
Thermodynamic and Kinetic Study of Carbon Dioxide Hydrogenation on the Metal-Terminated Tantalum-Carbide (111) Surface: A DFT Calculation

 , ,
, ,
Abstract
:1. Introduction
2. Computational Details
2.1. Method
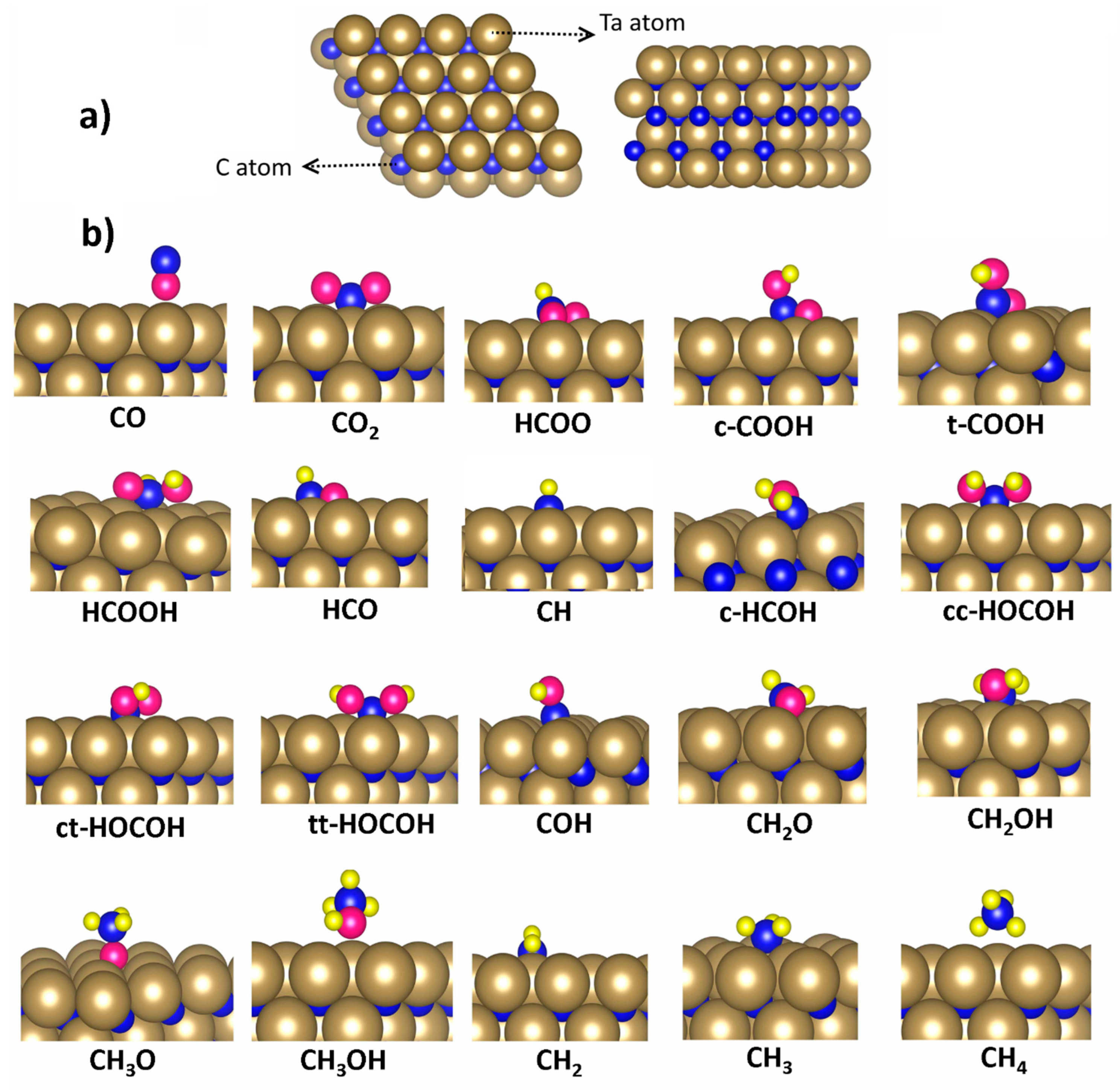
2.2. Model
3. Results and Discussion
3.1. Adsorption Configurations and Energies on TaC (111) Surface
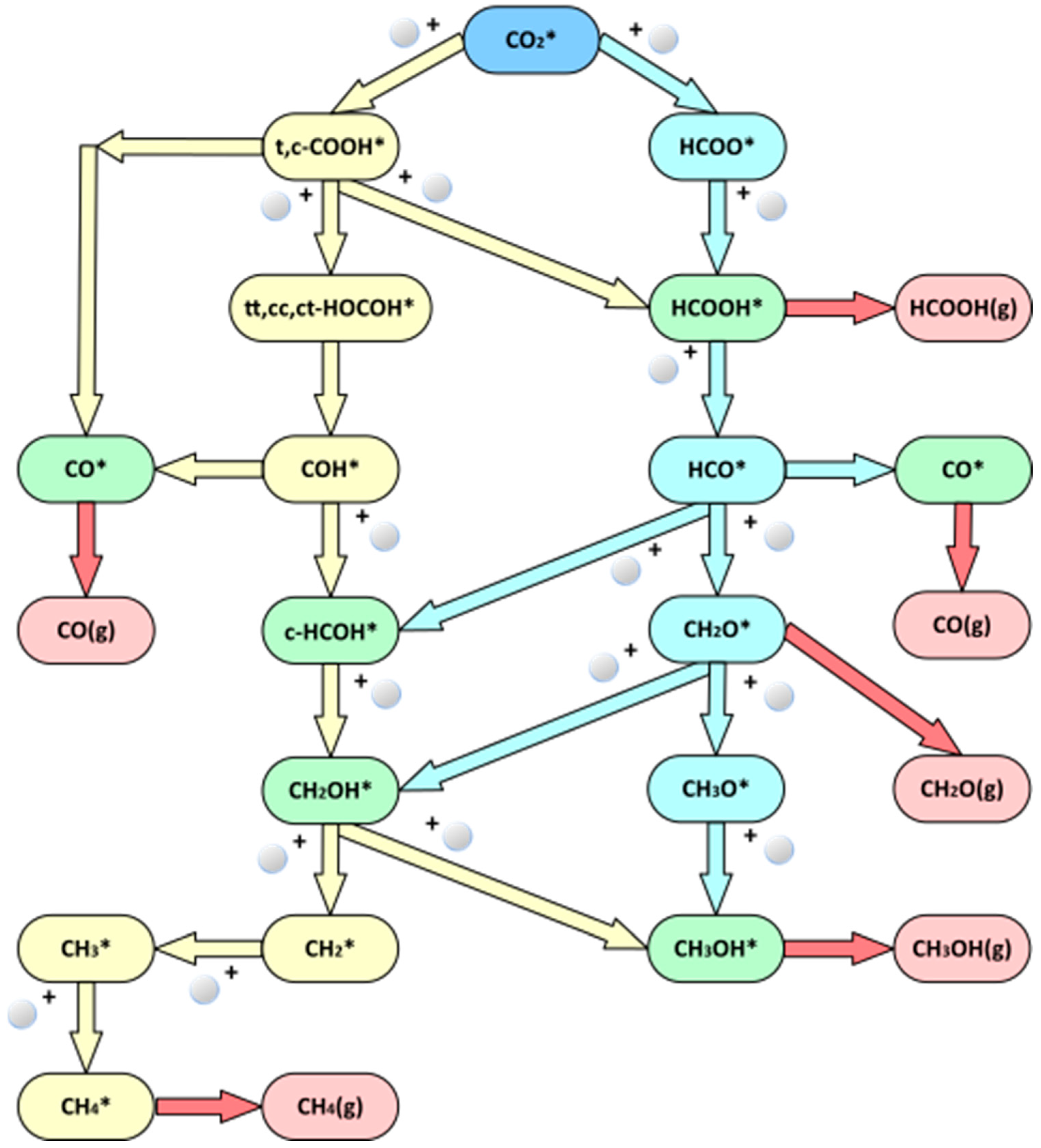
3.2. Reaction Networks of CO2 Hydrogenation
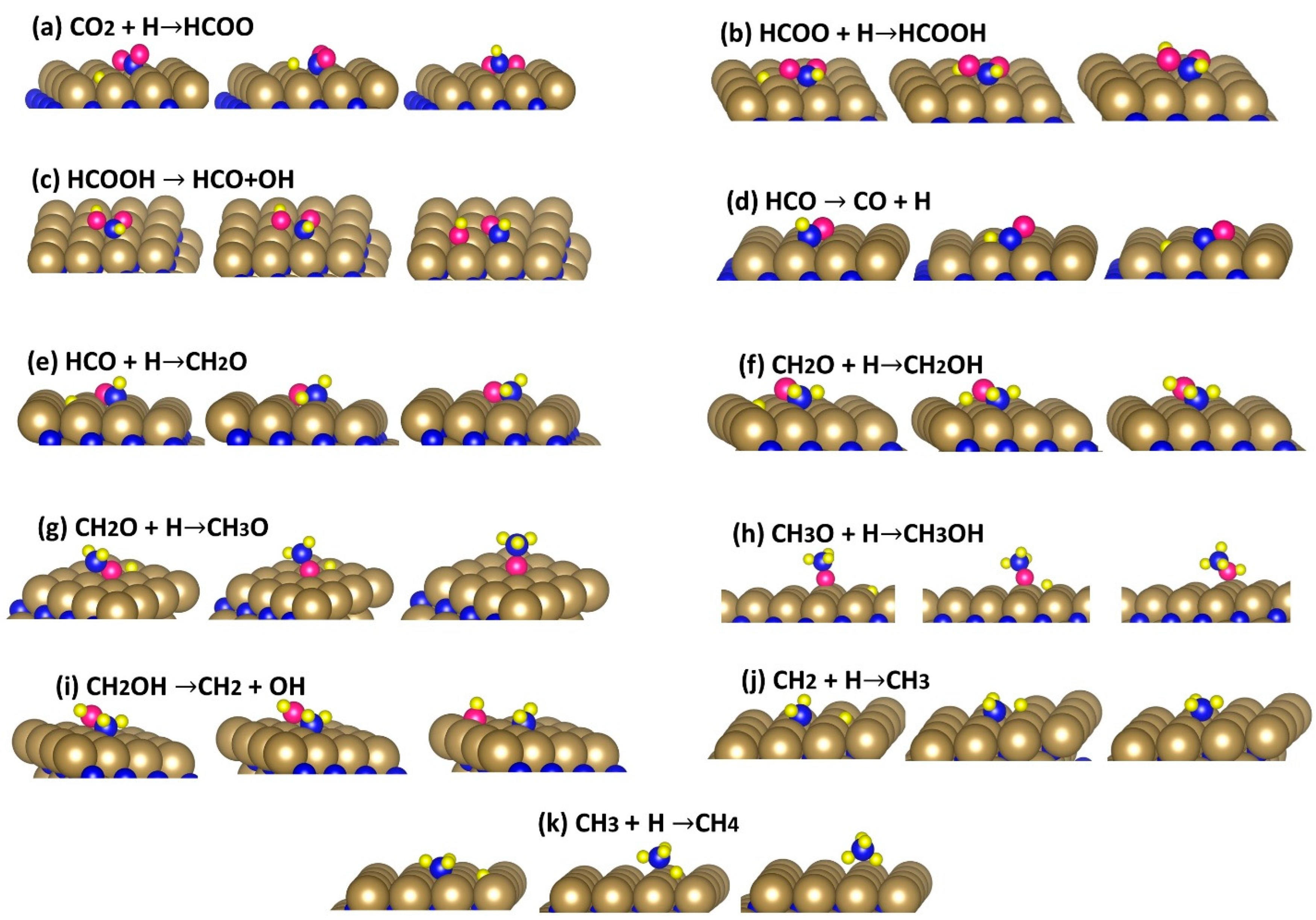
3.3. Chemical Reactions
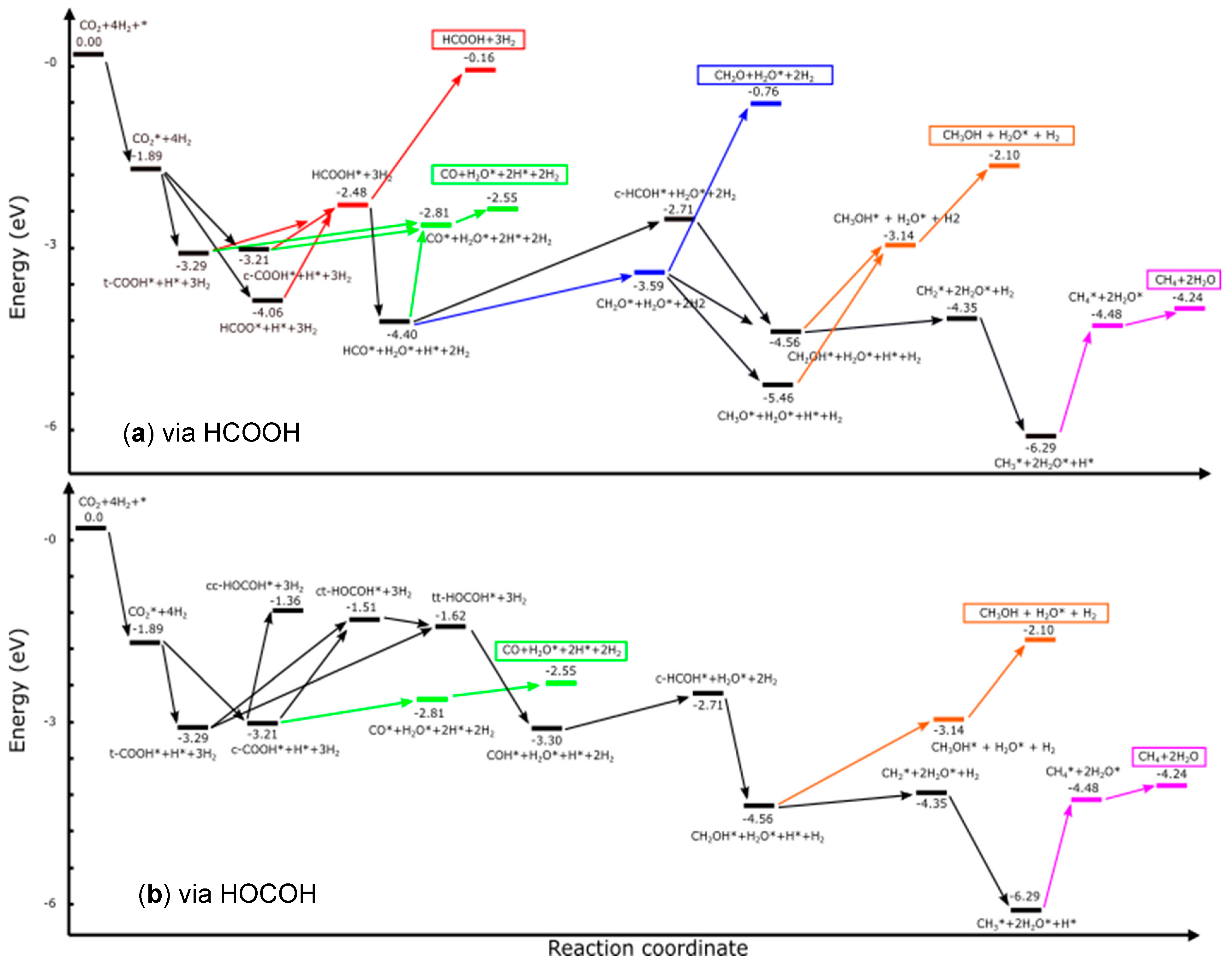
3.3.1. Reaction Pathways of CO2 to Possible Products through HCOOH
The Reaction Mechanisms for CO2 Hydrogenation to HCOOH
The Reaction Paths for CO2 Hydrogenation to CO via HCOOH
The Reaction Paths for the CO2 Hydrogenation to CH2O via HCOOH
The Reaction Paths for CO2 Hydrogenation to CH3OH via HCOOH
The Reaction Paths for CO2 Hydrogenation to CH4 via HCOOH
3.3.2. Reaction Pathways of CO2 to Possible Products through HOCOH
The Reaction Paths for CO2 Hydrogenation to CO via HOCOH
The Reaction Paths for CO2 Hydrogenation to CH3OH via HOCOH
The Reaction Paths for CO2 Hydrogenation to CH4 via HOCOH
3.4. Activation Energies
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Imyen, T.; Znoutine, E.; Suttipat, D.; Iadrat, P.; Kidkhunthod, P.; Bureekaew, S.; Wattanakit, C. Methane Utilization to Methanol by a Hybrid Zeolite@ Metal–Organic Framework. ACS Appl. Mater. Interfaces 2020, 12, 23812–23821. [Google Scholar] [CrossRef] [PubMed]
- Grasemann, M.; Laurenczy, G. Formic acid as a hydrogen source–recent developments and future trends. Energy Environ. Sci. 2012, 5, 8171–8181. [Google Scholar] [CrossRef]
- Ou, Z.; Qin, C.; Niu, J.; Zhang, L.; Ran, J. A comprehensive DFT study of CO2 catalytic conversion by H2 over Pt-doped Ni catalysts. Int. J. Hydrog. Energy 2019, 44, 819–834. [Google Scholar] [CrossRef]
