Nagy, F.; Tasnádi, G.; Balogh-Weiser, D.; Bell, E.; Hall, M.; Faber, K.; Poppe, L. Smart nanoparticles for selective immobilization of acid phosphatases. ChemCatChem 2018, 10, 3490–3499. [Google Scholar] [CrossRef] [Green Version]
- Macdonald, S.S.; Armstrong, Z.; Morgan-Lang, C.; Osowiecka, M.; Robinson, K.; Hallam, S.J.; Withers, S.G. Development and Application of a High-Throughput Functional Metagenomic Screen for Glycoside Phosphorylases. Cell Chem. Biol. 2019, 26, 1001–1012. [Google Scholar] [CrossRef]
- Franceus, J.; Pinel, D.; Desmet, T. Glucosylglycerate phosphorylase, an enzyme with novel specificity involved in compatible solute metabolism. Appl. Env. Microbiol. 2017, 83, e01434-17. [Google Scholar] [CrossRef] [Green Version]
- De Doncker, M.; De Graeve, C.; Franceus, J.; Beerens, K.; Křen, V.; Pelantová, H.; Vercauteren, R.; Desmet, T. Exploration of GH94 sequence space for enzyme discovery reveals a novel Glucosylgalactose phosphorylase specificity. ChemBioChem 2021, 22, 3319–3325. [Google Scholar] [CrossRef]
- Teze, D.; Coines, J.; Raich, L.; Kalichuk, V.; Solleux, C.; Tellier, C.; Andre-Miral, C.; Svensson, B.; Rovira, C. A single point mutation converts GH84 O-GlcNac hydrolases into phosphorylases: Experimental and theoretic evidence. J. Am. Chem. Soc. 2020, 142, 2120–2124. [Google Scholar] [CrossRef]
- Novick, S.J.; Dellas, N.; Mitchell, V.; Duan, D.; Nazor, J.; Alvizo, O.; Sowell-Kantz, A.A.; Moore, J.C.; Huffman, M.; Rodriguez-Granillo, A.; et al. Engineered Purine Nucleoside Phosphorylase Variant Enzymes. US 2022/0010316 A1, 2 November 2021. [Google Scholar]
- Kamel, S.; Thiele, I.; Neubauer, P.; Wagner, A. Thermophilic nucleoside phosphorylases: Their properties, characteristics and applications. BBA Proteins Proteom. 2020, 1868, 140304. [Google Scholar] [CrossRef]
- Vroom, J.; Sivaramakrishnan, S.; Hurtak, J.A. Engineered Phosphopentomutase Variant Enzymes. WO 2022/076454 A1, 15 June 2021. [Google Scholar]
- Dadashipour, M.; Iwamoto, M.; Hossain, M.M.; Akutsu, J.-I.; Zhang, Z.; Kawarabayasi, Y. Identification of a Direct Biosyn-thetic Pathway for UDP-N-Acetylgalactosamine from Glucosamine-6-phosphate in Thermophilic Crenarchaeon Sulfolobus tokodaii. J. Bacteriol. 2018, 200, e00048-18. [Google Scholar] [CrossRef] [PubMed]
- Bergmeyer, H.-U. Methods of Enzymatic Analysis; Academic Press: New York, NY, USA, 1965; pp. 407–410. [Google Scholar]
- Wieland, O. An enzymic method for estimating glycerol. Biochem. Ztschr. 1957, 329, 313–319. [Google Scholar]
- Hørder, M.; Elser, R.C.; Gerhardt, W.; Mathieu, M.; Sampson, E.J. International Federation of Clinical Chemistry (IFCC): Scientific Division, Committee on Enzymes. IFCC methods for the measurement of catalytic concentration enzymes. Part 7. IFCC method for creatine kinase (creatine N-phosphotransferase, EC 2.7.3.2). IFCC Recommendation. J. Autom. Chem. 1990, 12, 22–40. [Google Scholar] [CrossRef]
- Galburt, E.A.; Pelletier, J.; Wilson, G.; Stoddard, B.L. Structure of a tRNA repair enzyme and molecular biology workhorse: T4 polynucleotide kinase. Structure 2002, 10, 1249–1260. [Google Scholar] [CrossRef] [Green Version]
