
DFT Investigations on the Ring-Opening Polymerization of Trimethylene Carbonate Catalysed by Heterocyclic Nitrogen Bases
 , and
, and
Abstract
:1. Introduction Section
2. Results and Discussion
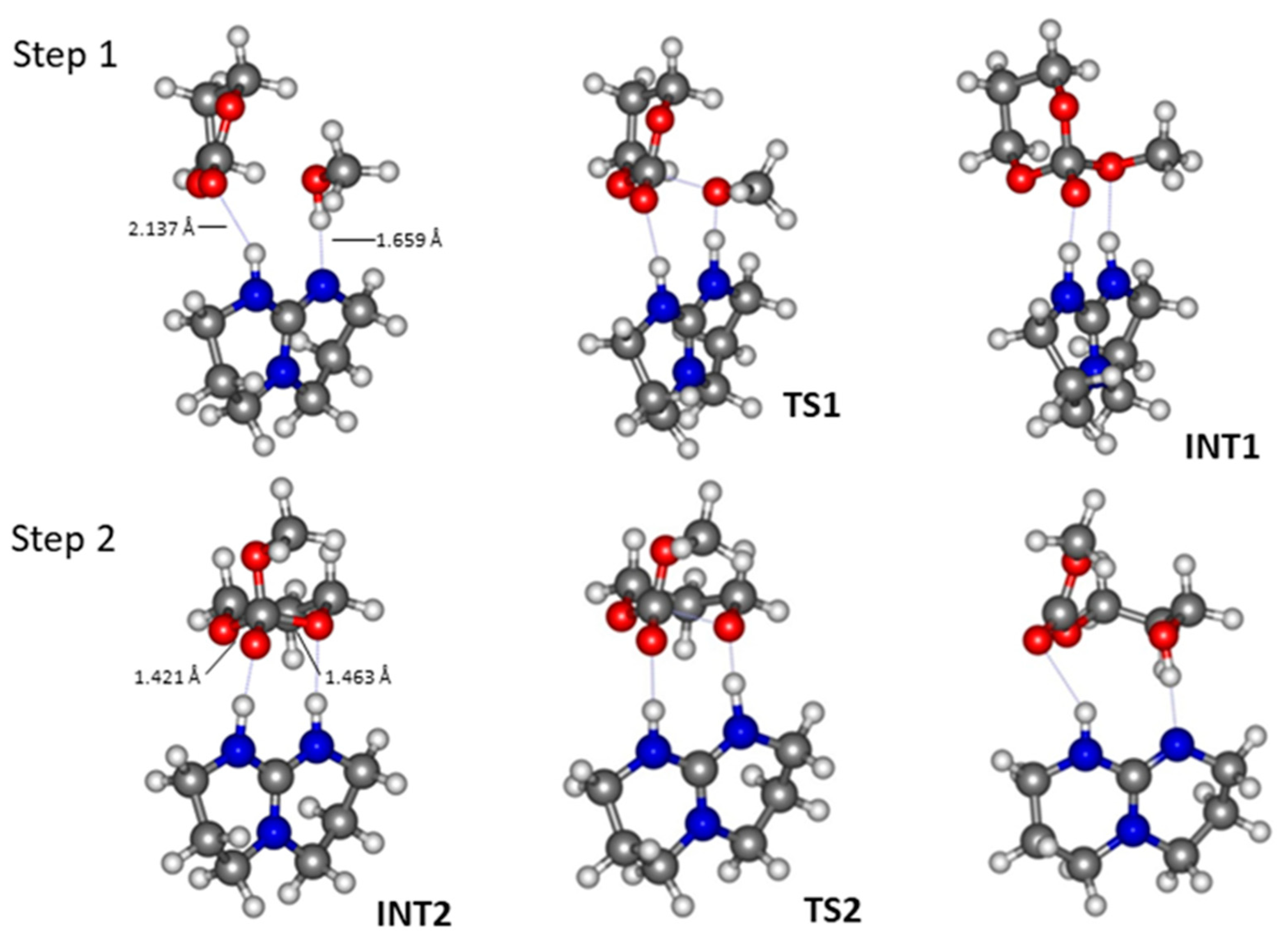
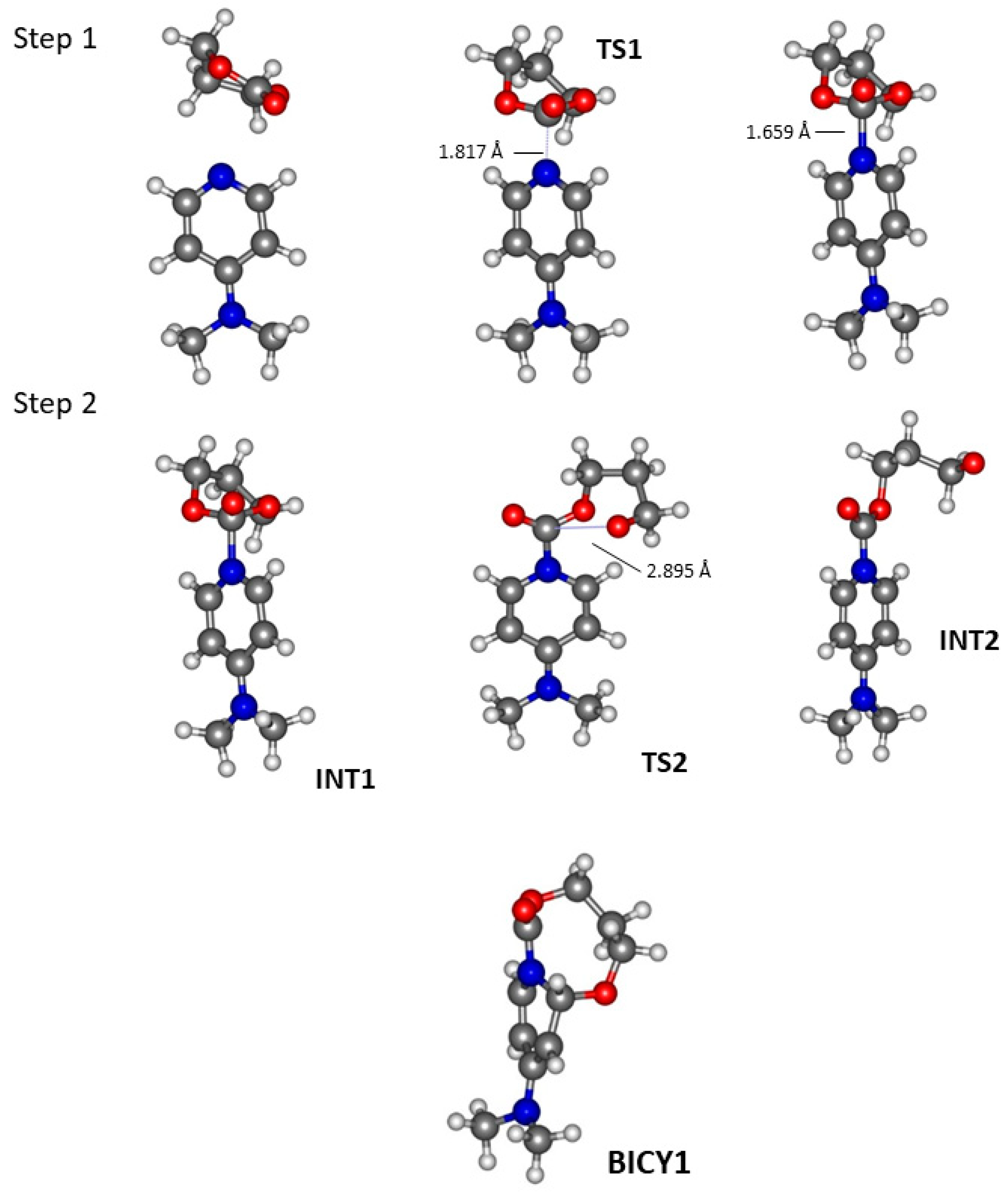
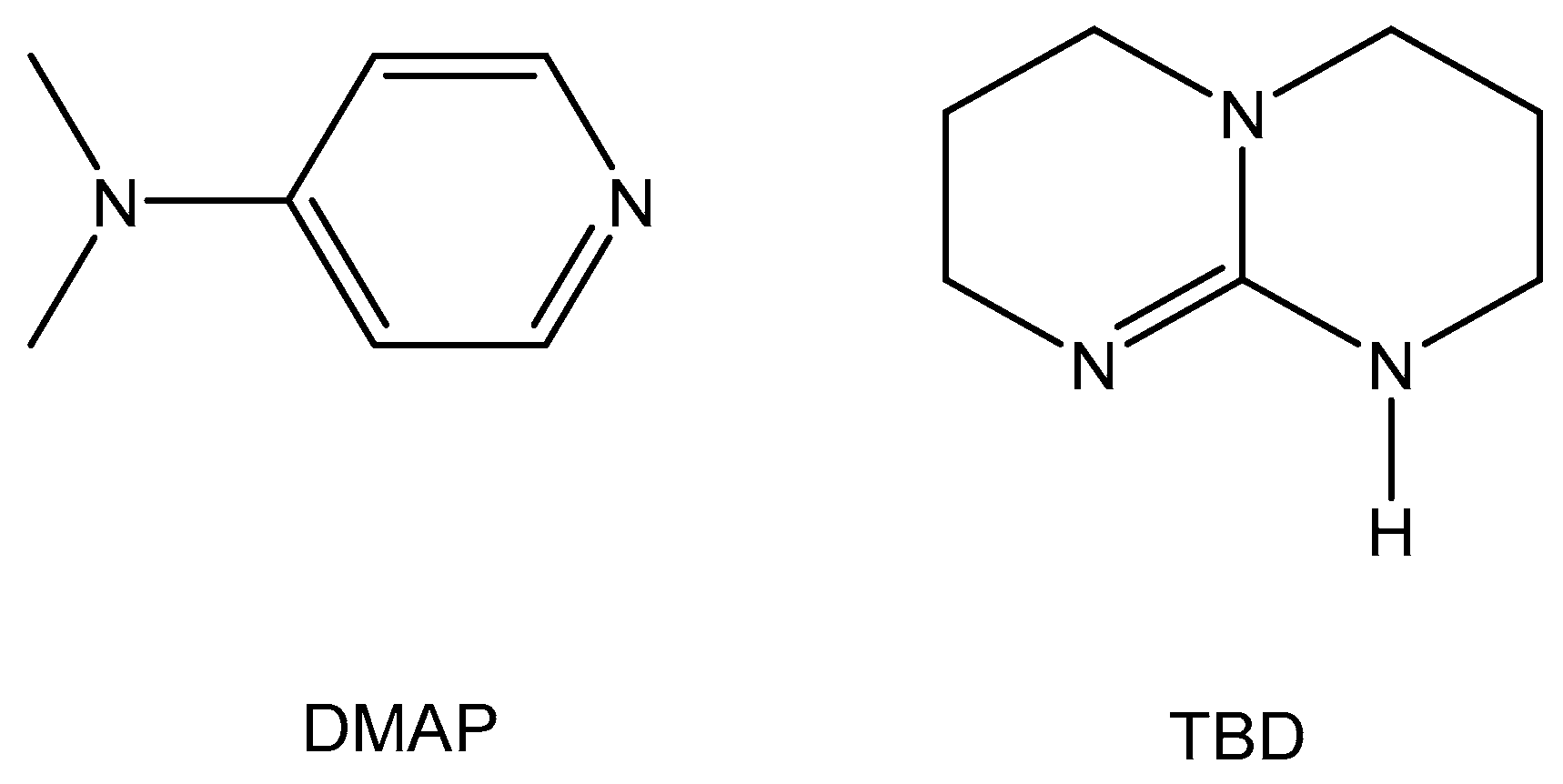
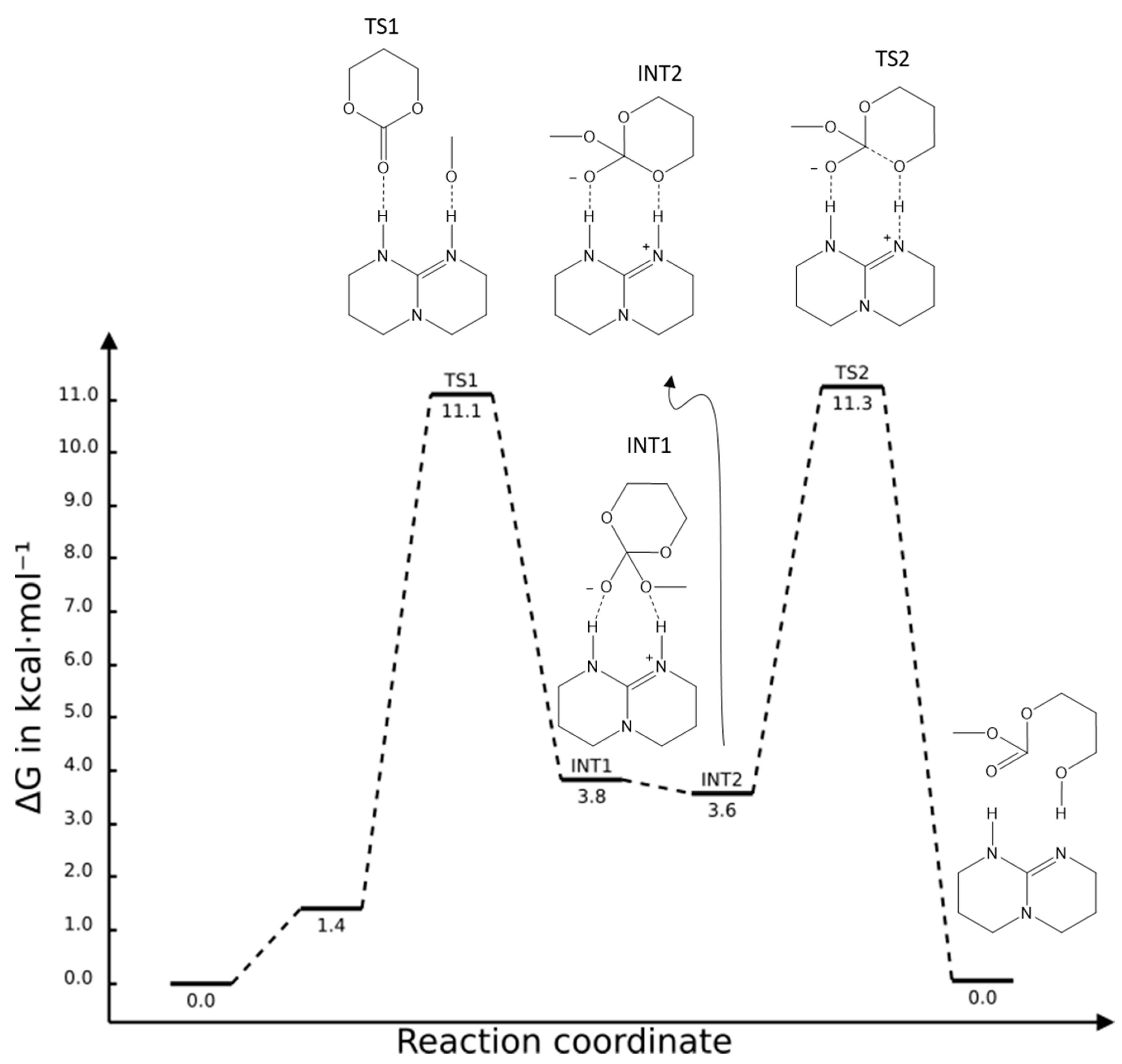
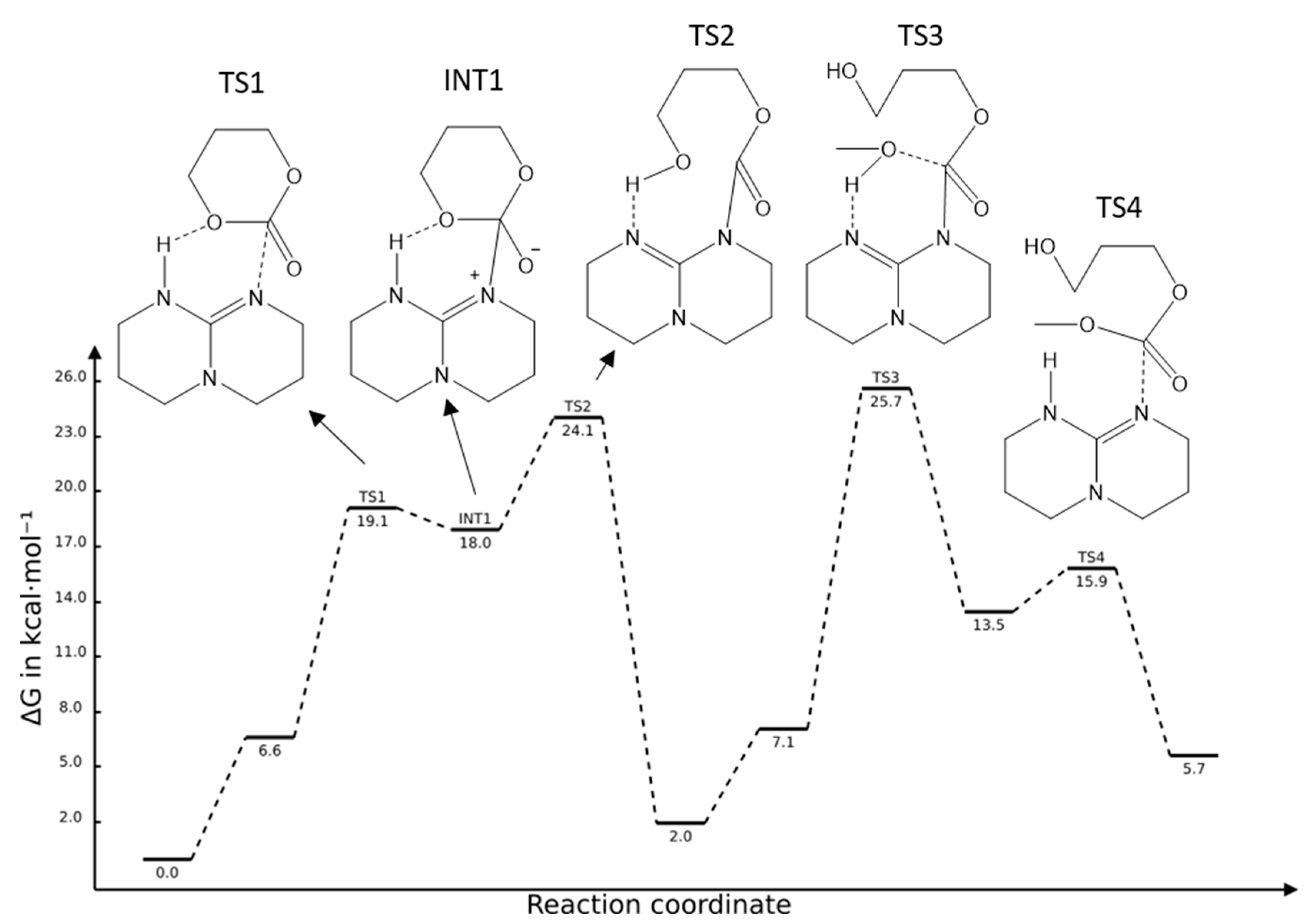
2.1. TBD Catalysed Ring-Opening Polymerization of TMC
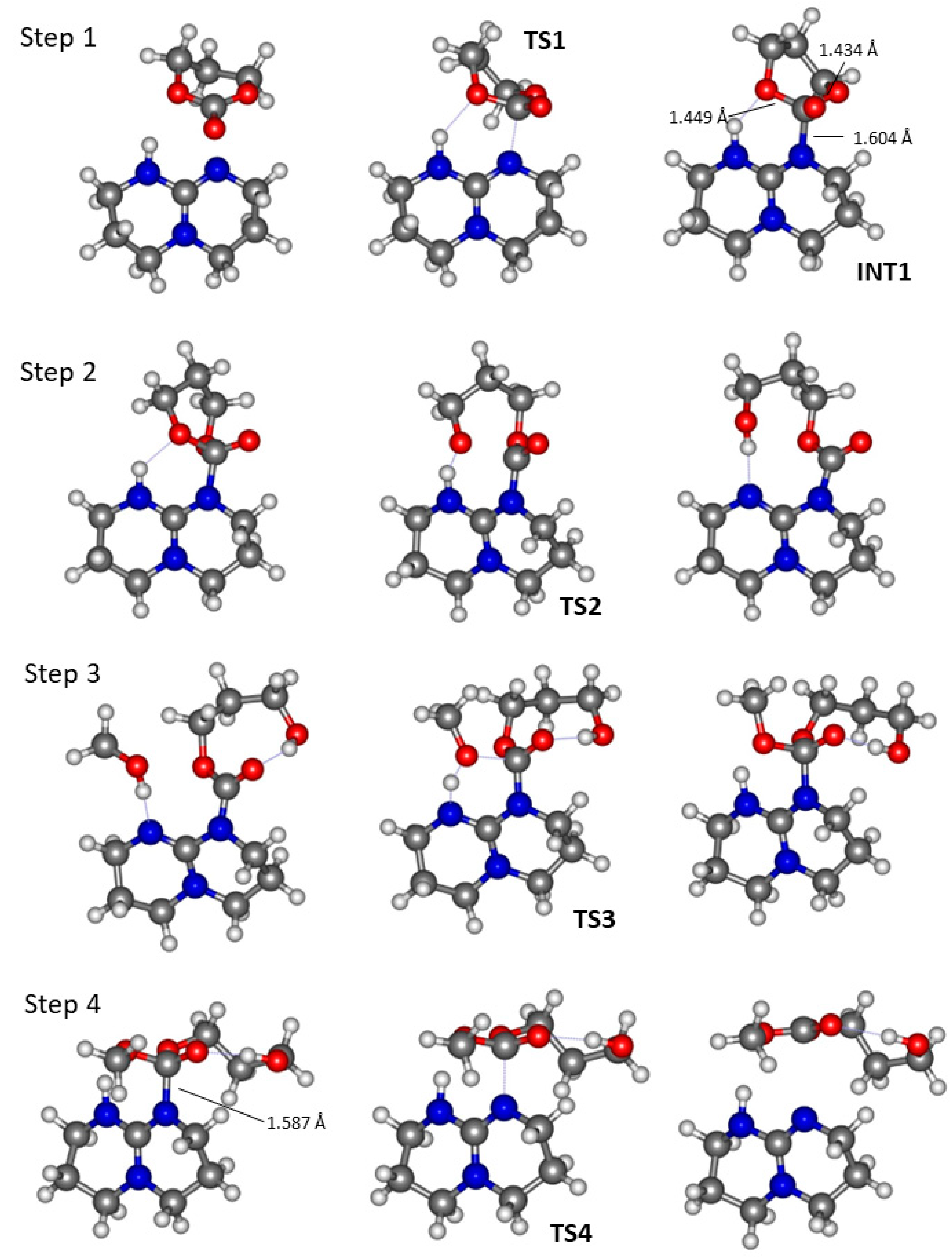
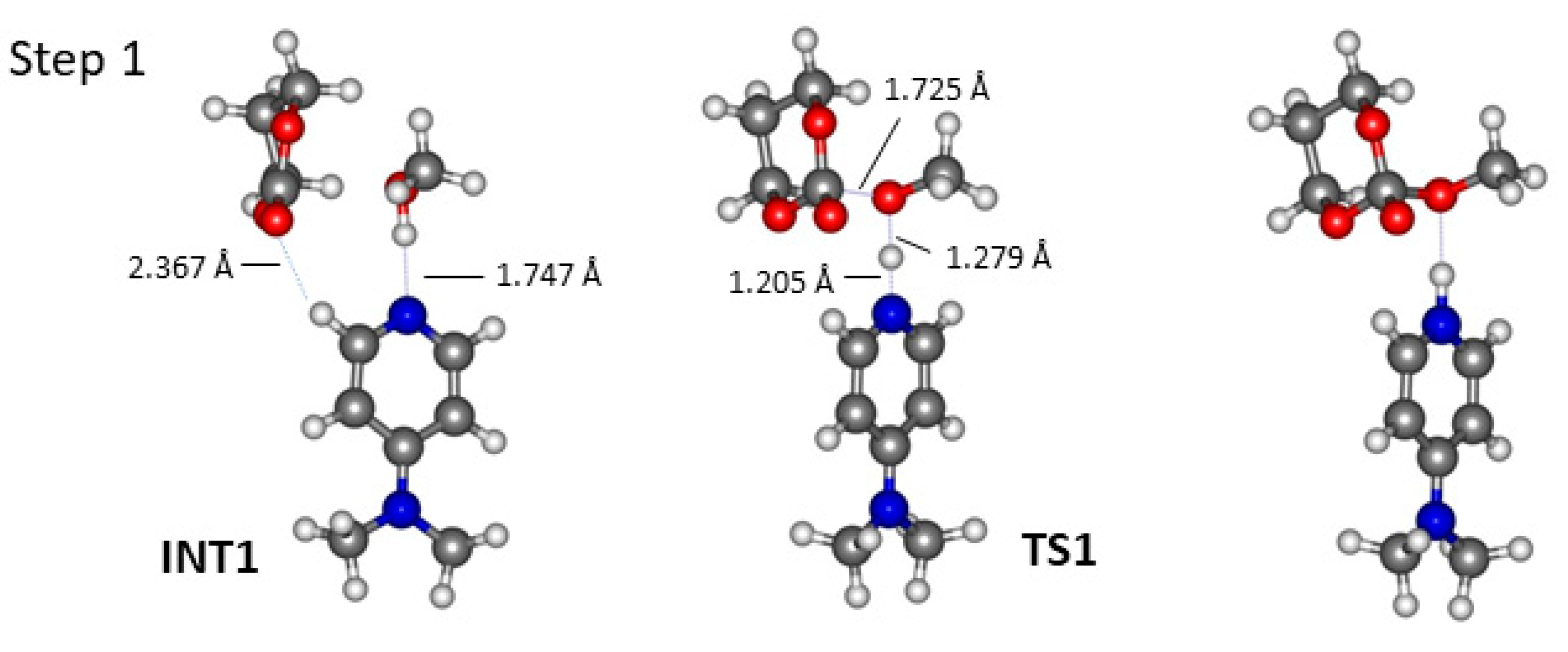
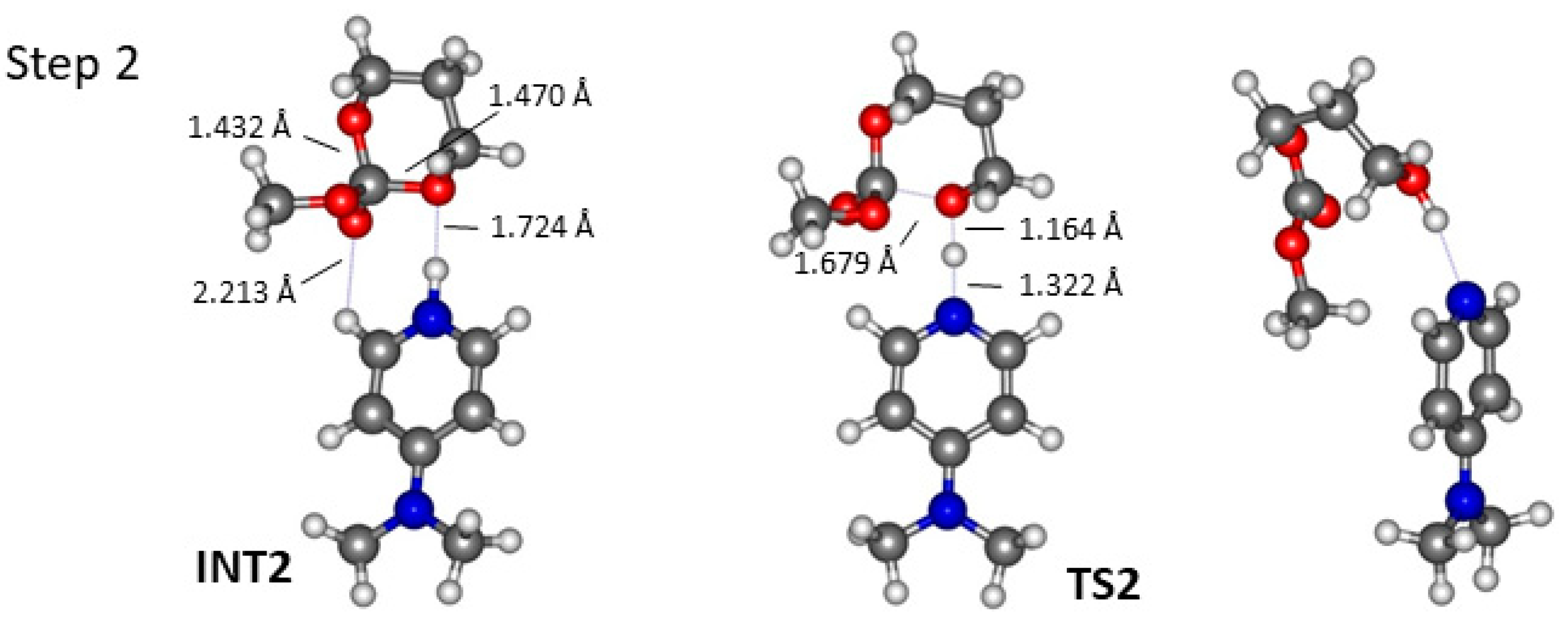
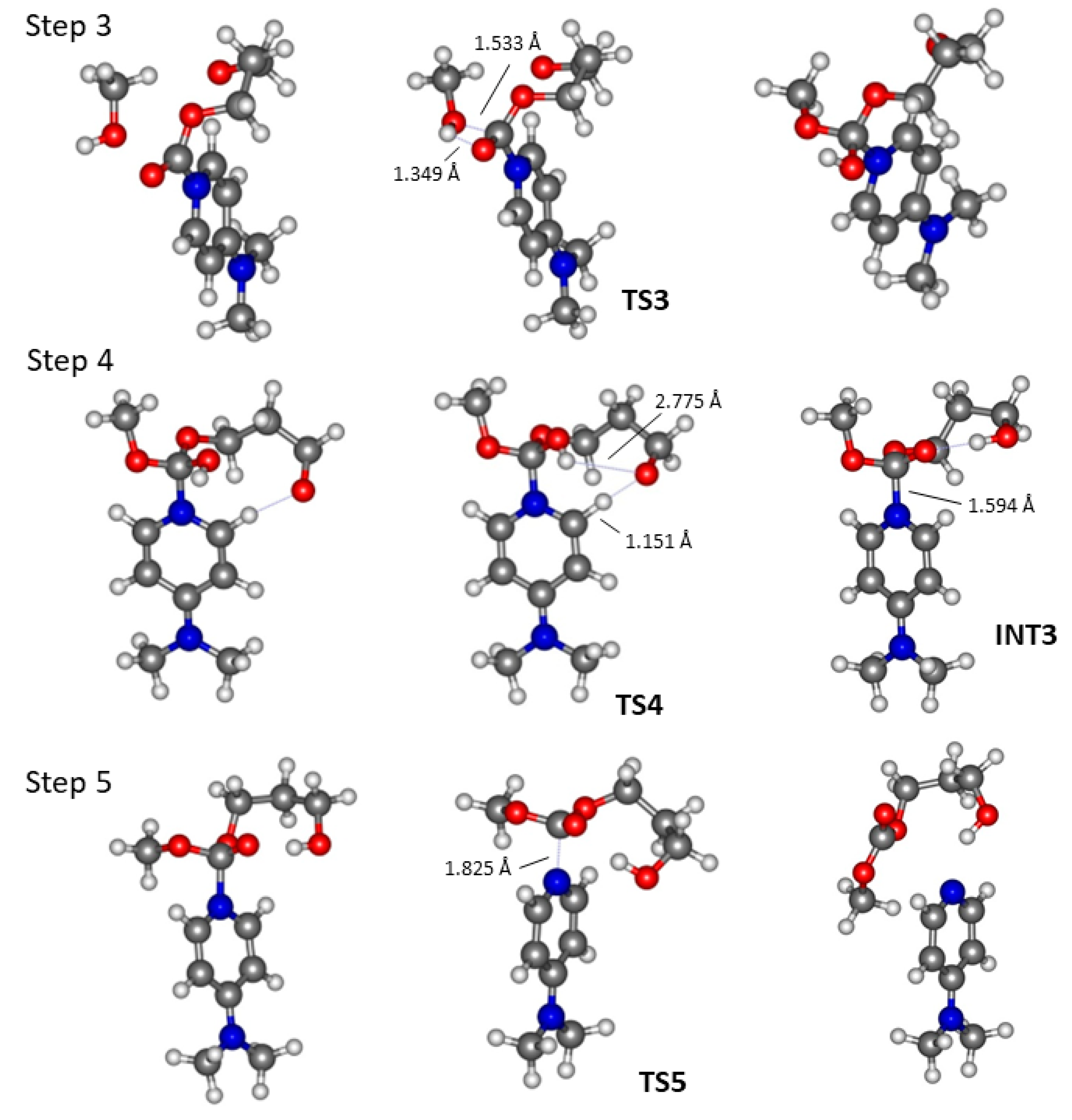
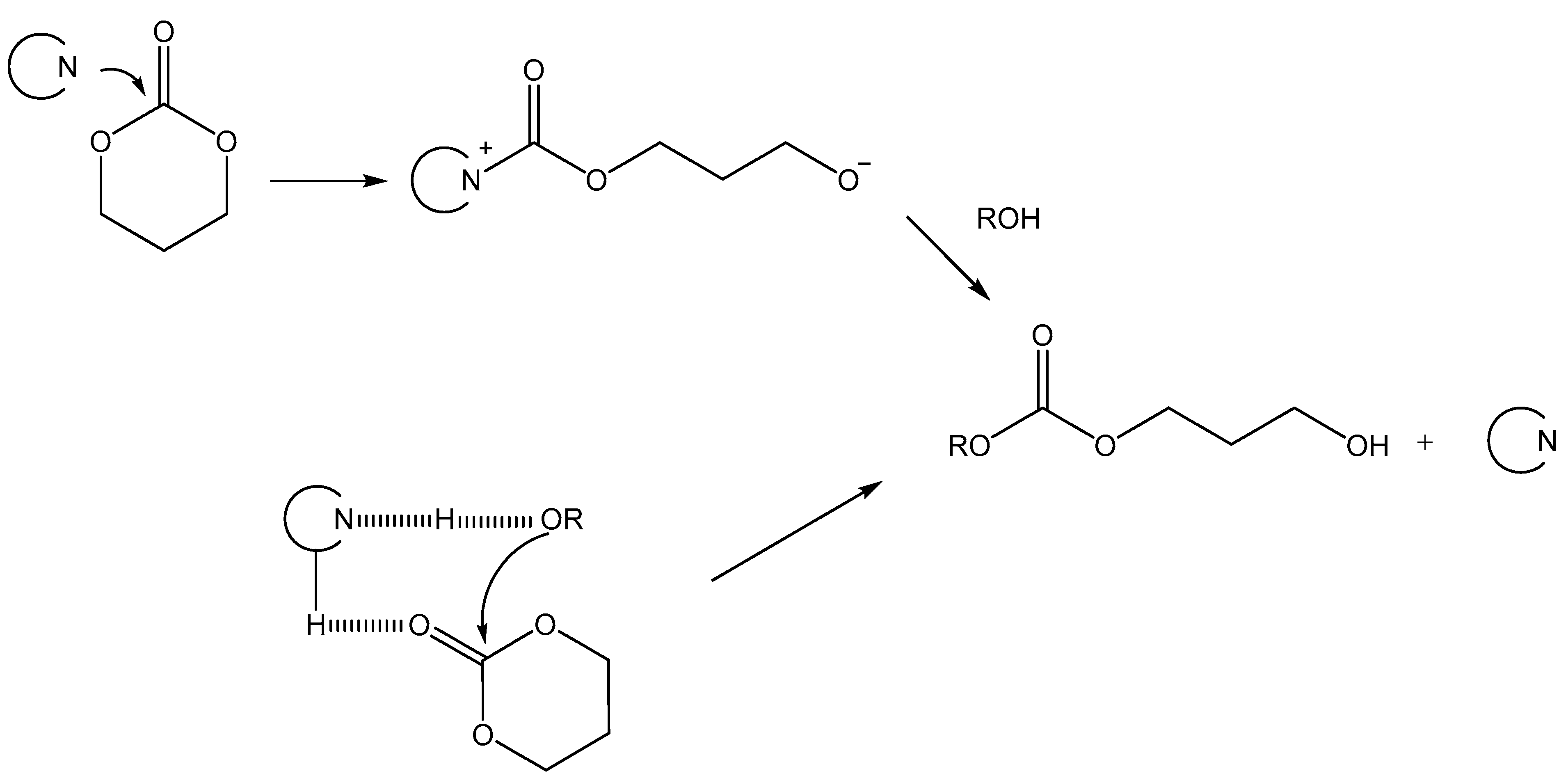
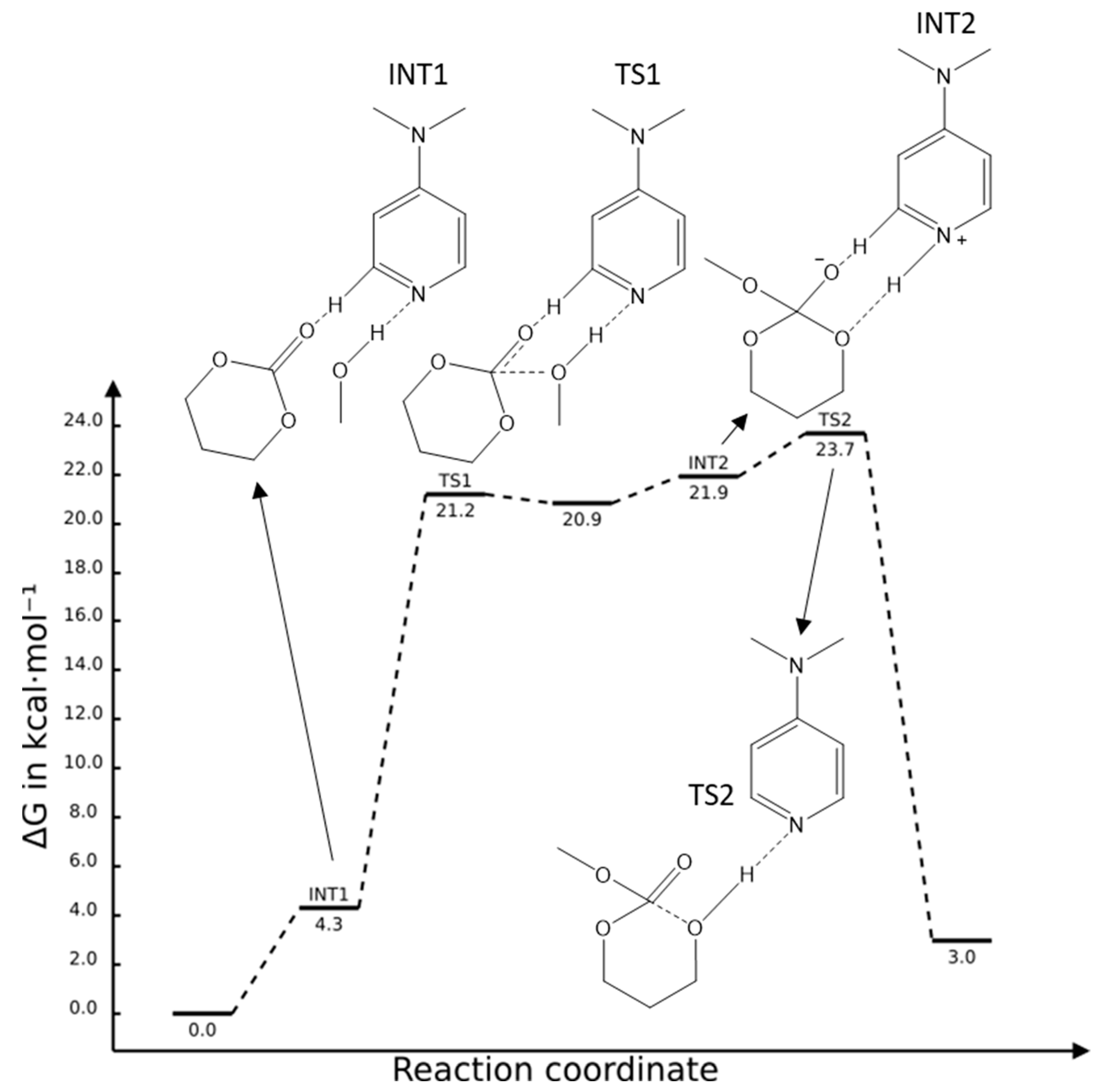
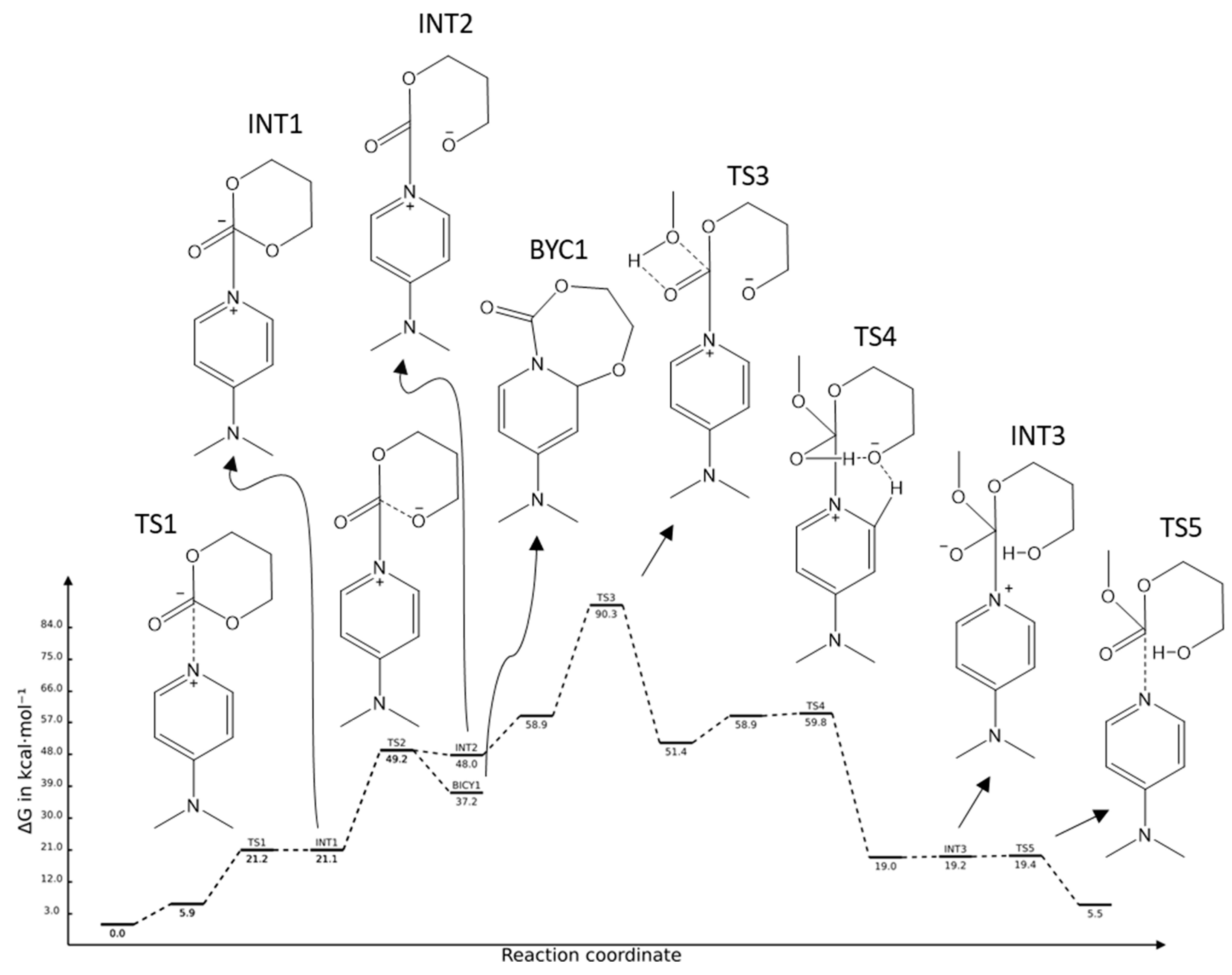
2.2. DMAP Catalysed Ring-Opening Polymerization of TMC
2.3. Experimental Confirmation
3. Materials and Methods
3.1. Methodological Details
3.2. Experimental Details
3.2.1. Reagents
3.2.2. Polymerizations
3.2.3. Analytics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Artham, T.; Doble, M. Biodegradation of Aliphatic and Aromatic Polycarbonates: Biodegradation of Aliphatic and Aromatic Polycarbonates. Macromol. Biosci. 