- Olah, G.A. Beyond oil and gas: The methanol economy. Angew. Chem. Int. Ed. 2005, 44, 2636–2639. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, S.G.; Vaidya, P.D.; Bhanage, B.M.; Joshi, J.B. Catalytic carbon dioxide hydrogenation to methanol: A review of recent studies. Chem. Eng. Res. Des. 2014, 92, 2557–2567. [Google Scholar] [CrossRef]
- Fiedler, E.; Grossmann, G.; Kersebohm, D.B.; Weiss, G.; Witte, C. Methanol. Ullmann’s Encycl. Ind. Chem. 2000, 21, 611–635. [Google Scholar]
- Mostaghimi, A.H.B.; Al-Attas, T.A.; Kibria, M.G.; Siahrostami, S. A review on electrocatalytic oxidation of methane to oxygenates. J. Mater. Chem. A 2020, 8, 15575–15590. [Google Scholar] [CrossRef]
- Gerberich, H.R.; Seaman, G.C. Formaldehyde. Kirk-Othmer Encycl. Chem. Technol. 2000, 11, 929–951. [Google Scholar]
- Zoller, B.; Zapp, J.; Huy, P.H. Rapid Organocatalytic Formation of Carbon Monoxide: Application towards Carbonylative Cross Couplings. Chem. Eur. J. 2020, 26, 9632. [Google Scholar]
- Dzade, N.Y.; de Leeuw, N.H. Activating the FeS (001) surface for CO2 adsorption and reduction through the formation of sulfur vacancies: A DFT-D3 study. Catalysts 2021, 11, 127. [Google Scholar] [CrossRef]
- Tafreshi, S.S.; Moshfegh, A.Z.; de Leeuw, N.H. Mechanism of Photocatalytic Reduction of CO2 by Ag3PO4 (111)/g-C3N4 Nanocomposite: A First-Principles Study. J. Phys. Chem. C 2019, 123, 22191–22201. [Google Scholar] [CrossRef]
- Porosoff, M.D.; Kattel, S.; Li, W.; Liu, P.; Chen, J.G. Identifying trends and descriptors for selective CO2 conversion to CO over transition metal carbides. Chem. Commun. 2015, 51, 6988–6991. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.-T.; De Smet, G.; Liao, Y.; Sun, R.; Zhou, C.; Beller, M.; Maes, B.U.W.; Sels, B.F. Homogeneous and heterogeneous catalysts for hydrogenation of CO2 to methanol under mild conditions. Chem. Soc. Rev. 2021, 50, 4259–4298. [Google Scholar] [CrossRef] [PubMed]
