
Computational Insight into Defective Boron Nitride Supported Double-Atom Catalysts for Electrochemical Nitrogen Reduction
Abstract
:
1. Introduction
2. Results and Discussion
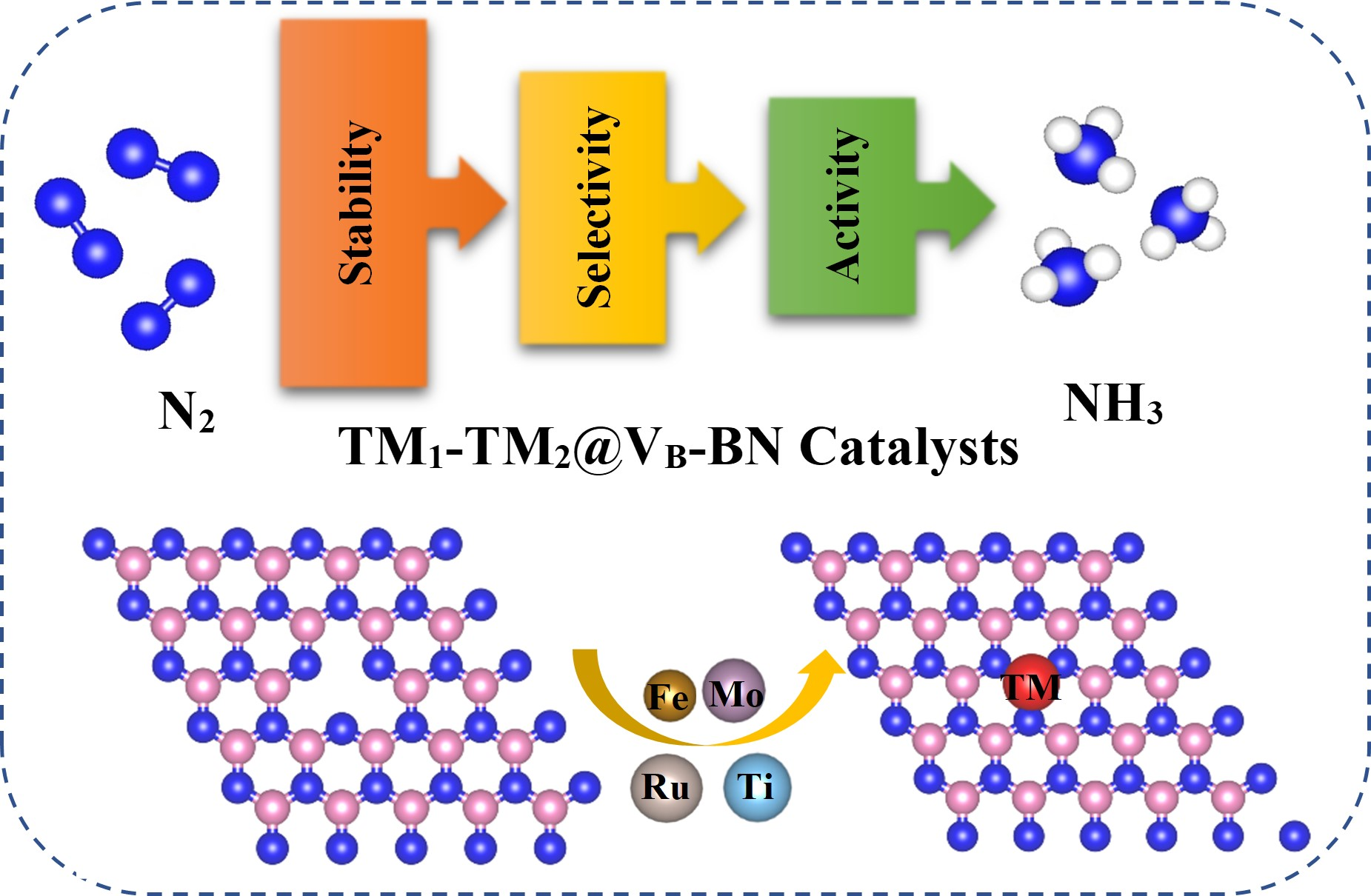
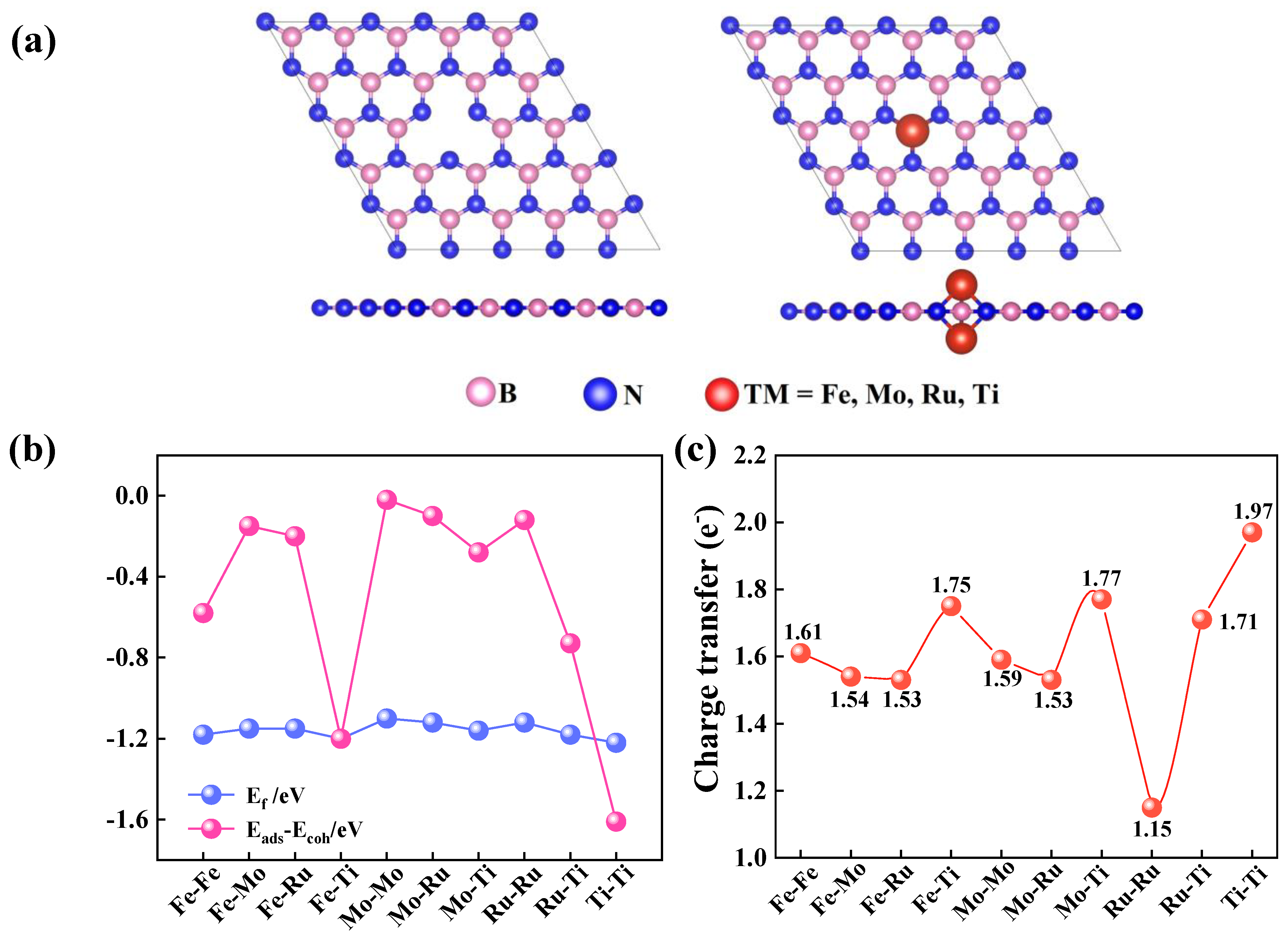
2.1. Structural and Electronic Properties of TM1-TM2@VB-BN
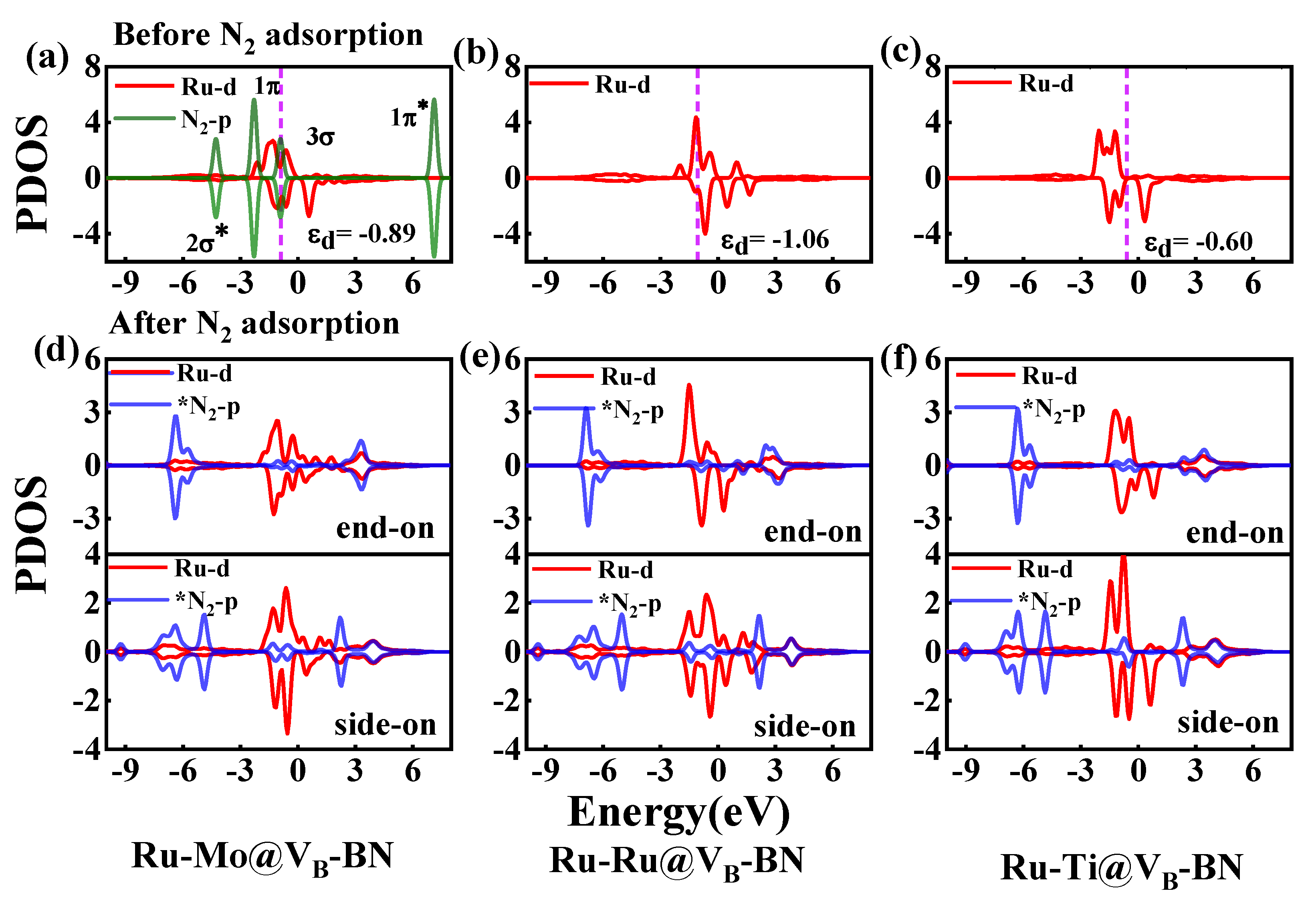
2.2. N2 Adsorption and Selectivity on TM1-TM2@VB-BN
2.3. Catalytic Activity of TM1-TM2@VB-BN
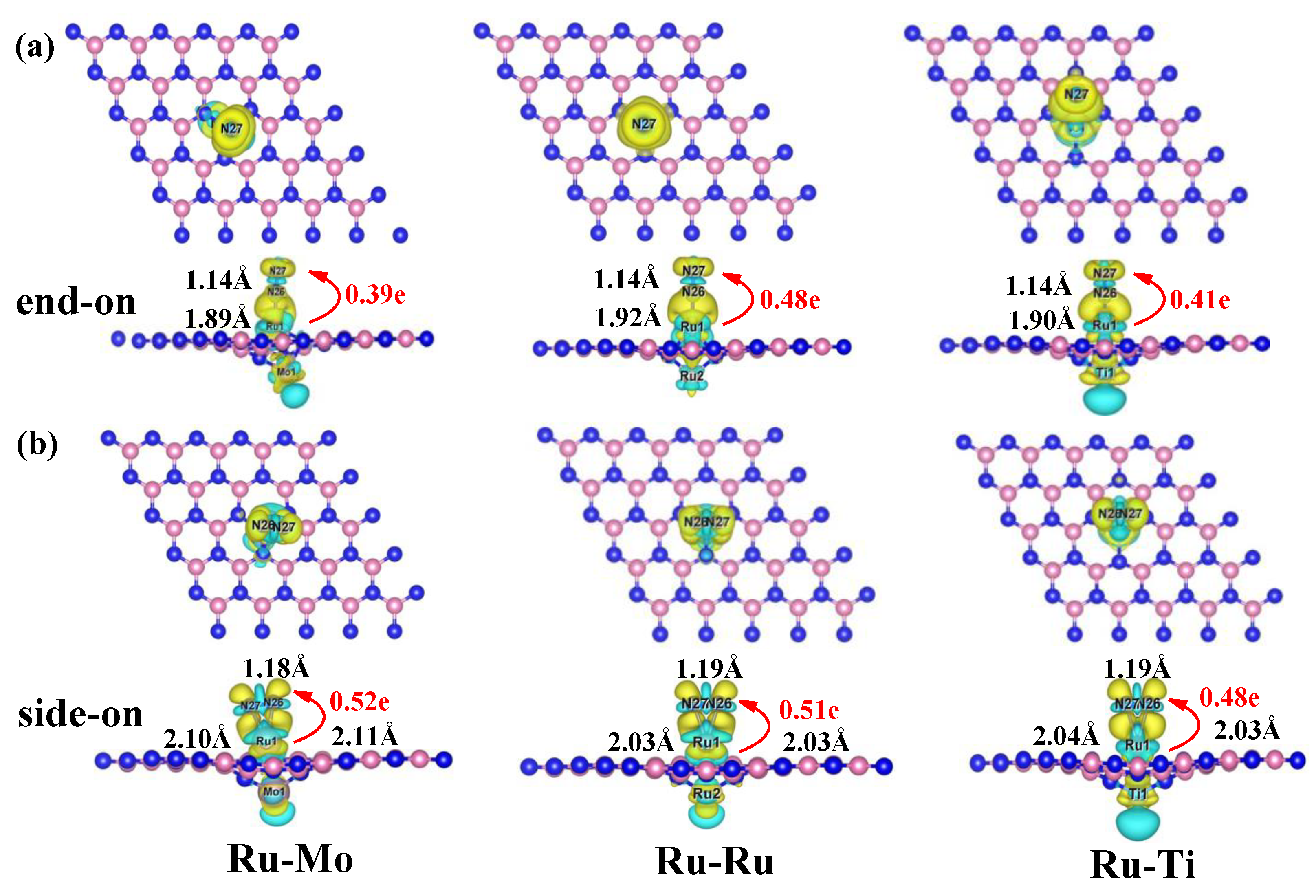
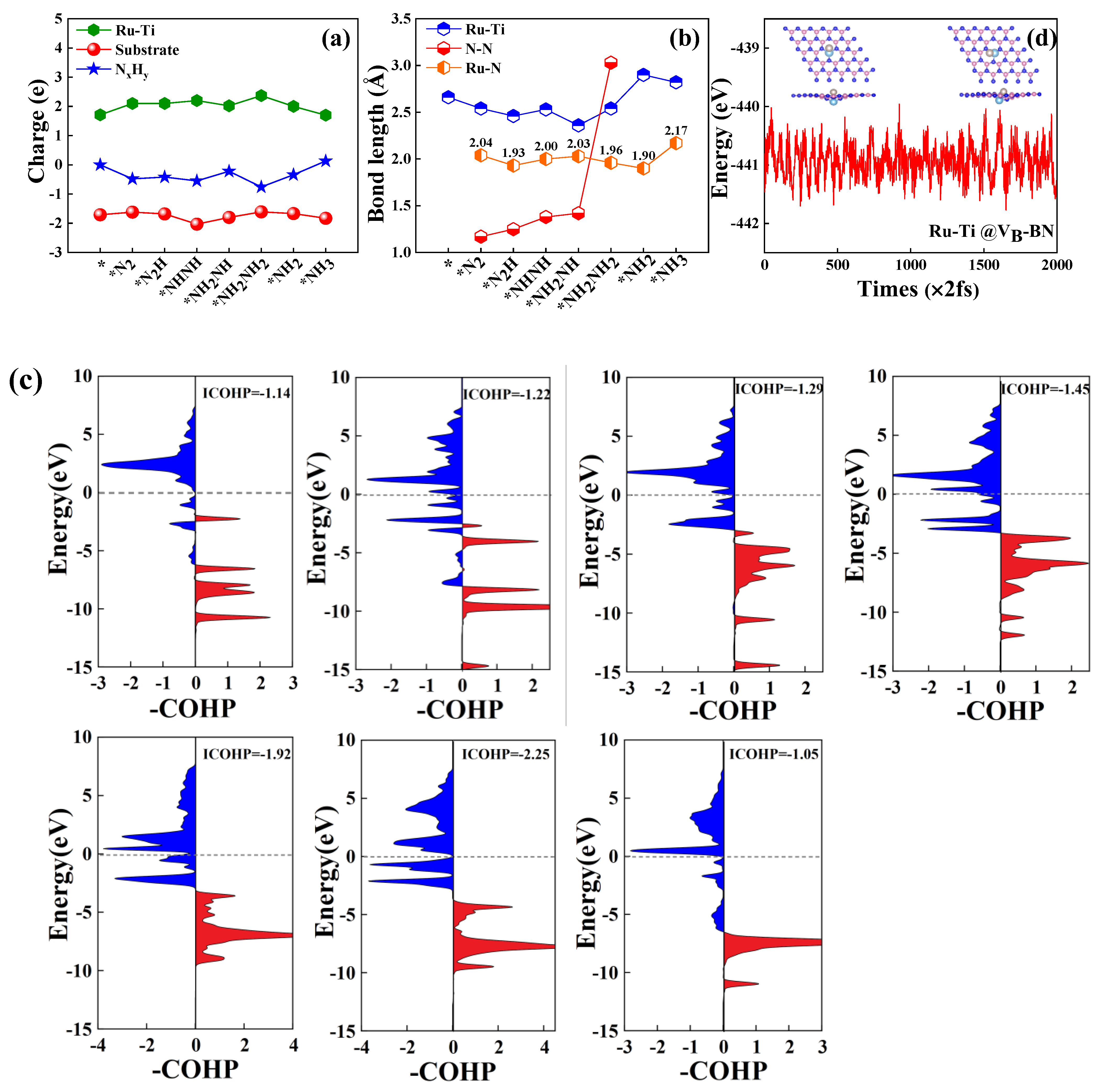
2.4. Activation Mechanisms of N2 on Ru-Mo/Ru/Ti@VB-BN
2.5. Full Conversion of N2 to NH3 on Ru-Mo/Ru/Ti @VB-BN
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Montoya, J.H.; Tsai, C.; Vojvodic, A.; Nørskov, J.K. The challenge of electrochemical ammonia synthesis: A new perspective on the role of nitrogen scaling relations. ChemSusChem 2015, 8, 2180–2186. [Google Scholar] [CrossRef]
- Licht, S.; Cui, B.; Wang, B.; Li, F.; Lau, J.; Liu, S. Ammonia synthesis by N2 and steam electrolysis in molten hydroxide suspensions of nanoscale Fe2O3. Science 2014, 345, 637–640. [Google Scholar] [CrossRef]
- Suryanto, B.H.; Matuszek, K.; Choi, J.; Hodgetts, R.Y.; Du, H.; Bakker, J.M.; Kang, C.S.; Cherepanov, P.V.; Simonov, A.N.; MacFarlane, D.R. Nitrogen reduction to ammonia at high efficiency and rates based on a phosphonium proton shuttle. Science 2021, 372, 1187–1191. [Google Scholar] [CrossRef]
- MacFarlane, D.R.; Cherepanov, P.V.; Choi, J.; Suryanto, B.H.; Hodgetts, R.Y.; Bakker, J.M.; Vallana, F.M.F.; Simonov, A.N. A roadmap to the ammonia economy. Joule 2020, 4, 1186–1205. [Google Scholar] [CrossRef]