2008, 8, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Feng, E.; Song, J. Renaissance of Aliphatic Polycarbonates: New Techniques and Biomedical Applications: Review. J. Appl. Polym. Sci. 2014, 131, 39822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.; Maynard, E.; Chiaradia, V.; Arno, M.C.; Dove, A.P. Aliphatic Polycarbonates from Cyclic Carbonate Monomers and Their Application as Biomaterials. Chem. Rev. 2021, 121, 10865–10907. [Google Scholar] [CrossRef]
- Nederberg, F.; Lohmeijer, B.G.G.; Leibfarth, F.; Pratt, R.C.; Choi, J.; Dove, A.P.; Waymouth, R.M.; Hedrick, J.L. Organocatalytic Ring Opening Polymerization of Trimethylene Carbonate. Biomacromolecules 2007, 8, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Helou, M.; Miserque, O.; Brusson, J.-M.; Carpentier, J.-F.; Guillaume, S.M. Organocatalysts for the Controlled “Immortal” Ring-Opening Polymerization of Six-Membered-Ring Cyclic Carbonates: A Metal-Free, Green Process. Chem.—A Eur. J. 2010, 16, 13805–13813. [Google Scholar] [CrossRef] [PubMed]
- Azemar, F.; Gimello, O.; Pinaud, J.; Robin, J.-J.; Monge, S. Insight into the Alcohol-Free Ring-Opening Polymerization of TMC Catalyzed by TBD. Polymers 2021, 13, 1589. [Google Scholar] [CrossRef] [PubMed]
- Lalanne-Tisné, M.; Eyley, S.; De Winter, J.; Favrelle-Huret, A.; Thielemans, W.; Zinck, P. Cellulose nanocrystals modification by grafting from ring opening polymerization of a cyclic carbonate. Carbohydr. Polym. 2022, 295, 119840. [Google Scholar] [CrossRef] [PubMed]