- Green, M.R.; Sambrook, J. Preparation of Labeled DNA, RNA, and Oligonucleotide Probes. Cold Spring Harb. Protoc. 2022, 1, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Anthony, T.M.; Pflum, M.K.H. Kinase-catalyzed biotinylation of DNA. Bioorg. Med. Chem. 2018, 26, 2331–2336. [Google Scholar] [CrossRef]
- Schumann, G.; Klauke, R.; Canalias, F.; Bossert-Reuther, S.; Franck, P.F.H.; Gella, F.-J.; Jørgensen, P.J.; Kang, D.; Lessinger, J.-M.; Panteghini, M.; et al. IFCC primary reference procedures for the measurement of catalytic activity concentrations of enzymes at 37 °C. Part 9: Reference procedure for the measurement of catalytic concentration of alkaline phosphatase. Clin. Chem. Lab. Med. 2011, 49, 1439–1446. [Google Scholar] [CrossRef] [Green Version]
- Shaban, S.M.; Jo, S.B.; Hafez, E.; Cho, J.H.; Kim, D.-H. A comprehensive overview on alkaline phosphatase targeting and reporting assays. Coord. Chem. Rev. 2022, 465, 214567. [Google Scholar] [CrossRef]
- Kitaoka, M.; Aoyagi, C.; Hayashi, K. Colorimetric Quantification of Cellobiose Employing Cellobiose Phosphorylase. Anal. Biochem. 2001, 292, 163–166. [Google Scholar] [CrossRef]
- Zhang, Z.; Jaffrezic-Renault, N.; Bessueille, F.; Leonard, D.; Xia, S.; Wang, X.; Chen, L.; Zhao, J. Development of a conductometric phosphate biosensor based on tri-layer maltose phosphorylase composite films. Ana. Chim. Acta 2008, 615, 73–79. [Google Scholar] [CrossRef]
- Apple, F.S.; Wu, A.H.B.; Mair, J.; Ravkilde, J.; Panteghini, M.; Tate, J.; Pagani, F.; Christenson, R.H.; Mockel, M.; Danne, O.; et al. Future Biomarker for Detection of Ischemia and Risk Stratification in Acute Coronary Syndrome. Clin. Chem 2005, 51, 810–824. [Google Scholar] [CrossRef] [PubMed]
- Hallows, W.C.; Yu, W.; Denu, J.M. Regulation of Glycolytic Enzyme Phosphoglycerate Mutase-1 by Sirt1 Protein-mediated Deacetylation. J. Biol. Chem. 2012, 287, 3850–3856. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Li, S.; Wang, Y.; Peng, H.; Zhang, X.; Zheng, Y.; Li, C.; Li, L.; Chen, R.; Chen, X.; et al. Phosphoglyceric acid mutase-1 contributes to oncogenic mTOR-mediated tumor growth and confers non-small cell lung cancer patients with poor prognosis. Cell Death Diff. 2018, 25, 1160–1173. [Google Scholar] [CrossRef] [Green Version]
- Wohlgemuth, R.; Littlechild, J. Complexity Reduction and Opportunities in the Design, Integration and Intensification of Biocatalytic Processes for Metabolite Synthesis. Front. Bioeng. Biotechnol. Bioprocess. Eng. 2022, 10, 958606. [Google Scholar] [CrossRef] [PubMed]
- Asano, Y.; Mihara, Y.; Yamada, H. A new enzymatic method of selective phosphorylation of nucleosides. J. Mol. Catal. B Enzym. 1999, 6, 271–277. [Google Scholar] [CrossRef]
- Suzuki, E.; Ishikawa, K.; Mihara, Y.; Shimba, N.; Asano, Y. Structural-based engineering for transferases to improve the industrial production of 5′-nucleotides. Bull. Chem. Soc. Jpn. 2007, 80, 276–286. [Google Scholar] [CrossRef] [Green Version]
- Van Herk, T.; Hartog, A.F.; van der Burg, A.M.; Wever, R. Regioselective phosphorylation of carbohydrates and various alcohols by bacterial acid phosphatases; Probing the substrate specificity of the enzyme from Shigella flexneri. Adv. Synth. Catal. 2005, 347, 1155–1162. [Google Scholar] [CrossRef]