- Soloveichik, G. Electrochemical synthesis of ammonia as a potential alternative to the Haber–Bosch process. Nat. Catal. 2019, 2, 377–380. [Google Scholar] [CrossRef]
- Yao, Y.; Wang, H.; Yuan, X.; Li, H.; Shao, M. Electrochemical nitrogen reduction reaction on ruthenium. ACS Energy Lett. 2019, 4, 1336–1341. [Google Scholar] [CrossRef]
- Rohr, B.A.; Singh, A.R.; Nørskov, J.K. A theoretical explanation of the effect of oxygen poisoning on industrial Haber-Bosch catalysts. J. Catal. 2019, 372, 33–38. [Google Scholar] [CrossRef]
- Martín, A.J.; Pérez-Ramírez, J. Heading to distributed electrocatalytic conversion of small abundant molecules into fuels, chemicals, and fertilizers. Joule 2019, 3, 2602–2621. [Google Scholar] [CrossRef]
- Yan, Z.; Ji, M.; Xia, J.; Zhu, H. Nitrogen reduction reaction: Recent advanced materials for electrochemical and photoelectrochemical synthesis of ammonia from dinitrogen: One step closer to a sustainable energy future. Adv. Energy Mater. 2020, 10, 2070049. [Google Scholar] [CrossRef]
- Guo, X.; Du, H.; Qu, F.; Li, J. Recent progress in electrocatalytic nitrogen reduction. J. Mater. Chem. A 2019, 7, 3531–3543. [Google Scholar] [CrossRef]
- Wang, D.; Azofra, L.M.; Harb, M.; Cavallo, L.; Zhang, X.; Suryanto, B.H.; MacFarlane, D.R. Energy-efficient nitrogen reduction to ammonia at low overpotential in aqueous electrolyte under ambient conditions. ChemSusChem 2018, 11, 3416–3422. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.R.; Rohr, B.A.; Statt, M.J.; Schwalbe, J.A.; Cargnello, M.; Nørskov, J.K. Strategies toward selective electrochemical ammonia synthesis. ACS Catal. 2019, 9, 8316–8324. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, Q.; Hao, W.; Fang, C.; Zhou, J.; Xu, J. A novel porous graphitic carbon nitride (g-C7N3) substrate: Prediction of metal-based π–d conjugated nanosheets toward the highly active and selective electrocatalytic nitrogen reduction reaction. J. Mater. Chem. A 2022, 10, 15036–15050. [Google Scholar] [CrossRef]
- Hu, X.; Xiong, L.; Fang, W.-H.; Su, N.Q. Computational insight into metallated graphynes as single atom electrocatalysts for nitrogen fixation. ACS Appl. Mater. Interfaces 2022, 14, 27861–27872. [Google Scholar] [CrossRef]
- Wu, Y.; He, C.; Zhang, W. “Capture-backdonation-recapture” mechanism for promoting N2 reduction by heteronuclear metal-free double-atom catalysts. J. Am. Chem. Soc. 2022, 44, 9344–9353. [Google Scholar] [CrossRef]
- Chen, Z.W.; Yan, J.M.; Jiang, Q. Single or double: Which is the altar of atomic catalysts for nitrogen reduction reaction? Small Methods 2019, 3, 1800291. [Google Scholar] [CrossRef]
- Zhang, Z.; Huang, X.; Xu, H. Anchoring an Fe dimer on nitrogen-doped graphene toward highly efficient electrocatalytic ammonia synthesis. ACS Appl. Mater. Interfaces 2021, 13, 43632–43640. [Google Scholar] [CrossRef]
- Ma, Z.; Cui, Z.; Xiao, C.; Dai, W.; Lv, Y.; Li, Q.; Sa, R. Theoretical screening of efficient single-atom catalysts for nitrogen fixation based on a defective BN monolayer. Nanoscale 2020, 12, 1541–1550. [Google Scholar] [CrossRef]