- Viñes, F.; Sousa, C.; Liu, P.; Rodriguez, J.; Illas, F. A systematic density functional theory study of the electronic structure of bulk and (001) surface of transition-metals carbides. J. Chem. Phys. 2005, 122, 174709. [Google Scholar] [CrossRef] [Green Version]
- Kitchin, J.R.; Nørskov, J.K.; Barteau, M.A.; Chen, J.G. Trends in the chemical properties of early transition metal carbide surfaces: A density functional study. Catal. Today 2005, 105, 66–73. [Google Scholar] [CrossRef]
- Hugosson, H.W.; Eriksson, O.; Jansson, U.; Ruban, A.V.; Souvatzis, P.; Abrikosov, I. Surface energies and work functions of the transition metal carbides. Surf. Sci. 2004, 557, 243–254. [Google Scholar] [CrossRef]
- Sharma, B.I.; Maibam, J.; Paul, R.; Thapa, R.; Singh, R.B. Studies on energy band structure of NbC and NbN using DFT. Indian J. Phys. 2010, 84, 671–674. [Google Scholar] [CrossRef]
- Gilles, R.; Mukherji, D.; Karge, L.; Strunz, P.; Beran, P.; Barbier, B.; Kriele, A.; Hofmann, M.; Eckerlebe, H.; Rösler, J. Stability of TaC precipitates in a Co-Re-based alloy being developed for ultra-high-temperature applications. J. Appl. Crystallogr. 2016, 49, 1253–1265. [Google Scholar] [CrossRef]
- Hocker, S.; Lipp, H.; Schmauder, S.; Bakulin, A.V.; Kulkova, S.E. Ab initio investigation of Co/TaC interfaces. J. Alloy. Compd. 2021, 853, 156944. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Evans, J.; Feria, L.; Vidal, A.B.; Liu, P.; Nakamura, K.; Illas, F. CO2 hydrogenation on Au/TiC, Cu/TiC, and Ni/TiC catalysts: Production of CO, methanol, and methane. J. Catal. 2013, 307, 162–169. [Google Scholar] [CrossRef]