- Babich, L.; Hartog, A.F.; van der Horst, M.A.; Wever, R. Continuous-flow reactor-based enzymatic synthesis of phosphorylated compounds on a large scale. Chem. Eur. J. 2012, 18, 6604–6609. [Google Scholar] [CrossRef]
- Van Herk, T.; Hartog, A.F.; Schoemaker, H.E.; Wever, R.; Faber, K. Simple Enzymatic in situ Generation of Dihydroxyacetone phosphate and its Use in a Cascade Reaction for the Production of Carbohydrates: Increased Efficiency by Phosphate Cycling. J. Org. Chem. 2006, 71, 6244–6247. [Google Scholar] [CrossRef]
- Tasnádi, G.; Hall, M.; Baldenius, K.; Ditrich, K.; Faber, K. Biocatalytic functionalization of hydroxyalkyl acrylates and phenoxy-ethanol via phosphorylation. J. Biotechnol. 2016, 233, 219–227. [Google Scholar] [CrossRef]
- Tasnádi, G.; Jud, W.; Hall, M.; Baldenius, K.; Ditrich, K.; Faber, K. Evaluation of Natural and Synthetic Phosphate Donors for the Improved Enzymatic Synthesis of Phosphate Monoesters. Adv. Synth. Catal. 2018, 360, 2394–2401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tasnádi, G.; Staśko, M.; Ditrich, K.; Hall, M.; Faber, K. Preparative-Scale Enzymatic Synthesis of rac-Glycerol-1-phosphate from Crude Glycerol Using Acid Phosphatases and Phosphate. ChemSusChem 2020, 13, 1759–1763. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Zheng, K.; Xu, X.; Gao, C.; Guo, L.; Liu, J.; Chen, X.; Liu, L.; Hu, G.; Wu, J. Enzymatic Production of Ascorbic Acid-2-Phosphate by Engineered Pseudomonas aeruginosa Acid Phosphatase. J. Agric. Food Chem. 2021, 69, 14215–14221. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.-L.; Dai, Y.-S.; Li, C.X.; Pan, J.; Xu, J.-H.; Mu, B. Enzymatic synthesis of high-titer nicotinamide mononucleotide with a new nicotinamide riboside kinase and an efficient ATP regeneration system. Bioresour. Bioprocess. 2022, 9, 26. [Google Scholar] [CrossRef]
- Gauss, D.; Schoenenberger, B.; Wohlgemuth, R. Chemical and enzymatic methodologies for the synthesis of enantiomerically pure glyceraldehyde 3-phosphates. Carbohydr. Res. 2014, 389, 18–24. [Google Scholar] [CrossRef]
- Wong, C.H.; Whitesides, G.M. Synthesis of Sugars by Aldolase-Catalyzed Condensation Reactions. J. Org. Chem. 1983, 48, 3199–3205. [Google Scholar] [CrossRef]
- Gauss, D.; Sánchez-Moreno, I.; Oroz-Guinea, I.; García-Junceda, E.; Wohlgemuth, R. Phosphorylation catalyzed by dihydro- xyacetone kinase. Eur. J. Org. Chem. 2018, 2018, 2892–2895. [Google Scholar] [CrossRef]
- Matsumi, R.; Hellriegel, C.; Schoenenberger, B.; Milesi, T.; Van Der Oost, J.; Wohlgemuth, R. Biocatalytic asymmetric phosphorylation of mevalonate. RSC Adv. 2014, 4, 12989–12994. [Google Scholar] [CrossRef]
- Hardt, N.; Kinfu, B.M.; Chow, J.; Schoenenberger, B.; Streit, W.R.; Obkircher, M.; Wohlgemuth, R. Biocatalytic Asymmetric Phosphorylation Catalyzed by Recombinant Glycerate-2-Kinase. ChemBioChem 2017, 18, 1518–1522. [Google Scholar] [CrossRef]