- Simón, L.; Goodman, J. The Mechanism of TBD-Catalyzed Ring-Opening Polymerization of Cyclic Esters. J. Org. Chem. 2007, 72, 9656–9662. [Google Scholar] [CrossRef]
- Bonduelle, C.; Martín-Vaca, B.; Cossío, F.P.; Bourissou, D. Monomer versus Alcohol Activation in the 4-Dimethylaminopyridine-Catalyzed Ring-Opening Polymerization of Lactide and LacticO-Carboxylic Anhydride. Chem.—A Eur. J. 2008, 14, 5304–5312. [Google Scholar] [CrossRef]
- Chuma, A.; Horn, H.W.; Swope, W.C.; Pratt, R.C.; Zhang, L.; Lohmeijer, B.G.G.; Wade, C.G.; Waymouth, R.M.; Hedrick, J.L.; Rice, J.E. The Reaction Mechanism for the Organocatalytic Ring-Opening Polymerization of l-Lactide Using a Guanidine-Based Catalyst: Hydrogen-Bonded or Covalently Bound? J. Am. Chem. Soc. 2008, 130, 6749–6754. [Google Scholar] [CrossRef]
- Katiyar, V.; Nanavati, H. Ring-opening polymerization of L-lactide using N-heterocyclic molecules: Mechanistic, kinetics and DFT studies. Polym. Chem. 2010, 1, 1491–1500. [Google Scholar] [CrossRef]
- Brown, H.A.; Waymouth, R.M. Zwitterionic Ring-Opening Polymerization for the Synthesis of High Molecular Weight Cyclic Polymers. Accounts Chem. Res. 2013, 46, 2585–2596. [Google Scholar] [CrossRef] [PubMed]