- Quesne, M.G.; Roldan, A.; de Leeuw, N.H.; Catlow, C.R.A. Bulk and surface properties of metal carbides: Implications for catalysis. Phys. Chem. Chem. Phys. 2018, 20, 6905–6916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunkel, C.; Vines, F.; Illas, F. Transition metal carbides as novel materials for CO2 capture, storage, and activation. Energy Environ. Sci. 2016, 9, 141–144. [Google Scholar] [CrossRef] [Green Version]
- Silveri, F.; Quesne, M.G.; Roldan, A.; De Leeuw, N.H.; Catlow, C.R.A. Hydrogen adsorption on transition metal carbides: A DFT study. Phys. Chem. Chem. Phys. 2019, 21, 5335–5343. [Google Scholar] [CrossRef] [PubMed]
- Levy, R.B.; Boudart, M. Platinum-Like Behavior of Tungsten Carbide in Surface Catalysis. Science 1973, 181, 547–549. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Kunkel, C.; Ramírez de la Piscina, P.; Homs, N.; Viñes, F.; Illas, F. Effective and Highly Selective CO Generation from CO2 Using a Polycrystalline α-Mo2C Catalyst. ACS Catal. 2017, 7, 4323–4335. [Google Scholar] [CrossRef] [Green Version]
- Posada-Pérez, S.; Ramírez, P.J.; Gutiérrez, R.A.; Stacchiola, D.J.; Viñes, F.; Liu, P.; Illas, F.; Rodriguez, J.A. The conversion of CO2 to methanol on orthorhombic β-Mo 2 C and Cu/β-Mo 2 C catalysts: Mechanism for admetal induced change in the selectivity and activity. Catal. Sci. Technol. 2016, 6, 6766–6777. [Google Scholar] [CrossRef]
- Quesne, M.G.; Roldan, A.; de Leeuw, N.H.; Catlow, C.R.A. Carbon dioxide and water co-adsorption on the low-index surfaces of TiC, VC, ZrC and NbC: A DFT study. Phys. Chem. Chem. Phys. 2019, 21, 10750–10760. [Google Scholar] [CrossRef] [Green Version]
- Posada-Pérez, S.; Ramírez, P.J.; Evans, J.; Viñes, F.; Liu, P.; Illas, F.; Rodriguez, J.A. Highly active Au/δ-MoC and Cu/δ-MoC catalysts for the conversion of CO2: The metal/C ratio as a key factor defining activity, selectivity, and stability. J. Am. Chem. Soc. 2016, 138, 8269–8278. [Google Scholar] [CrossRef] [Green Version]