- Schoenenberger, B.; Wszolek, A.; Meier, R.; Brundiek, H.; Obkircher, M.; Wohlgemuth, R. Recombinant AroL-Catalyzed Phosphorylation for the Efficient Synthesis of Shikimic Acid 3-Phosphate. Biotechnol. J. 2018, 13, 1700529. [Google Scholar] [CrossRef]
- Hardt, N.; Kind, S.; Schoenenberger, B.; Eggert, T.; Obkircher, M.; Wohlgemuth, R. Facile synthesis of D-xylulose-5-phosphate and L-xylulose-5-phosphate by xylulokinase-catalyzed phosphorylation. Biocat. Biotrans. 2020, 38, 35–45. [Google Scholar] [CrossRef]
- Schoenenberger, B.; Kind, S.; Meier, R.; Eggert, T.; Obkircher, M.; Wohlgemuth, R. Efficient biocatalytic synthesis of D-tagatose 1,6-diphosphate by LacC-catalysed phosphorylation of D-tagatose 6-phosphate. Biocat. Biotrans. 2020, 38, 53–63. [Google Scholar] [CrossRef]
- Wen, L.; Huang, K.; Liu, Y.; Wang, P.G. Facile enzymatic synthesis of phosphorylated ketopentoses. ACS Catal. 2016, 6, 1649–1654. [Google Scholar] [CrossRef]
- Wen, L.; Huang, K.; Wei, M.; Meisner, J.; Liu, Y.; Garner, K.; Zang, L.; Wang, X.; Li, X.; Fang, J.; et al. Facile Enzymatic Synthesis of Ketoses. Angew. Chem., Int. Ed. 2015, 54, 12654–12658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fricke, J.; Kargbo, R.; Regestein, L.; Lenz, C.; Peschel, G.; Rosenbaum, M.A.; Sherwood, A.; Hoffmeister, D. Scalable Hybrid Synthetic/Biocatalytic Route to Psilocybin. Chem. Eur. J. 2020, 26, 8281–8285. [Google Scholar] [CrossRef] [PubMed]
- Schoenenberger, B.; Wszolek, A.; Milesi, T.; Brundiek, H.; Obkircher, M.; Wohlgemuth, R. Synthesis of Nω-Phospho-L-arginine by Biocatalytic Phosphorylation of L-Arginine. ChemCatChem 2017, 9, 121–126. [Google Scholar] [CrossRef]
- De Winter, K.; Cerdobbel, A.; Soetaert, W.; Desmet, T. Operational stability of immobilized sucrose phosphorylase: Continuous production of α-glucose-1-phosphate at elevated temperatures. Proc. Biochem. 2011, 46, 2074–2078. [Google Scholar] [CrossRef]
- Bae, J.; Lee, D.; Kim, D.; Cho, S.-J.; Park, J.E.; Koh, S.; Kim, J.; Park, B.-H.; Choi, Y.; Shin, H.-J.; et al. Facile synthesis of glucose-1-phosphate from starch by Thermus thermophilus GK24 α-glucan phosphorylase. Proc. Biochem. 2005, 40, 3707–3713. [Google Scholar] [CrossRef]
- Kamel, S.; Weiss, M.; Klare, H.F.T.; Mikhailopoulo, I.A.; Neubauer, P.; Wagner, A. Chemo-enzymatic synthesis of α-D-pentofuranose-1-phosphates using thermostable pyrimidine nucleoside phosphorylases. Mol. Catal. 2018, 458, 52–59. [Google Scholar] [CrossRef]
- Heptinstall, J.; Ward, P.J.; Hancock, I.C. The enzymic synthesis of [32P]-N-acetylglucosamine. Anal. Biochem. 1978, 91, 158–165. [Google Scholar] [CrossRef]
- Hassan, M.I.; Lundgren, B.R.; Chaumun, M.; Whitfield, D.M.; Clark, B.; Schoenhofen, I.C.; Boddy, C.N. Total Biosynthesis of Legionaminic Acid, a Bacterial Sialic Acid Analogue. Angew. Chem. Intl. Ed. 2016, 55, 12018–12021. [Google Scholar] [CrossRef] [PubMed]
- Horinouchi, N.; Kawano, T.; Sakai, T.; Matsumoto, S.; Sasaki, M.; Mikami, Y.; Ogawa, J.; Shimizu, S. Screening and characterization of a phosphopentomutase useful for enzymatic production of 2’-deoxynucleoside. New Biotechnol. 2009, 26, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-H.; Kang, Y.-B.; Kim, D.-H.; Lee, T.-H.; Park, S.-H.; Lee, K.; Yoo, D.; Liou, K.-K.; Lee, H.-C.; Sohng, J.-K.; et al. One-pot enzymatic synthesis of deoxy-thymidine-diphosphate (TDP)-2-deoxy-α-D-glucose using phosphomannomutase. J. Mol. Catal. B Enzym. 2010, 62, 282–287. [Google Scholar] [CrossRef] [Green Version]