- Jin, C.; Lin, F.; Suenaga, K.; Iijima, S. Fabrication of a freestanding boron nitride single layer and its defect assignments. Phys. Rev. Lett. 2009, 102, 195505. [Google Scholar] [CrossRef] [Green Version]
- Hong, J.; Jin, C.; Yuan, J.; Zhang, Z. Atomic defects in two-dimensional materials: From single-atom spectroscopy to functionalities in opto-/electronics, nanomagnetism, and catalysis. Adv. Mater. 2017, 29, 1606434. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, Z. Single Mo atom supported on defective boron nitride monolayer as an efficient electrocatalyst for nitrogen fixation: A computational study. J. Am. Chem. Soc. 2017, 139, 12480–12487. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Wen, L.; Bian, X.; Zhang, Y.; Lin, W.; Huang, S.; Ding, K. Fe, Mo-Co-doped graphene for electrocatalytic N2-to-NH3 conversion: A DFT investigation. Appl. Surf. Sci. 2021, 569, 150921. [Google Scholar] [CrossRef]
- Yang, W.; Huang, H.; Ding, X.; Ding, Z.; Wu, C.; Gates, I.D.; Gao, Z. Theoretical study on double-atom catalysts supported with graphene for electroreduction of nitrogen into ammonia. Electrochim. Acta 2020, 335, 135667. [Google Scholar] [CrossRef]
- He, T.; Santiago, A.R.P.; Du, A. Atomically embedded asymmetrical dual-metal dimers on N-doped graphene for ultra-efficient nitrogen reduction reaction. J. Catal. 2020, 388, 77–83. [Google Scholar] [CrossRef]