- Acharya, A.K.; Chang, Y.A.; Jones, G.O.; Rice, J.E.; Hedrick, J.L.; Horn, H.W.; Waymouth, R.M. Experimental and Computational Studies on the Mechanism of Zwitterionic Ring-Opening Polymerization of δ-Valerolactone with N-Heterocyclic Carbenes. J. Phys. Chem. B 2014, 118, 6553–6560. [Google Scholar] [CrossRef] [PubMed]
- Sherck, N.J.; Kim, H.C.; Won, Y.-Y. Elucidating a Unified Mechanistic Scheme for the DBU-Catalyzed Ring-Opening Polymerization of Lactide to Poly(Lactic Acid). Macromolecules 2016, 49, 4699–4713. [Google Scholar] [CrossRef] [Green Version]
- Nogueira, G.; Favrelle, A.; Bria, M.; Prates Ramalho, J.P.; Mendes, P.J.; Valente, A.; Zinck, P. Adenine as an Organocatalyst for the Ring-Opening Polymerization of Lactide: Scope, Mechanism and Access to Adenine-Functionalized Polylactide. React. Chem. Eng. 2016, 1, 508–520. [Google Scholar] [CrossRef] [Green Version]
- Stanley, N.; Chenal, T.; Jacquel, N.; Saint-Loup, R.; Prates Ramalho, J.P.; Zinck, P. Organocatalysts for the Synthesis of Poly(Ethylene Terephthalate-Co-isosorbide Terephthalate): A Combined Experimental and DFT Study. Macromol. Mater. Eng. 2019, 304, 1900298. [Google Scholar] [CrossRef]
- Nifant’ev, I.; Ivchenko, P. DFT Modeling of Organocatalytic Ring-Opening Polymerization of Cyclic Esters: A Crucial Role of Proton Exchange and Hydrogen Bonding. Polymers 2019, 11, 2078. [Google Scholar] [CrossRef] [Green Version]