- Mahour, R.; Klapproth, J.; Rexer, T.F.T.; Schildbach, A.; Klamt, S.; Pietzsch, M.; Rapp, E.; Reichl, U. Establishment of a five-enzyme cell-free cascade for the synthesis of uridine diphosphate N-acetylglucosamine. J. Biotechnol. 2018, 283, 120–129. [Google Scholar] [CrossRef] [Green Version]
- Robescu, M.S.; Serra, I.; Terreni, M.; Ubiali, D.; Bavaro, T. A Multi-Enzymatic Cascade Reaction for the Synthesis of Vidarabine-5′-Monophosphate. Catalysts 2020, 10, 60. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Moreno, I.; Hélaine, V.; Poupard, N.; Charmantray, F.; Légeret, B.; Hecquet, L.; García-Junceda, E.; Wohlgemuth, R.; Guérard-Hélaine, C.; Lemaire, M. One-pot cascade reactions using fructose-6-phosphate aldolase: Efficient synthesis of D-arabinose 5-phosphate, D-fructose 6-phosphate and analogues. Adv. Synth. Catal. 2012, 354, 1725–1730. [Google Scholar] [CrossRef] [Green Version]
- Hélaine, V.; Mahdi, R.; Sudhir Babu, G.V.; de Berardinis, V.; Wohlgemuth, R.; Lemaire, M.; Guérard-Hélaine, C. Straightforward synthesis of terminally phosphorylated L-sugars via multienzymatic cascade reactions. Adv. Synth. Catal. 2015, 357, 1703–1708. [Google Scholar] [CrossRef]
- Huffman, M.A.; Fryszkowska, A.; Alvizo, O.; Borra-Garske, M.; Campos, K.R.; Canada, K.A.; Devine, P.N.; Duan, D.; Forstater, J.H.; Grosser, S.T.; et al. Design of an in vitro biocatalytic cascade for the manufacture of islatravir. Science 2019, 366, 1255–1259. [Google Scholar] [CrossRef]
- Schultheisz, H.L.; Szymczyna, B.R.; Scott, L.G.; Williamson, J.R. Pathway Engineered Enzymatic de Novo Purine Nucleotide Synthesis. ACS Chem. Biol. 2008, 3, 499–511. [Google Scholar] [CrossRef] [Green Version]
- Ren, S.; Huffman, M.A.; Whittaker, A.M.; Yang, H.; Nawrat, C.C.; Waterhouse, D.J.; Maloney, K.M.; Strotman, N.A. Synthesis of Isotopically Labeled Anti-HIV Nucleoside Islatravir though a One-Pot Biocatalytic Cascade Reaction. Org. Process Res. Dev. 2021, 25, 516–521. [Google Scholar] [CrossRef]
- McIntosh, J.A.; Benkovics, T.; Silverman, S.M.; Huffman, M.A.; Kong, J.; Maligres, P.E.; Itoh, T.; Yang, H.; Verma, D.; Pan, W.; et al. Engineered ribosyl-1-kinase enables concise synthesis of molnupiravir, an antiviral for COVID-19. ACS Centr. Sci. 2021, 7, 1980–1985. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, J.A.; Liu, Z.; Andresen, B.M.; Marzijarani, N.S.; Moore, J.C.; Marshall, N.M.; Borra-Garske, M.; Obligacion, J.V.; Fier, P.S.; Peng, F.; et al. A kinase-cGAS cascade to synthesize a therapeutic STING activator. Nature 2022, 603, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Murata, K.; Uchida, T.; Kato, J.; Chibata, I. Polyphosphate kinase: Distribution, Some Properties and Its Application as an ATP Regeneration System. Agric. Biol. Chem. 1988, 52, 1471–1477. [Google Scholar]
- Murata, K. Polyphosphate-dependent nicotinamide adenine dinucleotide (NAD) kinase: A novel missing link in human mito-chondria. Proc. Jpn. Acad., Ser. B 2021, 97, 479–498. [Google Scholar] [CrossRef] [PubMed]