- Zhao, W.; Chen, L.; Zhang, W.; Yang, J. Single Mo1 (W1, Re1) atoms anchored in pyrrolic-N3 doped graphene as efficient electrocatalysts for the nitrogen reduction reaction. J. Mater. Chem. A 2021, 9, 6547–6554. [Google Scholar] [CrossRef]
- Hellman, A.; Honkala, K.; Remediakis, I.; Logadottir, A.; Carlsson, A.; Dahl, S.; Christensen, C.H.; Nørskov, J.K. Ammonia synthesis and decomposition on a Ru-based catalyst modeled by first-principles. Surf. Sci. 2009, 603, 1731–1739. [Google Scholar] [CrossRef]
- Ghuman, K.K.; Tozaki, K.; Sadakiyo, M.; Kitano, S.; Oyabe, T.; Yamauchi, M. Tailoring widely used ammonia synthesis catalysts for H and N poisoning resistance. Phys. Chem. Chem. Phys. 2019, 21, 5117–5122. [Google Scholar] [CrossRef]
- Wang, Y.; Du, Y.; Meng, Y.; Xie, B.; Ni, Z.; Xia, S. Monatomic Ti doped on defective monolayer boron nitride as an electrocatalyst for the synthesis of ammonia: A DFT study. Appl. Surf. Sci. 2021, 563, 150277. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, X.; Qin, J.; Liu, R. High-throughput screening of single metal atom anchored on N-doped boron phosphide for N2 reduction. Nanoscale 2021, 13, 13437–13450. [Google Scholar] [CrossRef]
- Huang, B.; Wu, Y.; Chen, B.; Qian, Y.; Zhou, N.; Li, N. Transition-metal-atom-pairs deposited on g-CN monolayer for nitrogen reduction reaction: Density functional theory calculations. Chin. J. Catal. 2021, 42, 1160–1167. [Google Scholar] [CrossRef]
- Choi, C.; Back, S.; Kim, N.-Y.; Lim, J.; Kim, Y.-H.; Jung, Y. Suppression of hydrogen evolution reaction in electrochemical N2 reduction using single-atom catalysts: A computational guideline. ACS Catal. 2018, 8, 7517–7525. [Google Scholar] [CrossRef]