- Del Rosal, I.; Brignou, P.; Guillaume, S.M.; Carpentier, J.-F.; Maron, L. DFT Investigations on the Ring-Opening Polymerization of Cyclic Carbonates Catalyzed by Zinc-{β-Diiminate} Complexes. Polym. Chem. 2011, 2, 2564. [Google Scholar] [CrossRef]
- Del Rosal, I.; Brignou, P.; Guillaume, S.M.; Carpentier, J.-F.; Maron, L. DFT Investigations on the Ring-Opening Polymerization of Substituted Cyclic Carbonates Catalyzed by Zinc-{β-Diketiminate} Complexes. Polym. Chem. 2015, 6, 3336–3352. [Google Scholar] [CrossRef]
- Jitonnom, J.; Meelua, W. Effect of ligand structure in the trimethylene carbonate polymerization by cationic zirconocene catalysts: A “naked model” DFT study. J. Organomet. Chem. 2017, 841, 48–56. [Google Scholar] [CrossRef]
- Kazarina, O.V.; Gourlaouen, C.; Karmazin, L.; Morozov, A.G.; Fedushkin, I.L.; Dagorne, S. Low valent Al(ii)–Al(ii) catalysts as highly active ε-caprolactone polymerization catalysts: Indication of metal cooperativity through DFT studies. Dalton Trans. 2018, 47, 13800–13808. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Riffel, M.N.; Diaconescu, P.L. Redox Control of Aluminum Ring-Opening Polymerization: A Combined Experimental and DFT Investigation. Macromolecules 2017, 50, 1847–1861. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Luo, G.; Mehmood, A.; Zhao, Y.; Zhou, G.; Hou, Z.; Luo, Y. Theoretical Mechanistic Studies on Redox-Switchable Polymerization of Trimethylene Carbonate Catalyzed by an Indium Complex Bearing a Ferrocene-Based Ligand. Organometallics 2018, 37, 4599–4607. [Google Scholar] [CrossRef]