- Murata, K.; Uchida, T.; Kato, J.; Chibata, I. A Metaphosphate-dependent Nicotinamide Adenine Dinucleotide Kinase from Brevibacterium ammoniagenes. Agric. Biol. Chem. 1980, 44, 1165–1172. [Google Scholar] [CrossRef]
- Andexer, J.N.; Richter, M. Emerging Enzymes for ATP Regeneration in Biocatalytic Processes. ChemBioChem 2015, 16, 380–386. [Google Scholar] [CrossRef]
- Mordhorst, S.; Andexer, J.N. Round, round we go – strategies for enzymatic cofactor regeneration. Nat. Prod. Rep. 2020, 37, 1316–1333. [Google Scholar] [CrossRef]
- Hirschbein, B.L.; Mazenod, F.P.; Whitesides, G.M. Synthesis of Phosphoenolpyruvate and Its Use in Adenosine Triphosphate Cofactor Regeneration. J. Org. Chem. 1982, 47, 3765–3766. [Google Scholar] [CrossRef]
- Crans, D.C.; Whitesides, G.M. A Convenient Synthesis of Disodium Acetyl Phosphate for Use in in Situ ATP Cofactor Regeneration. J. Org. Chem. 1983, 48, 3130–3132. [Google Scholar] [CrossRef]
- Kazlauskas, R.J.; Whitesides, G.M. Synthesis of Methoxycarbonyl Phosphate, a New Reagent Having High Phosphoryl Donor Potential for Use in ATP Cofactor Regeneration. J. Org. Chem. 1985, 50, 1069–1076. [Google Scholar] [CrossRef]
- Tavanti, M.; Hosford, J.; Lloyd, R.C.; Brown, M.J.B. Recent Developments and Challenges for the Industrial Implementation of Polyphosphate Kinases. ChemCatChem 2021, 13, 3565–3580. [Google Scholar] [CrossRef]
- Kim, D.-M.; Swartz, J.R. Prolonging cell-free protein synthesis with a novel ATP regeneration system. Biotech. Bioeng. 1999, 66, 180–188. [Google Scholar] [CrossRef]
- Ruccolo, S.; Brito, G.; Christensen, M.; Itoh, T.; Mattern, K.; Stone, K.; Strotman, N.A.; Sun, A.C. Electrochemical Recycling of Adenosine Triphosphate in Biocatalytic Reaction Cascades. Chemrxiv 2022. [Google Scholar] [CrossRef]
- Molla, G.S.; Kinfu, B.M.; Chow, J.; Streit, W.; Wohlgemuth, R.; Liese, A. Bioreaction Engineering Leading to Efficient Synthesis of L-Glyceraldehyd-3-phosphate. Biotechnol. J. 2017, 12, 1600625. [Google Scholar] [CrossRef] [PubMed]
- Molla, G.S.; Himmelspach, A.; Wohlgemuth, R.; Haupt, E.T.K.; Liese, A. Mechanistic and kinetic elucidation of Mg2+/ATP molar ratio effect on glycerol kinase. Mol. Catal. 2018, 445, 36–42. [Google Scholar] [CrossRef]
- Wohlgemuth, R. Product Recovery. Compr. Biotechnol. 2011, 2, 591–601. [Google Scholar] [CrossRef]
- Knouse, K.W.; Flood, D.T.; Vantourout, J.C.; Schmidt, M.A.; Mcdonald, I.M.; Eastgate, M.D.; Baran, P.S. Nature chose phosphates and chemists should too: How emerging P (V) methods can augment existing strategies. ACS Cent. Sci. 2021, 7, 1473–1485. [Google Scholar] [CrossRef]
- Alcántara, A.R.; Domínguez de María, P.; Littlechild, J.A.; Schürmann, M.; Sheldon, R.A.; Wohlgemuth, R. Biocatalysis as Key to Sustainable Industrial Chemistry. ChemSusChem 2022, 15, e202102709. [Google Scholar] [CrossRef]
Figure 1.
Schematic representation of key classes of phosphorylation biocatalysts and the involved reactions.
Figure 1.
Schematic representation of key classes of phosphorylation biocatalysts and the involved reactions.