- Singh, A.R.; Rohr, B.A.; Schwalbe, J.A.; Cargnello, M.; Chan, K.; Jaramillo, T.F.; Chorkendorff, I.; Nørskov, J.K. Electrochemical ammonia synthesis the selectivity challenge. ACS Publ. 2017, 7, 706–709. [Google Scholar] [CrossRef] [Green Version]
- Azofra, L.M.; Sun, C.; Cavallo, L.; MacFarlane, D.R. Feasibility of N2 binding and reduction to ammonia on Fe-deposited MoS2 2d sheets: A DFT study. Chem. Eur. J. 2017, 23, 8275–8279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Huang, S. Tuning nitrogen reduction reaction activity via controllable Fe magnetic moment: A computational study of single Fe atom supported on defective graphene. Electrochim. Acta 2018, 284, 392–399. [Google Scholar] [CrossRef]
- Wang, C.; Zhao, Y.; Zhu, C.; Zhang, M.; Geng, Y.; Li, Y.; Su, Z. A two-dimensional conductive Mo-based covalent organic framework as an efficient electrocatalyst for nitrogen fixation. J. Mater. Chem. A 2020, 8, 23599–23606. [Google Scholar] [CrossRef]
- Seefeldt, L.C.; Hoffman, B.M.; Dean, D.R. Mechanism of Mo-dependent nitrogenase. Annu. Rev. Biochem. 2009, 78, 701. [Google Scholar] [CrossRef]
- Hinnemann, B.; Nørskov, J.K. Catalysis by enzymes: The biological ammonia synthesis. Top. Catal. 2006, 37, 55–70. [Google Scholar] [CrossRef]
- Le, Y.; Wei, C.; Xue, W.; Li, Y.; Zhang, Y.; Lin, W. Nitrogen reduction on crystalline carbon nitride supported by homonuclear bimetallic atoms. J. Chem. Phys. 2022, 157, 114704. [Google Scholar] [CrossRef]