- Venkataraman, S.; Ng, V.W.L.; Coady, D.J.; Horn, H.W.; Jones, G.O.; Fung, T.S.; Sardon, H.; Waymouth, R.M.; Hedrick, J.L.; Yang, Y.Y. A Simple and Facile Approach to Aliphatic N -Substituted Functional Eight-Membered Cyclic Carbonates and Their Organocatalytic Polymerization. J. Am. Chem. Soc. 2015, 137, 13851–13860. [Google Scholar] [CrossRef]
- Gregory, G.L.; Jenisch, L.M.; Charles, B.; Kociok-Köhn, G.; Buchard, A. Polymers from Sugars and CO2: Synthesis and Polymerization of a d -Mannose-Based Cyclic Carbonate. Macromolecules 2016, 49, 7165–7169. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Yang, X.; Song, Y.; Tran, D.K.; Wang, H.; Wilson, J.; Dong, M.; Vazquez, M.; Sun, G.; Wooley, K.L. Complexities of Regioselective Ring-Opening vs Transcarbonylation-Driven Structural Metamorphosis during Organocatalytic Polymerizations of Five-Membered Cyclic Carbonate Glucose Monomers. JACS Au 2022, 2, 515–521. [Google Scholar] [CrossRef]
- Song, Y.; Yang, X.; Shen, Y.; Dong, M.; Lin, Y.-N.; Hall, M.B.; Wooley, K.L. Invoking Side-Chain Functionality for the Mediation of Regioselectivity during Ring-Opening Polymerization of Glucose Carbonates. J. Am. Chem. Soc. 2020, 142, 16974–16981. [Google Scholar] [CrossRef]