- Sarabadani Tafreshi, S.; Ranjbar, M.; Taghizade, N.; Panahi, S.F.K.S.; Jamaati, M.; de Leeuw, N.H. A first-principles study of CO2 hydrogenation on Niobium-terminated NbC (111) surface. ChemPhysChem 2022, 23, e202100781. [Google Scholar] [CrossRef]
- Xu, W.; Ramírez, P.J.; Stacchiola, D.; Brito, J.L.; Rodriguez, J.A. The Carburization of Transition Metal Molybdates (MxMoO4, M = Cu, Ni or Co) and the Generation of Highly Active Metal/Carbide Catalysts for CO2 Hydrogenation. Catal. Lett. 2015, 145, 1365–1373. [Google Scholar] [CrossRef]
- Li, N.; Chen, X.; Ong, W.-J.; MacFarlane, D.R.; Zhao, X.; Cheetham, A.K.; Sun, C. Understanding of Electrochemical Mechanisms for CO2 Capture and Conversion into Hydrocarbon Fuels in Transition-Metal Carbides (MXenes). ACS Nano 2017, 11, 10825–10833. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Yang, H.; Gao, P.; Chen, X.; Liu, H.; Zhong, L.; Wang, H.; Wei, W.; Sun, Y. Effect of alkali metals on the performance of CoCu/TiO2 catalysts for CO2 hydrogenation to long-chain hydrocarbons. Chin. J. Catal. 2018, 39, 1294–1302. [Google Scholar] [CrossRef]
- Posada-Pérez, S.; Viñes, F.; Ramirez, P.J.; Vidal, A.B.; Rodriguez, J.A.; Illas, F. The bending machine: CO2 activation and hydrogenation on δ-MoC(001) and β-Mo2C(001) surfaces. Phys. Chem. Chem. Phys. 2014, 16, 14912–14921. [Google Scholar] [CrossRef]
- Porosoff, M.D.; Yang, X.; Boscoboinik, J.A.; Chen, J.G. Molybdenum Carbide as Alternative Catalysts to Precious Metals for Highly Selective Reduction of CO2 to CO. Angew. Chem. 2014, 126, 6823–6827. [Google Scholar] [CrossRef]
- Morales-García, Á.; Fernández-Fernández, A.; Viñes, F.; Illas, F. CO2 abatement using two-dimensional MXene carbides. J. Mater. Chem. A 2018, 6, 3381–3385. [Google Scholar] [CrossRef]
- Morales-García, Á.; Mayans-Llorach, M.; Viñes, F.; Illas, F. Thickness biased capture of CO2 on carbide MXenes. Phys. Chem. Chem. Phys. 2019, 21, 23136–23142. [Google Scholar] [CrossRef] [PubMed]