- Liu, A.; Gao, M.; Ren, X.; Meng, F.; Yang, Y.; Yang, Q.; Guan, W.; Gao, L.; Liang, X.; Ma, T. A two-dimensional Ru@ Mxene catalyst for highly selective ambient electrocatalytic nitrogen reduction. Nanoscale 2020, 12, 10933–10938. [Google Scholar] [CrossRef]
- Hu, L.G.; Wang, H.J.; Su, Y. Computational study of double transition metal atom anchored on graphdiyne monolayer for nitrogen electroreduction. ChemPhysChem 2022, 23, e202200149. [Google Scholar] [CrossRef]
- Azofra, L.M.; Li, N.; MacFarlane, D.R.; Sun, C. Promising prospects for 2d d2–d4 M3C2 transition metal carbides (Mxenes) in N2 capture and conversion into ammonia. Energy Environ. Sci. 2016, 9, 2545–2549. [Google Scholar] [CrossRef]
- Kour, G.; Mao, X.; Du, A. Computational screening of single-atom alloys TM@Ru(0001) for enhanced electrochemical nitrogen reduction reaction. J. Mater. Chem. A 2022, 10, 6204–6215. [Google Scholar] [CrossRef]
- Dronskowski, R.; Blochl, P.E. Crystal orbital hamilton populations (cohp)—Energy-resolved visualization of chemical bonding in solids based on density-functional calculations. J. Phys. Chem. 1993, 97, 8617–8624. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ernzerhof, M.; Burke, K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 1996, 105, 9982–9985. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-d) for the 94 elements h-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Tuckerman, M.; Laasonen, K.; Sprik, M.; Parrinello, M. Ab initio molecular dynamics simulation of the solvation and transport of hydronium and hydroxyl ions in water. J. Chem. Phys. 1995, 103, 150–161. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jonsson, H. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fe-Fe | Fe-Mo | Fe-Ru | Fe-Ti | Mo-Mo | Mo-Ru | |