- Li, S.; Lu, H.; Zhu, L.; Yan, M.; Kang, X.; Luo, Y. Ring-Opening Polymerization of l-Lactide Catalyzed by Food Sweetener Saccharin with Organic Base Mediated: A Computational Study. Polymer 2022, 246, 124747. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Piletic, I.R.; Edney, E.O.; Bartolotti, L.J. Barrierless Reactions with Loose Transition States Govern the Yields and Lifetimes of Organic Nitrates Derived from Isoprene. J. Phys. Chem. A 2017, 121, 8306–8321. [Google Scholar] [CrossRef]
- Peverati, R.; Truhlar, D.G. Quest for a Universal Density Functional: The Accuracy of Density Functionals across a Broad Spectrum of Databases in Chemistry and Physics. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2014, 372, 20120476. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Supramolecular Binding Thermodynamics by Dispersion-Corrected Density Functional Theory. Chem. Eur. J. 2012, 18, 9955–9964. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-P.; Gomes, J.; Mallikarjun Sharada, S.; Bell, A.T.; Head-Gordon, M. Improved Force-Field Parameters for QM/MM Simulations of the Energies of Adsorption for Molecules in Zeolites and a Free Rotor Correction to the Rigid Rotor Harmonic Oscillator Model for Adsorption Enthalpies. J. Phys. Chem. C 2015, 119, 1840–1850. [Google Scholar] [CrossRef]