- Hwu, H.H.; Chen, J.G. Surface Chemistry of Transition Metal Carbides. Chem. Rev. 2005, 105, 185–212. [Google Scholar] [CrossRef] [PubMed]
- Prats, H.; Stamatakis, M. Atomistic and electronic structure of metal clusters supported on transition metal carbides: Implications for catalysis. J. Mater. Chem. A 2022, 10, 1522–1534. [Google Scholar] [CrossRef]
- Johansson, L.I. Electronic and structural properties of transition-metal carbide and nitride surfaces. Surf. Sci. Rep. 1995, 21, 177–250. [Google Scholar] [CrossRef]
- Aizawa, T.; Souda, R.; Otani, S.; Ishizawa, Y.; Oshima, C. Bond softening in monolayer graphite formed on transition-metal carbide surfaces. Phys. Rev. B 1990, 42, 11469–11478. [Google Scholar] [CrossRef]
- Hulbert, S.L.; Kao, C.C.; Garrett, R.F.; Bartynski, R.A.; Yang, S.; Weinert, M.; Jensen, E.; Zehner, D.M. A comparison of the surface electronic structure of Ta(100) and TaC(111) using Auger-photoelectron coincidence spectroscopy. J. Vac. Sci. Technol. A 1991, 9, 1919–1923. [Google Scholar] [CrossRef]
- Zaima, S.; Shibata, Y.; Adachi, H.; Oshima, C.; Otani, S.; Aono, M.; Ishizawa, Y. Atomic chemical composition and reactivity of the TiC(111) surface. Surf. Sci. 1985, 157, 380–392. [Google Scholar] [CrossRef]
- Mavrikakis, M.; Hammer, B.; Nørskov, J.K. Effect of Strain on the Reactivity of Metal Surfaces. Phys. Rev. Lett. 1998, 81, 2819–2822. [Google Scholar] [CrossRef]
- Botana, A.S.; Norman, M.R. Electronic structure and magnetism of transition metal dihalides: Bulk to monolayer. Phys. Rev. Mater. 2019, 3, 044001. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Perdew, J.; Burke, K.; Ernzerhof, M. Perdew, burke, and ernzerhof reply. Phys. Rev. Lett. 1998, 80, 891. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Available online: https://icsd.products.fiz-karlsruhe.de/ (accessed on 3 September 2022).