|---|---|---|---|---|---|---|
| ΔG(*H) | −0.78 | −0.73 | −0.54 | −0.81 | −0.78 | −0.81 |
| ΔG(*N2)/end-on | −1.46 | −1.29 | −1.31 | −1.44 | −1.23 | −0.99 |
| ΔG(*N2)/side-on | −2.12 | −1.06 | −0.96 | −1.26 | −1.36 | −1.1 |
| Mo-Ti | Ru-Ru | Ru-Ti | Ti-Ti | Ti-Ru | ||
| ΔG(*H) | −0.80 | −0.55 | −0.62 | −0.93 | −0.87 | |
| ΔG(*N2)/end-on | −1.20 | −1.12 | −1.20 | −0.40 | −0.78 | |
| ΔG(*N2)/side-on | −1.31 | −0.68 | −0.89 | −1.33 | −0.90 | |
| Ti-Mo | Ti-Fe | Ru-Mo | Ru-Fe | Mo-Fe | ||
| ΔG(*H) | −1.00 | −0.89 | −0.57 | −0.60 | −0.87 | |
| ΔG(*N2)/end-on | −0.88 | −0.98 | −1.05 | −1.38 | −1.19 | |
| ΔG(*N2)/side-on | −1.07 | −1.12 | −0.70 | −0.83 | −1.23 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, R.; Xia, J.-Z.; Wu, Q. Computational Insight into Defective Boron Nitride Supported Double-Atom Catalysts for Electrochemical Nitrogen Reduction. Catalysts 2022, 12, 1404. https://doi.org/10.3390/catal12111404
Cao R, Xia J-Z, Wu Q. Computational Insight into Defective Boron Nitride Supported Double-Atom Catalysts for Electrochemical Nitrogen Reduction. Catalysts. 2022; 12(11):1404. https://doi.org/10.3390/catal12111404
Chicago/Turabian StyleCao, Rong, Jie-Zhen Xia, and Qi Wu. 2022. "Computational Insight into Defective Boron Nitride Supported Double-Atom Catalysts for Electrochemical Nitrogen Reduction" Catalysts 12, no. 11: 1404. https://doi.org/10.3390/catal12111404