- Breneman, C.M.; Wiberg, K.B. Determining Atom-Centered Monopoles from Molecular Electrostatic Potentials. The Need for High Sampling Density in Formamide Conformational Analysis. J. Comput. Chem. 1990, 11, 361–373. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Conv. a (%) | Mn b (g/mol) | ÐM c |
|---|---|---|---|
| DMAP | 19 | 5 500 | 1.7 |
| TBD d | >99 | 9 900 | 1.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lalanne-Tisné, M.; Favrelle-Huret, A.; Thielemans, W.; Prates Ramalho, J.P.; Zinck, P. DFT Investigations on the Ring-Opening Polymerization of Trimethylene Carbonate Catalysed by Heterocyclic Nitrogen Bases. Catalysts 2022, 12, 1280. https://doi.org/10.3390/catal12101280
Lalanne-Tisné M, Favrelle-Huret A, Thielemans W, Prates Ramalho JP, Zinck P. DFT Investigations on the Ring-Opening Polymerization of Trimethylene Carbonate Catalysed by Heterocyclic Nitrogen Bases. Catalysts. 2022; 12(10):1280. https://doi.org/10.3390/catal12101280
Chicago/Turabian StyleLalanne-Tisné, Michael, Audrey Favrelle-Huret, Wim Thielemans, João P. Prates Ramalho, and Philippe Zinck. 2022. "DFT Investigations on the Ring-Opening Polymerization of Trimethylene Carbonate Catalysed by Heterocyclic Nitrogen Bases" Catalysts 12, no. 10: 1280. https://doi.org/10.3390/catal12101280