- Heyden, A.; Bell, A.T.; Keil, F.J. Efficient methods for finding transition states in chemical reactions: Comparison of improved dimer method and partitioned rational function optimization method. J. Chem. Phys. 2005, 123, 224101. [Google Scholar] [CrossRef] [Green Version]
- Henkelman, G.; Jónsson, H. A dimer method for finding saddle points on high dimensional potential surfaces using only first derivatives. J. Chem. Phys. 1999, 111, 7010–7022. [Google Scholar] [CrossRef]
- Vojvodic, A.; Ruberto, C.; Lundqvist, B.I. Atomic and molecular adsorption on transition-metal carbide (111) surfaces from density-functional theory: A trend study of surface electronic factors. J. Phys. Condens. Matter 2010, 22, 375504. [Google Scholar] [CrossRef]
- Tafreshi, S.S.; Roldan, A.; de Leeuw, N.H. Density functional theory calculations of the hydrazine decomposition mechanism on the planar and stepped Cu(111) surfaces. Phys. Chem. Chem. Phys. 2015, 17, 21533–21546. [Google Scholar] [CrossRef] [Green Version]
- López, M.; Viñes, F.; Nolan, M.; Illas, F. Predicting the Effect of Dopants on CO2 Adsorption in Transition Metal Carbides: Case Study on TiC (001). J. Phys. Chem. C 2020, 124, 15969–15976. [Google Scholar] [CrossRef]
- Xu, W.; Ramirez, P.J.; Stacchiola, D.; Rodriguez, J.A. Synthesis of α-MoC1-x and β-MoCy Catalysts for CO2 Hydrogenation by Thermal Carburization of Mo-oxide in Hydrocarbon and Hydrogen Mixtures. Catal. Lett. 2014, 144, 1418–1424. [Google Scholar] [CrossRef]
- Kunkel, C.; Viñes, F.; Ramírez, P.J.; Rodriguez, J.A.; Illas, F. Combining Theory and Experiment for Multitechnique Characterization of Activated CO2 on Transition Metal Carbide (001) Surfaces. J. Phys. Chem. C 2019, 123, 7567–7576. [Google Scholar] [CrossRef]
- Li, Y.; Chen, Y.; Guo, Z.; Tang, C.; Sa, B.; Miao, N.; Zhou, J.; Sun, Z. Breaking the linear scaling relations in MXene catalysts for efficient CO2 reduction. Chem. Eng. J. 2022, 429, 132171. [Google Scholar] [CrossRef]
- Qi, K.-Z.; Wang, G.-C.; Zheng, W.-J. A first-principles study of CO hydrogenation into methane on molybdenum carbides catalysts. Surf. Sci. 2013, 614, 53–63. [Google Scholar] [CrossRef]
- Shi, X.-R.; Jiao, H.; Hermann, K.; Wang, J. CO hydrogenation reaction on sulfided molybdenum catalysts. J. Mol. Catal. A Chem. 2009, 312, 7–17. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Eads (eV) | Bond Length (Å) | Species | Eads (eV) | Bond Length (Å) |
|---|---|---|---|---|---|
| CO | −0.226 | d*Ta-C = 2.27, dO-C = 1.15 | c-HCOH | −0.458 | d*Ta-O = 2.21, dO-C = 1.51 |
| CO2 | −1.891 | d*Ta-O = 2.21, dTa-C = 2.27 | cc-HOCOH | −0.589 | d*Ta-O = 2.29, dO-C = 1.45 |
| HCOO | −0.477 | d*Ta-O = 2.14, d*Ta-O = 2.27, dO-C = 1.41 | ct-HOCOH | −0.739 | d*Ta-O = 2.27, dO-C = 1.46 |
| c-COOH | −0.620 | d*Ta-O = 2.23, d*Ta-O = 2.22, dO-C = 1.37, dO-C = 1.34 | tt-HOCOH | −0.850 | d*Ta-O = 2.25, dO-C = 1.46 |
| t-COOH | −0.702 | d*Ta-O = 2.21, d*Ta-O = 2.22, dO-C = 1.37, dO-C = 1.34 | COH | −0.233 | d*Ta-C = 2.05, dO-C = 1.36 |
| HCOOH | −0.711 | d*Ta-O = 2.06, dO-C = 1.49, dO-C = 1.35 | CH2O | −0.335 | d*Ta-O = 2.20, dO-C = 1.42 |