Figure 2.
Overview of major functions of phosphoryl-group transferring biocatalysts.

Figure 3.
Selected phosphotransferases for catalyzing phosphorylation reactions.

Figure 4.
Selected phosphohydrolases for catalyzing phosphorylation reactions.

Figure 5.
Selected phosphorylases for phosphorylation reactions.

Figure 6.
Selected phosphomutases for intramolecular phosphorylation reactions.

Figure 7.
Novel phosphotransferases for phosphorylation reactions.

Figure 8.
Novel phosphohydrolases for phosphorylation reactions.

Figure 9.
Novel phosphorylases for phosphorylation reactions.

Figure 10.
Novel phosphomutases for intramolecular phosphorylation reactions.

Figure 11.
Major analytical application areas for phosphorylation biocatalysts.

Figure 12.
Compound classes of starting materials for preparative phosphorylations using phosphorylation biocatalysts.
Figure 12.
Compound classes of starting materials for preparative phosphorylations using phosphorylation biocatalysts.

Figure 13.
Selected synthetic applications of phosphohydrolases in phosphorylation reactions.

Figure 14.
Synthetic applications of phosphotransferases in phosphorylation reactions.

Figure 15.
Synthetic applications of phosphorylases in phosphorylation reactions.

Figure 16.
Synthetic applications of phosphomutases in phosphorylation reactions.

Figure 17.
Synthetic reaction cascades involving phosphotransferases, phosphorylases and phosphomutases.
Figure 17.
Synthetic reaction cascades involving phosphotransferases, phosphorylases and phosphomutases.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wohlgemuth, R. The Power of Biocatalysts for Highly Selective and Efficient Phosphorylation Reactions. Catalysts 2022, 12, 1436. https://doi.org/10.3390/catal12111436
AMA Style
Wohlgemuth R. The Power of Biocatalysts for Highly Selective and Efficient Phosphorylation Reactions. Catalysts. 2022; 12(11):1436. https://doi.org/10.3390/catal12111436
Chicago/Turabian StyleWohlgemuth, Roland. 2022. "The Power of Biocatalysts for Highly Selective and Efficient Phosphorylation Reactions" Catalysts 12, no. 11: 1436. https://doi.org/10.3390/catal12111436
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.