| HCO | −0.330 | d*Ta-O = 2.18, d*Ta-O = 2.18, d*Ta-O = 4.18, dO-C = 1.43 | CH2OH | −0.752 | d*Ta-O = 2.23, dO-C = 1.15 |
| CH2 | −0.615 | d*Ta-C = 2.23 | CH3O | −0.645 | d*Ta-O = 2.24, dO-C = 1.45 |
| CH3 | −0.998 | d*Ta-C = 2.42, dH-C = 1.13 | CH3OH | −0.141 | d*Ta-O = 2.26, dO-C = 1.45 |
| CH4 | −0.095 | dTa-C = 2.80 |
| Elementary Reactions | ∆E (eV) | Elementary Reactions | ∆E (eV) |
|---|---|---|---|
| CO2 (g) → CO2* | −1.891 | c-COOH* + H* → cc-HOCOH* | 1.845 |
| CO2* + H* → HCOO* | 0.198 | t-COOH* + H* → tt-HOCOH* | 1.666 |
| CO2* + H* → c-COOH* | 1.056 | c-COOH* + H* → ct-HOCOH* | 1.695 |
| CO2* + H* → t-COOH* | 0.974 | t-COOH* + H* → ct-HOCOH* | 1.777 |
| c-COOH* → t-COOH* | −0.082 | cc-HOCOH* + H* → tt-HOCOH* | −0.261 |
| HCOO* + H* → HCOOH* | 1.580 | ct-HOCOH* + H* → tt-HOCOH* | −0.111 |
| c-COOH* → CO* + OH* | 0.702 | tt-HOCOH* → COH* + OH* | −1.375 |
| t-COOH* → CO* + OH* | 0.784 | ct-HOCOH* → COH* + OH* | −1.486 |
| c-COOH* + H* → HCOOH* | 0.723 | cc-HOCOH* → COH* + OH* | −1.636 |
| t-COOH* + H* → HCOOH* | 0.805 | COH* → CO* + H* | 0.493 |
| HCOOH* → HCO* + OH* | −1.611 | COH* + H* → c-HCOH* | 0.589 |
| HCOOH* → HCOOH(g) | 2.320 | c-HCOH* + H* → CH2OH* | 0.520 |
| HCO* + H* → c-HCOH* | 1.687 | CH2OH* + H* → CH3OH* | 1.424 |
| HCO* → CO* + H* | 1.590 | CH2OH* → CH2* + OH* | −1.855 |
| CO* → CO(g) | 0.263 | CH2* + H* → CH3* | 0.431 |
| HCO* + H* → CH2O* | 0.809 | CH3* + H* → CH4* | 1.818 |
| CH2O* → CH2O(g) | 2.834 | CH4* → CH4 (g) | 0.240 |
| CH2O* + H* → CH3O* | 0.504 | CH3OH* → CH3OH (g) | 1.037 |
| CH2O* + H* → CH2OH* | 1.398 | H2O* → H2O (g) | 0.894 |
| CH3O* + H* → CH3OH* | 2.318 |
| Elementary Reactions | Eb (eV) | Elementary Reactions | Eb (eV) |
|---|---|---|---|
| CO2* + H* → HCOO* | 0.808 | CH2O* + H* → CH3O* | 1.282 |
| HCOO* + H* → HCOOH* | 1.828 | CH3O* + H* → CH3OH* | 1.429 |
| HCOOH* → HCO* + OH* | 0.264 | CH2OH* → CH2* + OH* | 0.126 |
| HCO* → CO* + H* | 0.614 | CH2* + H* → CH3* | 1.210 |
| HCO* + H* → CH2O* | 1.304 | CH3* + H* → CH4* | 2.224 |
| CH2O* + H* → CH2OH* | 1.480 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarabadani Tafreshi, S.; Panahi, S.F.K.S.; Taghizade, N.; Jamaati, M.; Ranjbar, M.; de Leeuw, N.H. Thermodynamic and Kinetic Study of Carbon Dioxide Hydrogenation on the Metal-Terminated Tantalum-Carbide (111) Surface: A DFT Calculation. Catalysts 2022, 12, 1275. https://doi.org/10.3390/catal12101275
Sarabadani Tafreshi S, Panahi SFKS, Taghizade N, Jamaati M, Ranjbar M, de Leeuw NH. Thermodynamic and Kinetic Study of Carbon Dioxide Hydrogenation on the Metal-Terminated Tantalum-Carbide (111) Surface: A DFT Calculation. Catalysts. 2022; 12(10):1275. https://doi.org/10.3390/catal12101275
Chicago/Turabian StyleSarabadani Tafreshi, Saeedeh, S. Fatemeh. K. S. Panahi, Narges Taghizade, Maryam Jamaati, Mahkameh Ranjbar, and Nora H. de Leeuw. 2022. "Thermodynamic and Kinetic Study of Carbon Dioxide Hydrogenation on the Metal-Terminated Tantalum-Carbide (111) Surface: A DFT Calculation" Catalysts 12, no. 10: 1275. https://doi.org/10.3390/catal12101275